

Abstract
Six1 is a critical regulator of embryonic development that requires interaction with the Eya family of proteins (Eya1-4) to activate the transcription of genes involved in neurogenesis, myogenesis, and nephrogenesis. While expression of Six1 and Eya family members is predominantly observed in development, their overexpression is observed in numerous cancers. Importantly, both Six1 and Eya have independently been shown to mediate breast cancer metastasis, but whether they functionally interact during tumor progression has not been explored. Herein we demonstrate that knockdown of Eya2 in MCF7 mammary carcinoma cells reverses the ability of Six1 to induce TGF-β signaling, as well as to induce characteristics associated with epithelial-mesenchymal transition (EMT) and cancer stem cells (CSCs), suggesting that Six1 is dependent on Eya2 to mediate numerous pro-metastatic characteristics. The importance of the Six1/Eya interaction in human breast cancer is underscored by the finding that high levels of Six1 correlate with shortened time to relapse and metastasis as well as decreased survival only when co-expressed with high levels of Eya2. Overall, these data implicate Eya2 as a necessary cofactor for many of the metastasis promoting functions of Six1, suggesting that targeting the Six1/Eya interaction may inhibit breast cancer progression. Since Six1 and Eya2 are not highly expressed in most adult tissues, the Six1-Eya interaction may be a valuable future therapeutic target whose inhibition would be expected to impair breast cancer progression while conferring limited side effects.
Keywords: Six1, Eya, TGF-β, epithelial-mesenchymal transition, cancer stem cells
Introduction
Many processes necessary for early embryonic development are recapitulated in cancer, often as a result of homeobox gene re-expression (Cillo et al., 1999; Ford, 1998; Samuel and Naora, 2005). Such inappropriate expression of homeobox genes, which encode transcription factors, allows for the acquisition of early developmental phenotypes, such as proliferation, survival, invasion and migration in adult cells, thereby contributing to tumorigenesis and/or tumor progression (Abate-Shen, 2002). The Six1 homeobox gene plays a critical role in the development of a number of organs via promoting proliferation, survival, migration, and invasion of precursor cell populations (Laclef et al., 2003a; Laclef et al., 2003b; Xu et al., 2003; Li et al., 2003). In muscle development, Six1 contributes to the delamination and migration of myogenic precursor cells from the dermomyotome to the developing limb, in a process that entails an epithelial-mesenchymal transition (EMT)(Laclef et al., 2003a; Laclef et al., 2003b; Xu et al., 2003). In addition to the role of Six1 in development, recent work has identified misexpression of Six1 in numerous cancers including breast (Ford et al., 1998; Reichenberger et al., 2005), ovarian (Behbakht et al., 2007), cervical (Micalizzi et al., 2009), hepatocellular carcinoma (Ng et al., 2006), as well as Wilms’ tumors (Li et al., 2002) and alveolar rhabdomyosarcomas(Yu et al., 2004). Importantly, Six1 overexpression in cancer recapitulates the pro-proliferative, pro-survival, and pro-migratory phenotypes attributed to Six1 in development (Behbakht et al., 2007; Coletta et al., 2004; Micalizzi et al., 2009; Yu et al., 2004).
Six1 belongs to the evolutionarily conserved Six1-Eya-Dach transcriptional regulatory network that is critical during embryonic development. Members of the Eya family of Six1 coactivators (Eya1-4) play critical roles in Six1-mediated transcriptional activation. Six1 and Eya1 knockout mice phenocopy each other, and both molecules are necessary for the proper development of ears, muscle, kidney, thymus, and sensory neurons, as well as for overall neonatal survival (Ando et al., 2005; Laclef et al., 2003b; Li et al., 2003; Xu et al., 2003; Xu et al., 2002). The phenotypes observed in both KO mice are, at least in-part, due to poor progenitor cell proliferation and survival, underscoring the role of the Six1-Eya complex in the expansion of progenitor cell populations during normal development. The role of the Six1-Eya complex in human disease is also clear, as both Six1 and Eya1 mutations can be found in branchio-oto-renal syndrome, where mutations disrupt individual protein function, Six1-DNA binding, and Six1-Eya1 binding (Abdelhak et al., 1997a; Abdelhak et al., 1997b; Kochhar et al., 2008; Orten et al., 2008; Patrick et al., 2009; Ruf et al., 2004; Vincent et al., 1997). Additionally, Eya4 mutations are observed in sensorineural hearing loss (SNHL) (Schonberger et al., 2005; Wayne et al., 2001; Zhang et al., 2004) where Eya4 function and Six1-Eya4 interactions are lost. Together, data from both mouse models and human disease underscore the critical developmental function of the Six1-Eya interaction.
Although not highly expressed in most adult tissues, Six1 is misexpressed in multiple cancers and re-instates a pro-proliferative and pro-survival phenotype(Coletta et al., 2004; Yu et al., 2006). In breast cancer, Six1 also induces EMT- and stem cell-like phenotypes, the former of which is mediated through upregulating TGF-β signaling (Micalizzi et al., 2009) and is likely critical for Six1-induced metastasis. Six1 correlates with advanced disease in many types of cancer, including breast (Micalizzi et al., 2009), ovarian (Behbakht et al., 2007), and alveolar rhabdomyosarcomas (Yu et al., 2004). Overexpression of Eya1 and Eya2 family members have been identified in many of the same cancers as Six1(Li et al., 2002; Zhang et al., 2005). In ovarian cancer, two independent studies have demonstrated that Six1 and Eya2 each correlate with poor patient survival in advanced disease (Behbakht et al., 2007; Zhang et al., 2005); however, as of yet, the role of Six1 and Eya together in cancer has not been studied. Importantly, Eya was recently shown to increase proliferation, migration, invasion, transformation, and metastasis in mammary carcinoma cells (Pandey et al., 2010); suggesting that Eyas, like Six1, play an important role in breast cancer and that they may in fact cooperate with Six1 to confer pro-proliferative and migratory phenotypes. While a non-nuclear function of Eya was postulated to mediate its pro-tumorigenic phenotypes (Pandey et al., 2010), no formal experiments were conducted to test the relevance of the interaction between Six1 and Eya in breast tumorigenesis and/or metastasis.
