

Abstract
Enlargement of the vestibular aqueduct (EVA) is one of the most common inner ear malformations associated with sensorineural hearing loss in children. The delayed onset and progressive nature of this phenotype offer a window of opportunity to prevent or retard progression of hearing loss. EVA is not the direct cause of hearing loss in these patients, but rather is a radiologic marker for some underlying pathogenetic defect. Mutations of the SLC26A4 gene are a common cause of EVA. Studies of an Slc26a4 knockout mouse demonstrate that enlargement of the scala media is a key event in the pathogenesis of deafness. The enlargement is driven by fluid secretion in the vestibular labyrinth and a failure of fluid absorption in the embryonic endolymphatic sac. Elucidating the mechanism of hearing loss may offer clues to potential therapeutic strategies.
1. Clinical Phenotypes Associated with EVA
Enlargement of the vestibular aqueduct (EVA) is a common malformation identified in ears of children undergoing high-resolution imaging for sensorineural hearing loss (Fig. 1A). An enlarged vestibular aqueduct is also sometimes referred to as a dilated or large vestibular aqueduct (DVA or LVA). Valvassori and Clemis established the modern radiologic definition of EVA as a midpoint diameter of >1.5 mm or a grossly malformed overall morphology (Valvassori et al., 1978). These criteria have been adopted by a majority of studies. Computed tomography (CT) is the best radiologic modality to image bony structures such as the vestibular aqueduct. A single axial CT section can show the full length of the J-shaped vestibular aqueduct coursing from its aperture on the posterior aspect of the temporal bone to the medial aspect of the vestibule. The normal vestibular aqueduct is often so narrow that it is not visible in CT images. Magnetic resonance (MR) imaging provides complementary visualization of the soft tissue and fluid contents of an enlarged vestibular aqueduct: an enlarged endolymphatic sac and duct (Fig. 1B) (Phelps et al., 1998). The relationship of the vestibular aqueduct with the endolymphatic duct and sac is shown in Fig. 2.
Fig 1.
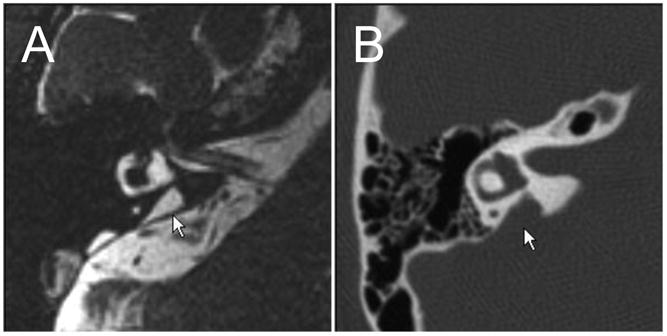
Radiologic imaging of an enlarged vestibular aqueduct. A) Axial computed tomography (CT) scan of an enlarged vestibular aqueduct (arrow). B) Axial MR (magnetic resonance) image of the soft tissue correlate of an enlarged vestibular aqueduct: an enlarged endolymphatic duct and sac (arrow). Reproduced from http://www.nidcd.nih.gov/health/hearing/eva-intro.htm.
Fig 2.

Schematic illustration of an enlarged vestibular aqueduct and endolymphatic sac and duct. Reproduced from http://www.nidcd.nih.gov/health/hearing/vestAque.htm.
Two studies published in 1989 described a distinctive auditory phenotype associated with isolated EVA (Jackler et al., 1989; Levenson et al., 1989). The hearing loss is predominantly sensorineural, variable in severity, asymmetric or unilateral, with a pre- or peri-lingual onset (before or near the time of speech and language acquisition). Many EVA patients have evidence of a conductive hearing loss component associated with normal middle ear findings (Arjmand et al., 2004; Govaerts et al., 1999; Nakashima et al., 2000). This is thought to be a cochlear conductive hearing loss due to a “third window” effect of the EVA upon sound transmission within the labyrinth (Merchant et al., 2007).
The sensorineural hearing loss associated with EVA can fluctuate or progress in a stepwise incremental fashion (Jackler et al., 1989; Levenson et al., 1989). In some patients, sudden hearing loss can be precipitated by minor head trauma or barotrauma. Although original reports emphasized EVA as the sole radiologic abnormality in these ears, this phenotype may also be observed in ears with EVA and cochlear anomalies. Associated cochlear anomalies can include a “Mondini” cochlea with reduced number of cochlear turns and an incomplete osseous partition of the turns. A more commonly observed anomaly in EVA ears is a hypoplastic cochlear modiolus (Lemmerling et al., 1997). There are differing conclusions on whether the presence or absence of cochlear malformations is related to the severity of hearing loss (Azaiez et al., 2007). However, in a study in which other underlying genotypic and phenotypic correlations were statistically accounted for, the presence of an associated cochlear anomaly was not independently associated with severity of hearing loss in ears with EVA (King et al., 2010).