In this study, we demonstrate a cooperative role between Six1 and Eya in mediating phenotypes associated with breast tumorigenesis and metastasis. Knockdown of Eya2 in Six1 overexpressing MCF7 mammary carcinoma cells reverses Six1-mediated induction of TGF-β signaling, as well as the ability of Six1 to induce EMT and an increase in cancer stem cell characteristics. The relevance of the Six1-Eya interaction in human breast cancer is underscored in gene expression datasets, where only co-expression of Six1 and Eya can predict poor patient prognosis. Taken together, these data strongly support the hypothesis that Eya is a critical activator of Six1 in human breast cancer, where it is required to mediate Six1-induced TGF-β-dependent EMT and expansion of the cancer stem-like cell population.
Results
Eya2 is the predominant Eya in MCF7 cells
We have previously shown that Six1 overexpression in the MCF7 mammary carcinoma cell line activates the TGF-β pathway (Micalizzi et al., 2009), leading to a loss of epithelial and gain of mesenchymal characteristics. To determine whether Six1 requires the Eya family of cofactors to mediate these phenotypes, we first examined which Eyas, if any, are expressed in MCF7 cells. Using qRT-PCR with plasmid standard curves and primers that specifically recognize individual Eya members, we determined the mRNA copy number of each Eya in MCF7 cells overexpressing Six1 (MCF7-Six1) and control (MCF7-Ctrl). Importantly, we identified Eya2 as the predominant Eya family member in MCF7 cells, exhibiting 5-fold greater expression than Eya3. In contrast, Eyas 1 and 4 were absent or expressed only at very low levels (Fig1a). To determine the dependence of Six1 phenotypes on Eya2 function, we stably and specifically knocked down Eya2 in MCF7-Six1 cells using two independent shRNA sequences. Independent control cell lines were also established in which a scramble shRNA control was introduced into MCF7-Six1 and MCF7-Ctrl cells. Two clonal isolates were propagated from each shRNA in each line to control for insertion site effects. Eya2 mRNA and protein levels were determined in each clone (Fig1b and c), and on average, we achieved a 75% knockdown.
Figure 1. Eya2 is the predominant Eya in MCF7 cells and can be efficiently knocked-down in MCF7-Six1 cells.

(a) Eya2 is the most abundant Eya family member in MCF7 cells. Quantitative real time PCR of Eyas1-4, indicating comparative Eya mRNA copy number in MCF7-Six1 or MCF7-Ctrl clones. Error bars represent the standard error of the mean of 3 grouped MCF7-Six1 or MCF7-Ctrl clones from a single RNA isolation. Data is a representative image of triplicate RNA isolations. (b) Real time PCR and (c) immunocytochemistry for Eya2 performed on MCF7 cells stably transfected with Eya2 shRNA or scramble shRNA, and then clonally selected. shRNA-a and shRNA-b are separate clonal isolates containing shRNA constructs targeting different regions of Eya2. Two clonal MCF7-Six1/shRNA isolates from each shRNA group were chosen for analysis. Error bars indicate the standard deviation of the mean of duplicate RNA isolations.
Eya2 knockdown in MCF7-Six1 cells reverses the ability of Six1 to induce TGF-β signaling
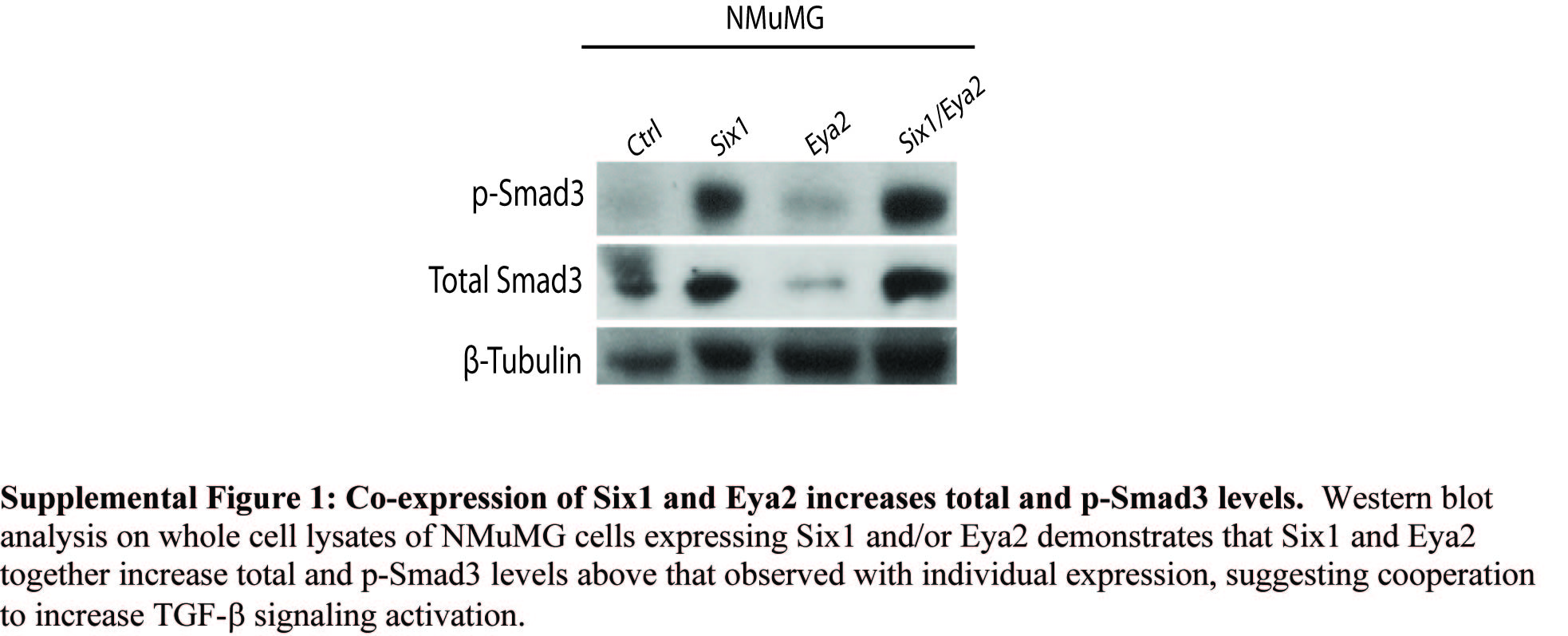
We have previously shown that Six1 activates the TGF-β signaling pathway when overexpressed in MCF7 cells (Micalizzi et al., 2009; Micalizzi et al., 2010). Using our Eya2 knockdown cells, we examined the dependence of Six1 on Eya2 in mediating increased TGF-β signaling. Because we have observed that Six1 overexpression leads to an increase both in the levels of the Type I TGF-β receptor (TβRI) (Micalizzi et al., 2010) and in total Smad3 levels (Micalizzi et al., 2009), each which may contribute to the overall increase in TGF-β signaling, we first examined whether loss of Eya2 would reverse the Six1-mediated increases in these protein levels. Indeed, Eya2 knockdown reverses the Six1-mediated increase in the levels of TβRI and Smad3 (Fig2b). Further, Eya2 knockdown in MCF7 cells leads to decreased signaling downstream of TGF-β as measured by 3TP-luciferase activity, a measurement of Smad-mediated TGF-β responsive transcription (Fig2c). Finally, co-expression of Six1 and Eya2 increases both total and p-Smad3 levels in NMuMG cells above that observed with expression of Six1 or Eya2 alone, suggesting that the two proteins cooperate to activate TGF-β signaling in multiple contexts (Supplemental Fig1). Together, these data demonstrate that Eya2 is necessary for the ability of Six1 to activate TGF-β signaling.