The delayed onset and progressive nature of hearing loss associated with EVA provides a therapeutic window for interventions to prevent or slow the progression of hearing loss. Such strategies could be of particular benefit during the critical period of speech and language acquisition in young children. Current strategies for patients with EVA include counseling to avoid head trauma and barotrauma, and rehabilitation of communication. The latter can be achieved with conventional hearing amplification or cochlear implantation according to the degree of hearing loss. Corticosteroids have been used to treat hearing loss associated with EVA (Lin et al., 2005). The results of these studies are difficult to interpret because the natural history of hearing loss associated with EVA is unpredictable and idiosyncratic. Rigorous clinical trials will be required to evaluate these or other interventions for EVA.
The distinctive hearing loss phenotype associated with EVA has spawned a variety of hypotheses for the mechanism of hearing loss. One early theory proposed that trauma or barotrauma increases intracranial pressure with reflux of the contents of the endolymphatic sac and duct into the scala media where it damages hair cells and hearing (Jackler et al., 1989). This theory lost favor because operations to obliterate or decompress the endolymphatic sac were either ineffective or detrimental to hearing (Wilson et al., 1997). Furthermore, there is no correlation of the size of the EVA with hearing loss (Griffith et al., 1996; King et al., 2010). A second theory proposed that hearing loss results from leakage of perilymph from an abnormal fistulous round window (Belenky et al., 1993), but this observation has not been reported by other authors. There are phenotypic similarities between EVA and the fluctuating hearing loss thought to be associated with endolymphatic hydrops, in which endolymph-containing spaces are dilated. Endolymphatic hydrops has been proposed to cause hearing loss via increased endolymph osmotic pressure, rupture of intracochlear membranes, alterations of endolymph composition due to mixing with perilymph, and damage to hair cells and hearing. However, recent analyses cast doubt on this mechanism of hearing loss and suggest that endolymphatic hydrops is a nonspecific marker for an underlying cellular or molecular lesion that is the direct cause of hearing loss (Merchant et al., 2005). Similarly, EVA is not thought to be a direct cause of hearing loss, but a radiologic marker for an underlying molecular or cellular defect (Griffith et al., 1996).
2. Genetics of EVA
EVA with hearing loss typically presents as a sole clinical abnormality, in which case it is termed nonsyndromic. EVA has been reported in association with congenital cytomegalovirus (CMV) infection (Bauman et al., 1994), which can cause a similar hearing loss phenotype (Dahle et al., 2000). However, congenital CMV infection is not a significant or common cause of EVA (Pryor et al., 2005a). EVA may also be associated with abnormalities of other organ systems as part of a genetic syndrome. Examples of syndromes that can include EVA are distal renal tubular acidosis with deafness, CHARGE syndrome, Waardenburg syndrome, and branchio-oto-renal syndrome. However, the most common syndrome associated with EVA is Pendred syndrome. Pendred syndrome has been phenotypically estimated to account for up to 10% of cases of hereditary hearing loss (Fraser, 1965).
Pendred syndrome is an autosomal recessive disorder that was originally described in 1896 as a combination of goiter (thyroid gland enlargement) and severe congenital deafness (Pendred, 1896). We now realize the phenotypic spectrum of Pendred syndrome is much broader. Although goiter is incompletely penetrant, there is an underlying, more penetrant, defect in the ability of the thyroid gland to organify iodine (i.e. incorporate inorganic iodine in thyroid hormone biosynthesis) (Morgans et al., 1958; Pryor et al., 2005b). The hearing loss is often milder and more delayed in onset than originally described and, in some cases, may even be unilateral. An important advance was recognition that EVA is a highly penetrant feature of Pendred syndrome (Phelps et al., 1998). Due to routine hearing screening and radiologic imaging of the temporal bones, Pendred syndrome now commonly presents as nonsyndromic EVA in children (Reardon et al., 2000). Vestibular dysfunction is incompletely penetrant and varies in severity from subclinical caloric hyporeflexia to severe vertiginous episodes (Bergstrom, 1980; Das, 1987).
Mutations in the SLC26A4 gene (formerly known as PDS) cause Pendred syndrome (Everett et al., 1997). Mutations in SLC26A4 can also be detected in some patients with nonsyndromic EVA (Usami et al., 1999). This genotypic and phenotypic overlap has caused confusion about the nosologic relationship of these disorders. Some authors consider Pendred syndrome and nonsyndromic EVA to be variants of the same disorder (Campbell et al., 2001) while others regard them as distinct entities based upon SLC26A4 genotypic and phenotypic correlations (Pryor et al., 2005b).