Figure 2. Eya2 knockdown in MCF7-Six1 cells reverses Six1-induced TGF-β signaling.

MCF7-Six1/Eya2 shRNA clones have decreased levels of (a) TβR1 and (b) total Smad3. (c) MCF7-Six1/Eya2 shRNA clones exhibit decreased TGF-β-responsive transcription compared to MCF7-Six1/scrambled controls as observed by performing luciferase activity assays with the 3TP reporter construct. All transfections were normalized to renilla luciferase activity. Data points show the mean of two individual clones and error bars represent the standard error of the mean for 2 experiments. P values represent unpaired t test statistical analysis.
Eya2 knockdown partially reverses the ability of Six1to induce EMT
In both cell culture and animal models, Six1 overexpression results in the loss of the epithelial phenotype with a concomitant gain in the mesenchymal phenotype (McCoy et al., 2009; Micalizzi et al., 2009). We have previously shown that this EMT requires Six1-induced TGF-β signaling (Micalizzi et al., 2009), and thus asked whether Six1 is also dependent on Eya2 to mediate EMT. In support of the hypothesis that Six1 requires Eya2 to mediate an EMT, knockdown of Eya2 in MCF7-Six1 cells reverses the Six1-induced increase in the mesenchymal marker fibronectin (Fig3a). Additionally, Eya2 is required for the Six1-induced re-localization of E-cadherin and β-catenin away from the insoluble (membranous) fraction and into the soluble (cytoplasmic/nuclear) fraction, a hallmark of EMT (Fig3b and 3c). As expected, the decreased levels of soluble β-catenin in the presence of Eya2 knockdown correlates with decreased Six1-induced β-catenin-responsive transcription as measured by TOP-FLASH reporter activity (Fig3d). These data demonstrate that Eya2 is required for the ability of Six1 to induce and/or maintain features of EMT associated with the induction of mesenchymal properties. However, loss of Eya2 did not restore cytokeratin 18 expression to MCF7 cells, which is normally downregulated in the presence of Six1 (data not shown), nor did it reverse the decrease in cell-matrix adhesion observed with Six1 overexpression (Fig3e). This suggests that either: 1) Eya2 is not globally required for Six1-induced EMT, or 2) that some Six1-induced characteristics of EMT are permanent once induced and no longer dependent on Six1 function.
Figure 3. Eya2 knockdown partially reverses Six1-induced EMT.

Eya2 knockdown in MCF7-Six1 cells reverses the ability of Six1 to (a) upregulate the mesenchymal marker Fibronectin and (b & c) to relocalize E-cadherin and β-catenin from the insoluble (membranous) fraction to the soluble (cytoplasmic) fraction as shown by western blot and quantitation following cell fractionation. (d) MCF7-Six1/Eya2 shRNA clones have decreased β-catenin responsive transcription compared to Six1 scrambled controls. β-catenin transcriptional activity was measured using the TOP-flash luciferase reporter construct normalized to renilla luciferase activity. Data points for fractionation and reporter activity show the mean of two individual clones and error bars represent the standard error of the mean for 2 experiments. P values represent unpaired t test statistical analysis. (e) Eya2 knockdown does not reverse the Six1 induced decrease in cell-matrix adhesion. Relative adherence measured by crystal violet staining.
Six1 is dependent on Eya2 for enhancement of cancer stem/progenitor cell characteristics
We recently demonstrated that Six1 promotes a stem/progenitor cell phenotype when overexpressed in mouse mammary glands and that tumors which arise in these transgenic mice appear to be derived from a stem/progenitor cell population (McCoy et al., 2009; Micalizzi et al., 2009). Additionally, Six1 drives TGF-β signaling and EMT, properties that are associated with cancer stem-like cells (CSCs) (McCoy et al., 2009; Micalizzi et al., 2009; Ouyang et al.; Taylor et al.). Thus, we set out to test whether Eya2, which is required for the ability of Six1 to induce TGF-β signaling and EMT, is also required for the ability of Six1 to induce a cancer stem cell-like phenotype. While Six1 overexpression of MCF7 cells leads to an increase in the population of cells that are CD44+ and CD24−, markers of mammary stem/progenitor cells (Al-Hajj et al., 2003), loss of Eya2 significantly reverses this increase (Fig4a and Supplemental Fig2). Furthermore, knockdown of Eya2 reverses the increase in functional cancer stem-like cells observed with Six1 overexpression, as demonstrated in secondary tumorsphere formation assays (Fig4b). Interestingly, Six1 overexpression not only increases the number of tumorspheres formed by MCF7 cells, but it significantly alters their morphology from a uniformed sphere to a less organized shape, a phenotype that is also reversed with Eya2 knockdown (Fig4c). Together, these results demonstrate that Eya2 is necessary for the ability of Six1 to induce characteristics of cancer stem/progenitor-like cells.
Figure 4. Eya2 knockdown reverses the Six1-induced increase in cancer stem cell characteristics.