Only one fourth of North American Caucasian EVA patients have two detectable mutant alleles of SLC26A4, one fourth have one detectable mutant allele, and one half of patients have no mutations (Campbell et al., 2001; Choi et al., 2009c). The causes of EVA in patients with only one or zero mutations of SLC26A4 are unknown. Undetected large genomic deletions or cryptic mutations in noncoding regions do not appear to account for this observation (Choi et al., 2009b). Digenic inheritance with mutations in the FOXI1 or KCNJ10 genes has been proposed for patients with one SLC26A4 mutation (Pryor et al., 2005b; Yang et al., 2007; Yang et al., 2009), but these findings have not been replicated in other studies (Jonard et al., 2010; Pera et al., 2008; Wu et al., 2010) and alternative hypotheses have not been excluded (Choi et al., 2009a). Mendelian genetic factors are unlikely in most EVA patients with no mutations since the proportion of siblings with EVA is much less than predicted for an autosomal recessive trait (Campbell et al., 2001; Choi et al., 2009c). In other populations such as Koreans, two mutant alleles can be found in 81% of EVA patients (Park et al., 2005).
In some of the reported genotypic surveys of childhood deafness among different populations, SLC26A4 mutations are the most common known genetic cause of childhood deafness (Anwar et al., 2009; Park et al., 2003). Genotypic surveys of large study populations have indicated that SLC26A4 mutations account for up to or more than 10% childhood deafness (Anwar et al., 2009; Park et al., 2003; Yuan et al., 2009). This comparatively high prevalence provides another impetus to develop new therapeutic or preventive strategies for EVA.
SLC26A4 encodes a multi-pass transmembrane protein called pendrin (Everett et al., 1997). Pendrin is expressed in a limited tissue distribution that includes the inner ear, thyroid, and kidney (Everett et al., 1997). Pendrin has been shown to exchange a variety of anions (Cl− and I−) and bases (e.g., OH− and HCO3−) across apical plasma membranes of epithelial cells (Royaux et al., 2001; Scott et al., 1999; Soleimani et al., 2001). In the thyroid follicle, pendrin is thought to mediate the transport of inorganic iodine across the apical membranes of follicular cells into the follicular lumen for biosynthesis of thyroid hormone (Royaux et al., 2000). In situ functional studies of pendrin have largely utilized a targeted deletion allele (“knockout”) of the mouse Slc26a4 gene (Everett et al., 2001).
3. Pathophysiological mechanisms of hearing loss in EVA
Our most significant mechanistic insights into the pathogenesis of hearing loss associated with EVA are based upon the Slc26a4 knockout (Slc26a4−/−) mouse that segregates a targeted deletion of exon 8 of Slc26a4 (Everett et al., 2001). Other mouse models include the Foxi1 knockout mouse (Hulander et al., 2003) and the loop mouse line segregating a chemically induced mutation of Slc26a4 (Dror et al., 2010).
The pathogenesis of EVA begins during the embryonic development of the inner ear. The inner ear develops from an invagination of the ectoderm that separates to form the initial otocyst. In mice the otocyst forms at embryonic day (E) 9.5 (Mansour et al., 2005). The otocyst is initially filled with amniotic fluid that has a plasma-like composition (Cheung et al., 2005). When and how the developing epithelia change the composition of the luminal fluid is currently unknown. At approximately E10.5, two protrusions begin to extend from the otocyst; one forms the cochlea and the other forms the endolymphatic sac. While the protrusions elongate and, in the case of the cochlea, coil, the center of the otocyst reorganizes into the vestibular labyrinth. The lumen of the cochlear protrusion opens at E14.5. Lumen formation depends on fluid secretion in the vestibular labyrinth and fluid absorption in the endolymphatic sac (Kim et al., 2010).
In the mouse inner ear, pendrin functions as a Cl−/HCO3− exchanger (Wangemann et al., 2007). Pendrin is expressed in the cochlea, the vestibular labyrinth and the endolymphatic sac. In the endolymphatic sac, pendrin is expressed in mitochondrial-rich cells that are interspersed among the principal ribosomal-rich cells (Dou et al., 2004; Royaux et al., 2003; Wangemann et al., 2004). In the cochlea, pendrin is expressed in a spiraling sheet of outer sulcus and spindle cells located in the lateral wall. In the vestibular labyrinth, pendrin is expressed in sheets of transitional cells that surround sensory cell patches (Wangemann et al., 2004). The earliest expression of pendrin occurs in the endolymphatic sac at E11.5 (Kim et al., 2011). Expression in the endolymphatic sac increases rapidly at E14.5. The onset of expression in the cochlea, utricle and saccule occurs at E13.5 to E16.5 (Kim et al., 2011).
The initial pathologic alteration in Slc26a4−/− mice includes an enlargement of the endolymphatic sac and cochlea that develops at E14.5, which is three days after the failed onset of expression in the endolymphatic sac (Kim et al., 2011). The enlargement leads to an approximately 10-fold increase in the cross-sectional area of the cochlear lumen that parallels normal cochlear growth (Fig. 2). The second pathologic alteration is an acidification of cochlear endolymph that develops at E15.5, which is one to two days after the failed onset of pendrin expression in the cochlea (Kim et al., 2011). Lack of pendrin expression also causes an acidification of the endolymphatic sac. However, this acidification develops later, at E17.5, which may reflect the stronger buffering power of the luminal fluid in the endolymphatic sac.
The enlargement and luminal acidification of the scala media spread the effect of pendrin deficiency from pendrin-expressing cells to a multitude of other cells. The enlargement may impair intercellular communication, possibly due to epithelial cell stretching and lengthening of diffusional distances between epithelial cells and between epithelial cells and mesenchymal cells such as fibrocytes. Intercellular communication plays a major role in cochlear development since impaired intercellular communication underlies the retarded development of the organ of Corti and may also contribute to the retarded development of stria vascularis (Kim et al., 2011; Wangemann et al., 2009). Thyroid hormone is a major factor in the retardation of the development of the organ of Corti. Fibrocytes located in the modiolus and in the lateral wall of the cochlea express, between P6 and P8, high levels of type 2 deiodinase (Dio2) to generate the biologically active hormone tri-iodothyronine from the prohormone thyroxine (Campos-Barros et al., 2000). Receptors for tri-iodothyronine are located in the organ of Corti and in other epithelial cells lining the cochlear duct (Bradley et al., 1994; Ng et al., 2009). The route taken by thyroid hormone between the hormone-generating cells and the receptor-bearing cells has not yet been delineated, although intercellular diffusion via gap junctions may be involved. Gap junctions may not only be the conduit for thyroid hormone but also for other growth and development-controlling factors as well as for nutritional substrates (Chang et al., 2008; Wang et al., 2009; Zhang et al., 2005). Lengthening of diffusion distances between fibrocytes and receptor-bearing epithelial cells may be responsible for the observed local hypothyroidism in Slc26a4−/− mice that leads to the observed retarded development of the organ of Corti (Wangemann et al., 2009).
The development of the stria vascularis in Slc26a4−/− cochleae is also retarded: the normal multilayered and highly vascularized anatomy is acquired with a delay (Kim et al., 2011). It is still unclear whether the retarded development of the stria vascularis in Slc26a4−/− mice is mainly a function of the approximately four-fold elevated H+ concentration in endolymph (Wangemann et al., 2007) or a function of the enlargement that is associated with an approximately 2.5-fold stretching of epithelial cells, including strial marginal cells, and with a displacement of neighboring fibrocytes. The premature onset of connexin 26 expression in basal cells of the stria vascularis is consistent with an impaired coordination of strial development (Kim et al., 2011). At P10, the stria vascularis is affected by oxidative stress (Singh et al., 2008) and fails to establish a normal endocochlear potential (Wangemann et al., 2007). Oxidative stress leads to the loss of expression of the K+ channel KCNJ10 protein, which is essential for the generation of the endocochlear potential (Singh et al., 2008; Wangemann et al., 2004). The endocochlear potential is essentially a K+ equilibrium potential that is generated by KCNJ10 in the intermediate cells of the stria vascularis, in conjunction with the very low K+ concentration of intrastrial fluid and a normally high K+ concentration in the cytosol of intermediate cells (Marcus et al., 2002; Wangemann, 2006). It is unclear whether this oxidative stress is a function of insufficient expression of defense mechanisms or whether oxidative stress is due to higher rates of metabolism necessary to support higher rates of K+ secretion to maintain a normal endolymphatic K+ concentration in an approximately 10-fold larger volume of scala media (Royaux et al., 2003). In addition, the acidification of cochlear endolymph may contribute to the loss of the endocochlear potential by enhancing oxidative stress through acid-activation of the K+ channel KCNQ1 (Unsold et al., 2000) and an increase in the rate of transepithelial K+ secretion across stria marginal cells, which would be associated with an increase in metabolism (Singh et al., 2008). Indeed, the endocochlear potential is reduced by experimental maneuvers that lead to an acute acidification of cochlear fluids (Ikeda et al., 1987a; Sterkers et al., 1984) and acidification of cochlear fluids has been shown to increase free radical stress, whereas alkalinization has a protective effect on hearing (Tanaka et al., 2004).
The luminal acidification and the loss of the endocochlear potential may jointly contribute to the approximately 100-fold elevation in the endolymphatic Ca2+ concentration (Ikeda et al., 1987b; Wangemann et al., 2007). Loss of the endocochlear potential may reduce the driving force for Ca2+ transport via cellular or paracellular pathways. Further, acidification inhibits transcellular Ca2+ absorption pathways that may include uptake of Ca2+ from endolymph via Ca2+-permeable TRPV4 and TRPV5 channels and export into perilymph via Ca2+-ATPases and Na+/Ca2+ exchangers. TRPV4 and TRPV5 channels are expressed in multiple epithelial cells of the cochlea and are inhibited by a luminal acidification (Vennekens et al., 2001; Wangemann et al., 2007). The resulting inhibition of Ca2+ absorption may lead to a failure to establish the normal endolymphatic Ca2+ concentration of 22 μM (Bosher et al., 1978; Wangemann et al., 2007). This low endolymphatic Ca2+ concentration is critical for normal auditory function. Elevated Ca2+ concentrations reduce microphonic potentials generated by the sensory cells (Tanaka et al., 1980) and excessive Ca2+ concentrations may damage hair cells through Ca2+ overload. Sensory hair cells in Slc26a4−/− mice degenerate between P15 and P30 after a history of thyroid hormone deprivation and under the burden of an elevated luminal Ca2+ concentration, luminal acidification and a deficient endocochlear potential (Everett et al., 2001).