(a) Flow cytometric analysis demonstrates loss of CD44+/CD24− cancer stem cells with knockdown of Eya2. (b) Secondary tumorsphere assays, a measurement of self renewal capacity, demonstrate decreased tumorsphere formation and (c) re-gained spherical shape with knockdown of Eya2. Antibodies used to perform flow cytometry include CD24 and CD44, markers found on human epithelial stem cells. Figure is a representative image of two experiments. Data points show the mean of two individual clones and error bars represent the standard error of the mean. P values represent unpaired t test statistical analysis. Original magnification, x200.
Co-expression of Eya2 and Six1 correlates with activated TGF-β signaling in human breast cancer
We previously demonstrated that Six1 induces metastasis via increasing TGF-β signaling. Additionally, we have shown that high levels of Six1 correlate with high levels of activated TGF-β signaling as well as with adverse outcomes in human breast cancer (Micalizzi et al., 2009). Based on our results suggesting that Eya2 is required for Six1-activated TGF-β signaling, we performed immunohistochemical analysis on breast cancer tissue arrays to determine if Six1 is dependent on Eya2 to mediate TGF-β signaling in human breast cancer. We first produced and characterized an Eya2-specific antibody using an N-terminal Eya2 peptide. Importantly, this newly developed antibody specifically recognizes Eya2 as opposed to other Eya family members (Supplemental Fig3). Using tissue arrays containing fifty malignant primary breast tissue samples, immunohistochemical analysis was performed with antibodies against Eya2, Six1, and Smad3, which when nuclear is an indicator of active TGF-β signaling. Following blinded scoring, the data were divided into above the mean and below the mean nuclear staining for Six1 and Eya2. Co-expression of high levels of Six1 and Eya2 significantly correlates with nuclear localized Smad3 (Fig5), supporting a critical cooperative role for Six1 and Eya in mediating TGF-β signaling in human breast cancer.
Figure 5. Co-expression of Eya2 and Six1 correlates with activated TGF-β signaling in human breast cancer.

(a) Representative images of human breast cancer tissue arrays stained with anti-Six1, anti-Eya2, and anti-Smad3 antibodies show that low Six1 and low Eya2 correlate with little nuclear Smad3, whereas tumors that express both high Six1 and Eya2 show high levels of nuclear Smad3. (b) Quantitation of Smad3 staining. Staining of tissues was scored on a scale from 0-4 for each antibody. Each sample was categorized as having above (high) or below (low) the mean Eya2 and/or Six1 staining and values were plotted against the average Smad3 scores within the group. Statistical analysis performed using Anova. Original magnification, x400. Scale bars: 20um.
Co-expression of Six1 and Eya2 correlates with advanced disease and poor prognosis in human breast cancer
Because Six1 requires Eya2 to mediate increased TGF-β signaling and to promote EMT and cancer stem cell-like properties, and because Six1 and Eya2 correlate with activated TGF-β signaling in human breast cancer, which we have previously shown is essential for Six1-induced metastasis, we further examined whether Six1 is dependent on Eya2 to predict poor prognosis in human breast cancer. Using the van de Vijver gene expression dataset, profiling tumor mRNA from 295 women with early-stage invasive breast carcinoma (van ’t Veer et al., 2002), we previously demonstrated that high Six1 levels significantly correlate with poor patient prognosis as measured by shortened time to relapse, shortened time to metastasis, and decreased breast cancer specific survival (McCoy et al., 2009; Micalizzi et al., 2009). However, in this previous analysis, the correlation with Eya2 was not investigated. We now show that high levls of Eya2 also significantly correlate with shortened time to relapse, time to metastasis, and decreased survival (Supplemental Fig4). However, in breast cancer patients whose tumors expressed high Six1 levels in the absence of high Eya2, as well as high Eya2 levels in the absence of high Six1, there is NO correlation with any of these parameters. In contrast, only when tumors express BOTH high levels of Six1 and Eya2 together, is a strong significant correlation found with shortened time to relapse, shortened time to metastasis, and with shortened survival (Fig6). These results can be corroborated in the independent Wang dataset (Wang et al., 2005), consisting of 286 lymph-node negative breast cancer patients (Supplemental Fig5). These data strongly suggest that Six1 and Eya2 cooperate in human breast cancer and reinforce the hypothesis that Eya2 is required for the ability of Six1 to mediate tumor progression.
Figure 6. Coordinate high Six1 and Eya2 expression are required to predict poor prognosis in human breast cancer.

In a gene expression dataset of 295 women with early-stage invasive breast carcinoma (van ’t Veer et al., 2002), patient samples expressing high Six1 in the absence of high Eya2, and high Eya2 in the absence of high Six1, do not correlate with reduced time to metastasis, reduced time to relapse, and shortened breast cancer-specific survival while high Six1 and high Eya2 together in the same patient sample significantly correlates with shortened time to relapse and to metastasis, and with shortened breast cancer specific survival. The mean value for Six1 and/or Eya2 expression was used to divide the samples into high (above the mean) and low (below the mean) Six1 and Eya2 expression. P-values were calculated by log-rank analysis.
As Eya1, Eya2, and Eya4 have all been implicated in cancer, we extended our analysis of the van de Vijver dataset to examine whether any Eya family member is able to cooperate with Six1 in human breast cancer, or whether this cooperation is specific to Eya2. Importantly, in the van de Vijver dataset, Eya1 in conjunction with Six1 also significantly associates with shortened time to relapse and to metastasis, as well as with shortened survival (Fig7), and although not significant, Eya1 does show a cooperative trend with Six1 in the Wang dataset (Supplemental Fig6). In comparison, Eya3 only correlates with survival in the van de Vijver dataset and Eya4 does not correlate with any adverse outcome in conjunction with Six1 in either dataset (data not shown). Together, these data strongly suggest that Eya1 and Eya2 are both able to cooperate with Six1 in human breast cancer.
Figure 7. Coordinate high Six1 and Eya1 expression are required to predict poor prognosis in human breast cancer.

In a gene expression dataset of 295 women with early-stage invasive breast carcinoma (van ’t Veer et al., 2002), patient samples expressing high Six1 in the absence of high Eya1, and high Eya1 in the absence of high Six1, do NOT correlate with reduced time to metastasis, time to relapse and shortened breast cancer specific survival while high Six1 and high Eya1 together in the same patient sample significantly correlates with the aforementioned prognostic indicators. The mean value for Six1 and/or Eya1 expression was used to divide the samples into high (above the mean) and low (below the mean) Six1 and Eya1 expression. P-values were calculated by log-rank analysis.