How might these observations in Slc26a4−/− mice explain the etiology of fluctuating hearing loss in EVA patients? It is conceivable that fluctuation is due to the sensitivity of the endocochlear potential to oxidative stress. The endocochlear potential and oxidative stress may comprise a negative feedback system that oscillates and generates fluctuations in the endocochlear potential, which is required for hearing (Fig. 4). The hypothesized feedback loop is comprised of three elements. First, reactive oxygen species (ROS) are generated by marginal cells of stria vascularis as a byproduct of metabolism, which is necessary to support K+ secretion (Wangemann et al., 1995). Second, the ROS-sensitive K+ channel KCNJ10 that generates the endocochlear potential and supplies K+ to the marginal cells (Singh et al., 2008), and third, K+ induced stimulation of K+ secretion (Wangemann et al., 1995; Wangemann et al., 1996). ROS-induced loss of KCNJ10 would abolish the endocochlear potential and hearing and the associated reduction in K+ flux toward marginal cells would limit the rate of K+ secretion, metabolism and ROS production. The reduced ROS production would then permit restoration of KCNJ10 expression, KCNJ10 channel function would restore the endocochlear potential and restore hearing but also supply increased amounts of K+ to marginal cells, which again would stimulate K+ secretion, metabolism and ROS production. Irreversible hearing loss would result when endolymphatic Ca2+ concentrations rise and hair cells succumb to Ca2+ overload (Everett et al., 2001; Wangemann et al., 2007).
Fig 4.

Hypothetical mechanism for fluctuating hearing loss. A) Diagram based on a cochlea obtained from a P7 Slc26a4+/− mouse. B) Diagram of the stria vascularis illustrating a negative feedback mechanism that leads to fluctuating loss of KCNJ10, the K+ channel that generates the endocochlear potential. Fluctuating loss of the endocochlear potential can be expected to the lead to fluctuating loss of hearing.
4. Conclusions
Enlargement of the vestibular aqueduct (EVA) is a comparatively common but enigmatic sensorineural hearing loss disorder in children. Studies in mouse models demonstrate that enlargement and acidification of the scala media are early events in the pathogenesis of hearing loss. Future work to elucidate the mechanism of hearing loss should focus on fluid transport in cochlear development and alterations of cellular and molecular function and signaling in the lateral wall of the cochlea. The results of these studies may lead to treatment strategies to preserve hearing in humans with mutations of SLC26A4.
Fig 3.

Cochlear enlargement. Reproduced from Kim et al. (Kim et al., 2010). A) Diagram based on a cochlea obtained from an E18.5 Slc26a4+/− mouse. B) Diagram based on the enlarged cochlea obtained from an E18.5 Slc26a4−/− mouse. C) Measurements of cross-sectional areas of scala media from the basal turn of the cochlea in Slc26a4+/− and Slc26a4−/− mice. Note that the growth of the lumen is parallel between Slc26a4+/− and Slc26a4−/− mice and that a ∼10-fold enlargement is maintained throughout development. Abbreviations: C, otic capsule; S, stria vascularis; H, sensory hair cells; M, modiolus; N, cochlear nerve. Spaces occupied by mesenchymal cell (green) are compressed in Slc26a4−/− mice, and fibrocytes in the modiolus (M) and between the otic capsule (C) and stria vascularis (S) are displaced.
Acknowledgments
The authors are supported by NIH intramural research fund Z01-DC-000060 (A.J.G.) and Kansas State University (P.W.). We thank our colleagues for critical review of this manuscript. Figures 1 and 2 were provided by the National Institute on Deafness and Other Communication Disorders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anwar S, Riazuddin S, Ahmed ZM, Tasneem S, Ateeq ul J, Khan SY, Griffith AJ, Friedman TB. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J Hum Genet. 2009;54:266–70. doi: 10.1038/jhg.2009.21. [DOI] [PubMed] [Google Scholar]
- Arjmand EM, Webber A. Audiometric findings in children with a large vestibular aqueduct. Arch Otolaryngol Head Neck Surg. 2004;130:1169–74. doi: 10.1001/archotol.130.10.1169. [DOI] [PubMed] [Google Scholar]
- Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith RJ. Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet. 2007;122:451–7. doi: 10.1007/s00439-007-0415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman NM, Kirby-Keyser LJ, Dolan KD, Wexler D, Gantz BJ, McCabe BF, Bale JF., Jr Mondini dysplasia and congenital cytomegalovirus infection. J Pediatr. 1994;124:71–8. doi: 10.1016/s0022-3476(94)70256-x. [DOI] [PubMed] [Google Scholar]
- Belenky WM, Madgy DN, Leider JS, Becker CJ, Hotaling AJ. The enlarged vestibular aqueduct syndrome (EVA syndrome) Ear Nose Throat J. 1993;72:746–51. [PubMed] [Google Scholar]
- Bergstrom L. Pendred’s syndrome with atypical features. Ann Otol Rhinol Laryngol. 1980;89:135–9. doi: 10.1177/000348948008900209. [DOI] [PubMed] [Google Scholar]
- Bosher SK, Warren RL. Very low calcium content of cochlear endolymph, an extracellular fluid. Nature. 1978;273:377–8. doi: 10.1038/273377a0. [DOI] [PubMed] [Google Scholar]
- Bradley DJ, Towle HC, Young WS., 3rd Alpha and beta thyroid hormone receptor (TR) gene expression during auditory neurogenesis: evidence for TR isoform-specific transcriptional regulation in vivo. Proc Natl Acad Sci U S A. 1994;91:439–43. doi: 10.1073/pnas.91.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, Karniski LP, Sheffield VC, Smith RJ. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001;17:403–11. doi: 10.1002/humu.1116. [DOI] [PubMed] [Google Scholar]
- Campos-Barros A, Amma LL, Faris JS, Shailam R, Kelley MW, Forrest D. Type 2 iodothyronine deiodinase expression in the cochlea before the onset of hearing. Proc Natl Acad Sci U S A. 2000;97:1287–92. doi: 10.1073/pnas.97.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Tang W, Ahmad S, Zhou B, Lin X. Gap junction mediated intercellular metabolite transfer in the cochlea is compromised in connexin30 null mice. PLoS One. 2008;3:e4088. doi: 10.1371/journal.pone.0004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CY, Brace RA. Amniotic fluid volume and composition in mouse pregnancy. J Soc Gynecol Investig. 2005;12:558–62. doi: 10.1016/j.jsgi.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Choi BY, Alper SL, Griffith AJ. Response to: The c.-103T>C Variant in the 5′-UTR of SLC26A4 Gene: A Pathogenic Mutation or Coincidental Polymorphism? Hum Mutat. 2009a;30:1471. doi: 10.1002/humu.21097. [DOI] [PubMed] [Google Scholar]
- Choi BY, Madeo AC, King KA, Zalewski CK, Pryor SP, Muskett JA, Nance WE, Butman JA, Brewer CC, Griffith AJ. Segregation of enlarged vestibular aqueducts in families with non-diagnostic SLC26A4 genotypes. J Med Genet. 2009b;46:856–61. doi: 10.1136/jmg.2009.067892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BY, Stewart AK, Madeo AC, Pryor SP, Lenhard S, Kittles R, Eisenman D, Kim HJ, Niparko J, Thomsen J, Arnos KS, Nance WE, King KA, Zalewski CK, Brewer CC, Shawker T, Reynolds JC, Butman JA, Karniski LP, Alper SL, Griffith AJ. Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum Mutat. 2009c;30:599–608. doi: 10.1002/humu.20884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahle AJ, Fowler KB, Wright JD, Boppana SB, Britt WJ, Pass RF. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J Am Acad Audiol. 2000;11:283–90. [PubMed] [Google Scholar]
- Das VK. Pendred’s syndrome with episodic vertigo, tinnitus and vomiting and normal bithermal caloric responses. J Laryngol Otol. 1987;101:721–2. doi: 10.1017/s0022215100102592. [DOI] [PubMed] [Google Scholar]
- Dou H, Xu J, Wang Z, Smith AN, Soleimani M, Karet FE, Greinwald JH, Jr, Choo D. Co-expression of pendrin, vacuolar H+-ATPase alpha4-subunit and carbonic anhydrase II in epithelial cells of the murine endolymphatic sac. J Histochem Cytochem. 2004;52:1377–84. doi: 10.1177/002215540405201014. [DOI] [PubMed] [Google Scholar]
- Dror AA, Politi Y, Shahin H, Lenz DR, Dossena S, Nofziger C, Fuchs H, Hrabe de Angelis M, Paulmichl M, Weiner S, Avraham KB. Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J Biol Chem. 2010;285:21724–35. doi: 10.1074/jbc.M110.120188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153–61. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–22. [Google Scholar]