Discussion
The Six homeobox transcription factor reactivates developmental pathways out of context in numerous tumor types, likely contributing to both tumor initiation and tumor progression. Downstream pathways involved in, and required for, Six1-activated tumor initiation and tumor progression have been studied intensely over the past few years (Coletta et al., 2004; Micalizzi et al., 2009; Micalizzi et al., 2010; Yu et al., 2006). Since Six1 has no intrinsic activation or repression domains, we hypothesized that it would require a co-factor(s) to mediate its tumor promotional characteristics. Because members of the developmentally required Eya coactivator family are overexpressed in many of the same cancers as Six1, including ovarian cancer where both Six1 and Eya2 correlate with advanced-disease and poor patient survival (Behbakht et al., 2007; Zhang et al., 2005), we pursued Eya proteins as relevant required co-factors of Six1 in human breast cancer.
To establish if Six1 requires Eya to mediate its tumorigenic effects, we knocked down Eya2 in MCF7 mammary carcinoma cells overexpressing Six1. In this study, we demonstrate for the first time, a cooperative role for Six1 and Eya in mediating the pro-metastatic phenotypes induced by Six1. Eya2 is required by Six1 to activate TGF-β signaling, as well as to induce both EMT and cancer stem cell-like characteristics. These data are supported clinically by the demonstration that Six1 and Eya2 are co-expressed with activated TGF-β signaling, and that the two proteins together, but not individually, correlate with shortened time to relapse and metastasis and with decreased survival. These results strongly suggest that Eya2 and Six1 act in concert to promote breast tumor progression.
Although all Eya family members cooperate with Six1 to mediate Six1-induced transcription in cell culture models (Li et al., 2003; Ohto et al., 1999; Zhang et al., 2004), we demonstrate that only Eya1 and Eya2 strongly collaborate with Six1 in human breast cancer. This association of Six1 with Eya1 and Eya2 is not surprising as Eya1 and Six1 knockout mice phenocopy each other (Ando et al., 2005; Laclef et al., 2003b; Li et al., 2003; Xu et al., 1999; Xu et al., 2003) and as Eya2 and Six1 both correlate with advanced disease and decreased survival in ovarian cancer (Behbakht et al., 2007; Zhang et al., 2005). In contrast, overexpression of Eya3 has not been observed in cancer, nor has an endogenous role for Eya3 with Six1 been demonstrated. Interestingly, Eya4 is methylated and downregulated in gastrointestinal cancers (Osborn et al., 2006; Zou et al., 2005), but is overexpressed in malignant peripheral nerve sheath tumors along with other Eya and Six family members (Miller et al., 2010), suggesting that Eya4 may play roles in both tumor suppression and progression, depending on the context. Thus, while Eya1 and Eya2 appear to be the key Eya family members necessary for Six1-mediated tumor progression in the breast, we hypothesize that, given the correct context, any Eya family member may be sufficient to cooperate with Six1 to mediate tumorigenesis.
Interestingly, a recent study demonstrates that overexpression of Eya in MCF7 mammary carcinoma cells promotes proliferation, transformation, migration, and invasion of breast cancer cells, while knockdown of Eya3 in MDA-231 metastatic mammary carcinoma cells inhibits invasion, migration, and metastasis in nude mice (Pandey et al., 2010). The authors of this recent study suggest that Eya promotes tumorigenic phenotypes largely through cytoplasmic functions, since nuclear targeted Eya did not induce transformation, migration or invasion of breast cancer cells to the same extent as wild type Eya protein. While a cytoplasmic function for Eya likely contributes to its ability to mediate tumorigenic and metastatic phenotypes, the cell lines used in this study contain low levels of Six1, making it difficult to assess the dependence of Eya on Six1 in this context. It has previously been shown that the presence of Six family members significantly affects the percentage of Eya proteins located in the nucleus versus the cytoplasm (Ohto et al., 1999), and that the cytoplasmic/nuclear Eya ratio is further influenced by expression of Abl and G-alphai proteins, which function with Eya in the cytoplasm (Embry et al., 2004; Fan et al., 2000; Xiong et al., 2009). Interestingly, complete eye restoration in Drosophila requires balanced nuclear and cytoplasmic eya expression (Xiong et al., 2009), suggesting that full Eya function may require spatial regulation between the two Eya pools. Indeed, while the cytoplasmic function of non-targeted Eya was deemed critical for the pro-tumorigenic and metastatic properties observed in the aforementioned study, nuclear targeted Eya still increased pro-tumorigenic/metastatic phenotypes 3-fold above baseline (Pandey et al., 2010), suggesting that the cytoplasmic and nuclear functions of Eya may cooperate to impart the tumorigenic response. Importantly, the requirement of a direct Six1-Eya interaction in mediating metastatic phenotypes, as opposed to the two proteins working in parallel cooperative pathways, is not yet known. Nonetheless, the cell culture models used in this study are strongly supported by the clinical data published herein, and indicate that Six1 and Eya indeed require each other to mediate breast cancer progression.
In addition to its transactivation activity, Eya contains two phosphatase activities, N-terminal serine/threonine phosphatase activity and C-terminal tyrosine phosphatase activity. The C-terminal tyrosine phosphatase activity acts in the nucleus to activate a repair and survival pathway by dephosphorylating H2AX in response to DNA damage (Cook et al., 2009; Krishnan et al., 2009) and in the cytoplasm to mediate phosphotyrosine signaling networks during Drosophila development by interacting with the Abelson tyrosine kinase (abl)(Xiong et al., 2009). The N-terminal serine/threonine phosphatase activity plays a role in the cytoplasm, where it is required for the ability of Eya4 to enhance the innate immune response (Okabe et al., 2009). Significantly, the tyrosine phosphatase activity of Eya is necessary for Eya-induced migration, invasion, transformation, and metastasis (Pandey et al., 2010). The requirement for Eya phosphatase activity in mediating transcriptional activation, specifically through Six1, has not been completely elucidated, particularly in mammalian cells. Early in-vitro studies analyzing activation of Six1 reporter promoters suggest that the Eya phosphatase activity is important to co-activate transcription through Six1 (Li et al., 2003). However, a more recent study in Drosophila suggests Eya phosphatase activity is only required for a subset of Six1 transcriptional targets (Jemc and Rebay, 2007). Thus, the role of Eya phosphatase activity in mediating Six1-induced pro-metastatic phenotypes remains to be determined.