- Fraser GR. Association of congenital deafness with goitre (Pendred’s syndrome): a study of 207 families. Ann Hum Genet. 1965;28:201–249. doi: 10.1111/j.1469-1809.1964.tb00479.x. [DOI] [PubMed] [Google Scholar]
- Govaerts PJ, Casselman J, Daemers K, De Ceulaer G, Somers T, Offeciers FE. Audiological findings in large vestibular aqueduct syndrome. Int J Pediatr Otorhinolaryngol. 1999;51:157–64. doi: 10.1016/s0165-5876(99)00268-2. [DOI] [PubMed] [Google Scholar]
- Griffith AJ, Arts A, Downs C, Innis JW, Shepard NT, Sheldon S, Gebarski SS. Familial large vestibular aqueduct syndrome. Laryngoscope. 1996;106:960–5. doi: 10.1097/00005537-199608000-00009. [DOI] [PubMed] [Google Scholar]
- Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, Johansson BR, Steel KP, Enerback S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development. 2003;130:2013–25. doi: 10.1242/dev.00376. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kusakari J, Takasaka T, Saito Y. Early effects of acetazolamide on anionic activities of the guinea pig endolymph: evidence for active function of carbonic anhydrase in the cochlea. Hear Res. 1987a;31:211–6. doi: 10.1016/0378-5955(87)90189-4. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kusakari J, Takasaka T, Saito Y. The Ca2+ activity of cochlear endolymph of the guinea pig and the effect of inhibitors. Hear Res. 1987b;26:117–25. doi: 10.1016/0378-5955(87)90040-2. [DOI] [PubMed] [Google Scholar]
- Jackler RK, De La Cruz A. The large vestibular aqueduct syndrome. Laryngoscope. 1989;99:1238–42. doi: 10.1288/00005537-198912000-00006. discussion 1242–3. [DOI] [PubMed] [Google Scholar]
- Jonard L, Niasme-Grare M, Bonnet C, Feldmann D, Rouillon I, Loundon N, Calais C, Catros H, David A, Dollfus H, Drouin-Garraud V, Duriez F, Eliot MM, Fellmann F, Francannet C, Gilbert-Dussardier B, Gohler C, Goizet C, Journel H, Mom T, Thuillier-Obstoy MF, Couderc R, Garabedian EN, Denoyelle F, Marlin S. Screening of SLC26A4, FOXI1 and KCNJ10 genes in unilateral hearing impairment with ipsilateral enlarged vestibular aqueduct. Int J Pediatr Otorhinolaryngol. 2010;74:1049–53. doi: 10.1016/j.ijporl.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Kim HM, Wangemann P. Failure of fluid absorption in the endolymphatic sac initiates cochlear enlargement that leads to deafness in mice lacking pendrin expression. PLoS One. 2010;5:e14041. doi: 10.1371/journal.pone.0014041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Wangemann P. Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS One. 2011 doi: 10.1371/journal.pone.0017949. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KA, Choi BY, Zalewski C, Madeo AC, Manichaikul A, Pryor SP, Ferruggiaro A, Eisenman D, Kim HJ, Niparko J, Thomsen J, Butman JA, Griffith AJ, Brewer CC. SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope. 2010;120:384–9. doi: 10.1002/lary.20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmerling MM, Mancuso AA, Antonelli PJ, Kubilis PS. Normal modiolus: CT appearance in patients with a large vestibular aqueduct. Radiology. 1997;204:213–9. doi: 10.1148/radiology.204.1.9205250. [DOI] [PubMed] [Google Scholar]
- Levenson MJ, Parisier SC, Jacobs M, Edelstein DR. The large vestibular aqueduct syndrome in children. A review of 12 cases and the description of a new clinical entity. Arch Otolaryngol Head Neck Surg. 1989;115:54–8. doi: 10.1001/archotol.1989.01860250056026. [DOI] [PubMed] [Google Scholar]
- Lin CY, Lin SL, Kao CC, Wu JL. The remediation of hearing deterioration in children with large vestibular aqueduct syndrome. Auris Nasus Larynx. 2005;32:99–105. doi: 10.1016/j.anl.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Mansour SL, Schoenwolf GC. Morphogenesis of the inner ear. In: Kelley MW, Wu D, Popper AN, Fay RR, editors. Development of the inner ear. Springer; 2005. pp. 43–84. [Google Scholar]
- Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol. 2002;282:C403–7. doi: 10.1152/ajpcell.00312.2001. [DOI] [PubMed] [Google Scholar]
- Merchant SN, Adams JC, Nadol JB., Jr Pathophysiology of Meniere’s syndrome: are symptoms caused by endolymphatic hydrops? Otol Neurotol. 2005;26:74–81. doi: 10.1097/00129492-200501000-00013. [DOI] [PubMed] [Google Scholar]
- Merchant SN, Nakajima HH, Halpin C, Nadol JB, Jr, Lee DJ, Innis WP, Curtin H, Rosowski JJ. Clinical investigation and mechanism of air-bone gaps in large vestibular aqueduct syndrome. Ann Otol Rhinol Laryngol. 2007;116:532–41. doi: 10.1177/000348940711600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgans ME, Trotter WR. Association of congenital deafness with goitre; the nature of the thyroid defect. Lancet. 1958;1:607–9. doi: 10.1016/s0140-6736(58)90866-3. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Ueda H, Furuhashi A, Sato E, Asahi K, Naganawa S, Beppu R. Air-bone gap and resonant frequency in large vestibular aqueduct syndrome. Am J Otol. 2000;21:671–4. [PubMed] [Google Scholar]