In conclusion, we have found that the Eya co-activator is required for the ability of Six1 to mediate a number of pro-metastatic properties, and that the two molecules together significantly predict adverse outcomes in human breast cancer. Understanding the necessity of Six1 on both a direct interaction with Eya and on the Eya phosphatase activity is important for future development of anti-cancer agents that target the Six1-Eya complex. Six1 and Eya2 are implicated in advanced breast cancer and are not normally expressed in most adult tissues. Thus, inhibiting these proteins in breast, ovarian, and other carcinomas may result in a therapeutic agent that would target tumor progression with limited side effects to patients.
Methods
Cell culture
One MCF7-Six1 and one MCF7-Ctrl stable clone (Ford et al., 1998) was stably transfected using Effectene (Qiagen, Valencia, CA, USA) with 5 shRNA constructs and one scramble negative control in the SureSilencing pGeneClip vector (SABiosciences, Frederick, MD, USA). Cells were selected with 2.5ug/ml puromycin and 2 individual clonal isolates chosen from each of two working Eya2 shRNA constructs (shRNA1: CGTGCGCATTGGCCTTATGAT; shRNA2: GGGTTCTATCAAGGAGGAAAT), as well as 2 scrambled control clones (GGAATCTCATTCGATGCATAC).
Real Time PCR
Total RNA was TRIzol isolated and RNeasy mini kit purified (Qiagen). Quantification to compare Eya mRNA levels was performed using plasmid standard curves and calculation of copy number for each Eya mRNA. Relative expression was used for comparison of Eya2 levels between shRNA clones and determined by ΔΔCT method (Livak and Schmittgen, 2001). Supplemental Figure7 lists primer and probes.
Antibody Production
An Eya2 antibody was produced by Proteintech Group, Inc (Chicago, IL). An N-terminal Eya2 peptide (aa17-37: LDKLKFNRADAAVWTLSDRQG) was KLH conjugated. 1mg of peptide was injected on day1 with boosts on days 28, 40, 58, and 76 and bleeds on days 72 and 102. The antibody was antigen affinity purified and tested on lysates from MCF7 cells transfected with Eya2 or Eyas 1,3,4 for control.
Immunofluorescence
Cells were grown on glass slides, fixed with formaldehyde, permeablized with 0.5% Triton X-100/PBS, incubated with Eya2 antibody (1:100; Ford Lab), then with Texas Red-conjugated antirabbit IgG antibody (Sigma-Aldrich, St. Louis, MO) and stained with DAPI.
Western blot analysis
Western blot analysis was performed on whole cell lysates made with RIPA buffer(Ford et al., 2000). Primary antibodies used were: E-cadherin, β-catenin, and Fibronectin (BD Biosciences, San Diego, CA, USA), β-actin (Sigma-Aldrich), Smad3 (Invitrogen, Carlsbad, CA, USA) Eyas 1, 3, 4 (SantaCruz Biotechnology, Santa Cruz, USA). Cell fractionation performed as previously described (Shtutman et al., 2006).
3TP and TOP-flash luciferase
Eya2 and scramble shRNA clones were plated in triplicate at 110,000 cells/well in a 12-well plate. After 24 hours, cells were cotransfected with 3TP (Wrana et al., 1992) or pTOP-flash plasmid (Korinek et al., 1997), and a renilla luciferase construct containing a cryptic promoter, using FuGENE 6 (Roche, Indianapolis, IN, USA). After 48 hours, lysates were prepared and analyzed with the Dual Luciferase Kit (Promega, Madison, WI, USA) on a Modulus Microplate (Turner Biosystems, Sunnyvale, CA, USA).
Cell adhesion
For cell adhesion, 96-well plates coated with laminin, fibronectin, collagen I, or collagen IV (BD Biosciences, Biocoat) were blocked with 1%BSA for 1 hour. 2.5 × 104 cells were added, incubated 1 hour at 37°, and then washed 3 times with PBS, fixed for 10 minutes with cold methanol, stained with 0.04% crystal violet, and solubilized with 10% glacial acetic acid. Absorbance was determined at 570 nm on a microplate reader (Molecular Devices, Sunnyvalle, CA, USA).
Cancer Stem Cell Assays
7.5×105 cells were plated and grown for 48 hours. For flow cytometry analysis, cells were trypsinized, washed with 0.5%BSA-PBS, stained with anti-CD44-APC and anti-CD24-biotin (BD Biosciences) (1:100) for 30 minutes, then stained with anti-streptavidin-V450 (BD Biosciences) (1:1000) for 30 minutes, and resuspended in 400 μl of 0.06 μg/ul PI/0.5% BSA-PBS. Fluorescence was detected with CyAn (Beckman Coulter). For tumorsphere assays, cells were trypsinized and re-plated at 1,000 cells/ml in 6-well, ultra-low attachment plates (Fisher-Scientific) in 2 mls of serum-free DMEM/F12 media (Hyclone), supplemented with 20ng/ml bFGF (BD Biosciences), 20ng/ml EGF (BD Biosciences), 4 μg/ml heparin (Sigma), penicillin-streptomycin (Hyclone), and B27 (Gibco). The cells were fed 1 ml of media every 2-3 days. On day 10, all cells/spheres were collected, digested using 0.05% trypsin with 0.53mm EDTA-4Na (Invitrogen), and single cells plated as above at 2,000 cells per well to perform secondary assays. Tumorspheres were counted on day 8 of second passage.
Immunohistochemistry
Tumor array BRC711 was obtained from US Biomax (Rockville, MD, USA). Paraffin embedded sections were stained as previously described (Harrell et al., 2006) following the ImmPRESS kit protocol (Vector Laboratories, Burlingame, CA). Primary antibodies included Eya2 (1:300; Ford Lab), Six1 (1:75; Atlas Antibodies, Stockholm, Sweden), and Smad3 (5ug/ml; Invitrogen). Nuclear staining intensity in tumor cells was scored in a blinded fashion by a pathologist (PJ) on a 0-4 scale with 4 corresponding to most intense staining.
Analysis of microarray data
Gene expression and clinical outcome data were obtained from the van de Vijver and Wang datasets (van ’t Veer et al., 2002; Wang et al., 2005). Six1 and Eya1-4 gene expression was obtained for each tumor sample. All samples were mean-centered for each gene and divided into 2 groups: samples that express the denoted gene or gene combination above (high) or below the mean (low) and the rest of the samples that did not fit the denoted gene expressions (other). Each data set was analyzed separately. Kaplan-Meier survival curves were generated using WinStat for Excel (R. Fitch Software). Normalization was obtained from the Stanford Microarray Database (van de Vijver) and NCBI GEO (Wang: GSE2034).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was funded by grants from the National Cancer Institute (2RO1-CA095277) and The American Cancer Society (#RSG-07-183-01-DDC) to H.L.F, and The Department of Defense Breast Cancer Synergistic Idea Award (BC084105) to H.L.F. and R.Z. S.M.F. and D.S.M. were funded by predoctoral fellowships from the Department of Defense Breast Cancer Research Program (W81XWH-08-1-0332 and W81XWH-06-1-0757, respectively). We would like to thank Alana Welm for training in microarray datamining techniques, database sharing, and helpful datamining discussions, and Katherine Martin, of Bioarray Therapeutics, for data analysis of the Wang dataset.
Financial Support: This work was funded by grants from the National Cancer Institute (2RO1-CA095277) and The American Cancer Society (#RSG-07-183-01-DDC) to H.L.F, and The Department of Defense Breast Cancer Synergistic Idea Award (BC084105) to H.L.F. and R.Z. S.M.F. and D.S.M. were funded by predoctoral fellowships from the Department of Defense Breast Cancer Research Program (W81XWH-08-1-0332 and W81XWH-06-1-0757, respectively).
Footnotes
Conflict of Interest The authors declare no conflict of interest.
Supplementary information is available at Oncogene’s website.
References
- Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2:777–85. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. Clustering of mutations responsible for branchio-oto-renal (BOR) syndrome in the eyes absent homologous region (eyaHR) of EYA1. Hum Mol Genet. 1997a;6:2247–55. doi: 10.1093/hmg/6.13.2247. [DOI] [PubMed] [Google Scholar]
- Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997b;15:157–64. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando Z, Sato S, Ikeda K, Kawakami K. Slc12a2 is a direct target of two closely related homeobox proteins, Six1 and Six4. Febs J. 2005;272:3026–41. doi: 10.1111/j.1742-4658.2005.04716.x. [DOI] [PubMed] [Google Scholar]
- Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A, et al. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007;67:3036–42. doi: 10.1158/0008-5472.CAN-06-3755. [DOI] [PubMed] [Google Scholar]
- Cillo C, Faiella A, Cantile M, Boncinelli E. Homeobox genes and cancer. Exp Cell Res. 1999;248:1–9. doi: 10.1006/excr.1999.4451. [DOI] [PubMed] [Google Scholar]
- Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L, et al. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci U S A. 2004;101:6478–83. doi: 10.1073/pnas.0401139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–6. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embry AC, Glick JL, Linder ME, Casey PJ. Reciprocal signaling between the transcriptionalco-factor Eya2 and specific members of the Galphai family. Mol Pharmacol. 2004;66:1325–31. doi: 10.1124/mol.104.004093. [DOI] [PubMed] [Google Scholar]
- Fan X, Brass LF, Poncz M, Spitz F, Maire P, Manning DR. The alpha subunits of Gz and Giinteract with the eyes absent transcription cofactor Eya2, preventing its interaction with the sixclass of homeodomain-containing proteins. J Biol Chem. 2000;275:32129–34. doi: 10.1074/jbc.M004577200. [DOI] [PubMed] [Google Scholar]
- Ford HL. Homeobox genes: a link between development, cell cycle, and cancer? Cell Biol Int. 1998;22:397–400. doi: 10.1006/cbir.1998.0329. [DOI] [PubMed] [Google Scholar]
- Ford HL, Kabingu EN, Bump EA, Mutter GL, Pardee AB. Abrogation of the G2 cell cyclecheckpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc Natl Acad Sci U S A. 1998;95:12608–13. doi: 10.1073/pnas.95.21.12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford HL, Landesman-Bollag E, Dacwag CS, Stukenberg PT, Pardee AB, Seldin DC. Cell cycle-regulated phosphorylation of the human SIX1 homeodomain protein. J Biol Chem. 2000;275:22245–54. doi: 10.1074/jbc.M002446200. [DOI] [PubMed] [Google Scholar]
- Harrell JC, Dye WW, Allred DC, Jedlicka P, Spoelstra NS, Sartorius CA, et al. Estrogen receptor positive breast cancer metastasis: altered hormonal sensitivity and tumor aggressiveness in lymphatic vessels and lymph nodes. Cancer Res. 2006;66:9308–15. doi: 10.1158/0008-5472.CAN-06-1769. [DOI] [PubMed] [Google Scholar]
- Jemc J, Rebay I. Identification of transcriptional targets of the dual-function transcription factor/phosphatase eyes absent. Dev Biol. 2007;310:416–29. doi: 10.1016/j.ydbio.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochhar A, Orten DJ, Sorensen JL, Fischer SM, Cremers CW, Kimberling WJ, et al. SIX1 mutation screening in 247 branchio-oto-renal syndrome families: a recurrent missense mutation associated with BOR. Hum Mutat. 2008;29:565. doi: 10.1002/humu.20714. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Jeong DG, Jung SK, Ryu SE, Xiao A, Allis CD, et al. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A. X is mediated by the protein phosphatase eyes absent. J Biol Chem. 2009;284:16066–70. doi: 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laclef C, Hamard G, Demignon J, Souil E, Houbron C, Maire P. Altered myogenesis in Six1-deficient mice. Development. 2003a;130:2239–52. doi: 10.1242/dev.00440. [DOI] [PubMed] [Google Scholar]
- Laclef C, Souil E, Demignon J, Maire P. Thymus, kidney and craniofacial abnormalities in Six1 deficient mice. Mech Dev. 2003b;120:669–79. doi: 10.1016/s0925-4773(03)00065-0. [DOI] [PubMed] [Google Scholar]
- Li CM, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, et al. Gene expression in Wilms’ tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol. 2002;160:2181–90. doi: 10.1016/S0002-9440(10)61166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activityregulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426:247–54. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, et al. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors thatundergo epithelial-mesenchymal transition. J Clin Invest. 2009;119:2663–77. doi: 10.1172/JCI37691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Christensen KL, Jedlicka P, Coletta RD, Baron AE, Harrell JC, et al. The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymaltransition and metastasis in mice through increasing TGF-beta signaling. J Clin Invest. 2009;119:2678–90. doi: 10.1172/JCI37815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Wang CA, Farabaugh SM, Schiemann WP, Ford H. Homeoprotein Six1 Increases TGF-{beta} Type I Receptor and Converts TGF-{beta} Signaling from Suppressive toSupportive for Tumor Growth. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-10-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, et al. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010;29:368–79. doi: 10.1038/onc.2009.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KT, Man K, Sun CK, Lee TK, Poon RT, Lo CM, et al. Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br J Cancer. 2006;95:1050–5. doi: 10.1038/sj.bjc.6603399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohto H, Kamada S, Tago K, Tominaga SI, Ozaki H, Sato S, et al. Cooperation of six and eya in activation of their target genes through nuclear translocation of Eya. Mol Cell Biol. 1999;19:6815–24. doi: 10.1128/mcb.19.10.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, Sano T, Nagata S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature. 2009;460:520–4. doi: 10.1038/nature08138. [DOI] [PubMed] [Google Scholar]
- Orten DJ, Fischer SM, Sorensen JL, Radhakrishna U, Cremers CW, Marres HA, et al. Branchio-oto-renal syndrome (BOR): novel mutations in the EYA1 gene, and a review of the mutational genetics of BOR. Hum Mutat. 2008;29:537–44. doi: 10.1002/humu.20691. [DOI] [PubMed] [Google Scholar]
- Osborn NK, Zou H, Molina JR, Lesche R, Lewin J, Lofton-Day C, et al. Aberrant methylation of the eyes absent 4 gene in ulcerative colitis-associated dysplasia. Clin Gastroenterol Hepatol. 2006;4:212–8. doi: 10.1016/j.cgh.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Ouyang G, Wang Z, Fang X, Liu J, Yang CJ. Molecular signaling of the epithelial to mesenchymal transition in generating and maintaining cancer stem cells. Cell Mol Life Sci. 67:2605–18. doi: 10.1007/s00018-010-0338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey RN, Rani R, Yeo EJ, Spencer M, Hu S, Lang RA, et al. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene. 2010;29:3715–22. doi: 10.1038/onc.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick AN, Schiemann BJ, Yang K, Zhao R, Ford HL. Biochemical and functional characterization of six SIX1 Branchio-oto-renal syndrome mutations. J Biol Chem. 2009;284:20781–90. doi: 10.1074/jbc.M109.016832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules RS, Levedakou EN, Wilson SJ, Innes CL, Rhodes N, Tlsty TD, et al. Defective G2 checkpoint function in cells from individuals with familial cancer syndromes. Cancer Res. 1995;55:1763–73. [PubMed] [Google Scholar]
- Reichenberger KJ, Coletta RD, Schulte AP, Varella-Garcia M, Ford HL. Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 2005;65:2668–75. doi: 10.1158/0008-5472.CAN-04-4286. [DOI] [PubMed] [Google Scholar]
- Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A. 2004;101:8090–5. doi: 10.1073/pnas.0308475101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel S, Naora H. Homeobox gene expression in cancer: insights from developmentalregulation and deregulation. Eur J Cancer. 2005;41:2428–37. doi: 10.1016/j.ejca.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Schonberger J, Wang L, Shin JT, Kim SD, Depreux FF, Zhu H, et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet. 2005;37:418–22. doi: 10.1038/ng1527. [DOI] [PubMed] [Google Scholar]
- Shtutman M, Levina E, Ohouo P, Baig M, Roninson IB. Cell adhesion molecule L1 disrupts E-cadherin-containing adherens junctions and increases scattering and motility of MCF7 breastcarcinoma cells. Cancer Res. 2006;66:11370–80. doi: 10.1158/0008-5472.CAN-06-2106. [DOI] [PubMed] [Google Scholar]
- Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 15:169–90. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Vincent C, Kalatzis V, Abdelhak S, Chaib H, Compain S, Helias J, et al. BOR and BO syndromes are allelic defects of EYA1. Eur J Hum Genet. 1997;5:242–6. [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–9. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Wayne S, Robertson NG, DeClau F, Chen N, Verhoeven K, Prasad S, et al. Mutations in the transcriptional activator EYA4 cause late-onset deafness at the DFNA10 locus. Hum Mol Genet. 2001;10:195–200. doi: 10.1093/hmg/10.3.195. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–14. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- Xiong W, Dabbouseh NM, Rebay I. Interactions with the abelson tyrosine kinase reveal compartmentalization of eyes absent function between nucleus and cytoplasm. Dev Cell. 2009;16:271–9. doi: 10.1016/j.devcel.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears andkidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23:113–7. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130:3085–94. doi: 10.1242/dev.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Zheng W, Laclef C, Maire P, Maas RL, Peters H, et al. Eya1 is required for themorphogenesis of mammalian thymus, parathyroid and thyroid. Development. 2002;129:3033–44. doi: 10.1242/dev.129.13.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Davicioni E, Triche TJ, Merlino G. The homeoprotein six1 transcriptionally activates multiple protumorigenic genes but requires ezrin to promote metastasis. Cancer Res. 2006;66:1982–9. doi: 10.1158/0008-5472.CAN-05-2360. [DOI] [PubMed] [Google Scholar]
- Yu Y, Khan J, Khanna C, Helman L, Meltzer PS, Merlino G. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med. 2004;10:175–81. doi: 10.1038/nm966. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang N, Huang J, Buckanovich RJ, Liang S, Barchetti A, et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005;65:925–32. [PubMed] [Google Scholar]
- Zhang Y, Knosp BM, Maconochie M, Friedman RA, Smith RJ. A comparative study of Eya1 and Eya4 protein function and its implication in branchio-oto-renal syndrome and DFNA10. J Assoc Res Otolaryngol. 2004;5:295–304. doi: 10.1007/s10162-004-4044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Osborn NK, Harrington JJ, Klatt KK, Molina JR, Burgart LJ, et al. Frequent methylation of eyes absent 4 gene in Barrett’s esophagus and esophageal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2005;14:830–4. doi: 10.1158/1055-9965.EPI-04-0506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.