- Ng L, Hernandez A, He W, Ren T, Srinivas M, Ma M, Galton VA, St Germain DL, Forrest D. A protective role for type 3 deiodinase, a thyroid hormone-inactivating enzyme, in cochlear development and auditory function. Endocrinology. 2009;150:1952–60. doi: 10.1210/en.2008-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, Jang HS, Moon SK, Lee SC, Chun YM, Lee HK, Choi JY, Jung SC, Griffith AJ, Koo SK. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet. 2005;67:160–5. doi: 10.1111/j.1399-0004.2004.00386.x. [DOI] [PubMed] [Google Scholar]
- Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami SI, Nance WE, Wilcox ER, Griffith AJ. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40:242–8. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendred V. Deaf-Mutism and Goitre. Lancet. 1896;ii:532. [Google Scholar]
- Pera A, Villamar M, Vinuela A, Gandia M, Meda C, Moreno F, Hernandez-Chico C. A mutational analysis of the SLC26A4 gene in Spanish hearing-impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss. Eur J Hum Genet. 2008;16:888–96. doi: 10.1038/ejhg.2008.30. [DOI] [PubMed] [Google Scholar]
- Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE, Kendall-Taylor P, Graham JM, Cadge BC, Stephens SG, Pembrey ME, Reardon W. Radiological malformations of the ear in Pendred syndrome. Clin Radiol. 1998;53:268–73. doi: 10.1016/s0009-9260(98)80125-6. [DOI] [PubMed] [Google Scholar]
- Pryor SP, Demmler GJ, Madeo AC, Yang Y, Zalewski CK, Brewer CC, Butman JA, Fowler KB, Griffith AJ. Investigation of the role of congenital cytomegalovirus infection in the etiology of enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg. 2005a;131:388–92. doi: 10.1001/archotol.131.5.388. [DOI] [PubMed] [Google Scholar]
- Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, Griffith AJ. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005b;42:159–65. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon W, CFOM, Trembath R, Jan H, Phelps PD. Enlarged vestibular aqueduct: a radiological marker of pendred syndrome, and mutation of the PDS gene. QJM. 2000;93:99–104. doi: 10.1093/qjmed/93.2.99. [DOI] [PubMed] [Google Scholar]
- Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology. 2000;141:839–45. doi: 10.1210/endo.141.2.7303. [DOI] [PubMed] [Google Scholar]
- Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A. 2001;98:4221–6. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royaux IE, Belyantseva IA, Wu T, Kachar B, Everett LA, Marcus DC, Green ED. Localization and functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome. J Assoc Res Otolaryngol. 2003;4:394–404. doi: 10.1007/s10162-002-3052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet. 1999;21:440–3. doi: 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- Singh R, Wangemann P. Free radical stress-mediated loss of Kcnj10 protein expression in stria vascularis contributes to deafness in Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2008;294:F139–48. doi: 10.1152/ajprenal.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burnham CE. Pendrin: an apical Cl-/OH-/HCO3- exchanger in the kidney cortex. Am J Physiol Renal Physiol. 2001;280:F356–64. doi: 10.1152/ajprenal.2001.280.2.F356. [DOI] [PubMed] [Google Scholar]
- Sterkers O, Saumon G, Tran Ba Huy P, Ferrary E, Amiel C. Electrochemical heterogeneity of the cochlear endolymph: effect of acetazolamide. Am J Physiol. 1984;246:F47–53. doi: 10.1152/ajprenal.1984.246.1.F47. [DOI] [PubMed] [Google Scholar]
- Tanaka F, Whitworth CA, Rybak LP. Round window pH manipulation alters the ototoxicity of systemic cisplatin. Hear Res. 2004;187:44–50. doi: 10.1016/s0378-5955(03)00330-7. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Asanuma A, Yanagisawa K. Potentials of outer hair cells and their membrane properties in cationic environments. Hear Res. 1980;2:431–8. doi: 10.1016/0378-5955(80)90079-9. [DOI] [PubMed] [Google Scholar]
- Unsold B, Kerst G, Brousos H, Hubner M, Schreiber R, Nitschke R, Greger R, Bleich M. KCNE1 reverses the response of the human K+ channel KCNQ1 to cytosolic pH changes and alters its pharmacology and sensitivity to temperature. Pflugers Arch. 2000;441:368–78. doi: 10.1007/s004240000434. [DOI] [PubMed] [Google Scholar]
- Usami S, Abe S, Weston MD, Shinkawa H, Van Camp G, Kimberling WJ. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188–92. doi: 10.1007/s004390050933. [DOI] [PubMed] [Google Scholar]
- Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope. 1978;88:723–8. doi: 10.1002/lary.1978.88.5.723. [DOI] [PubMed] [Google Scholar]
- Vennekens R, Prenen J, Hoenderop JG, Bindels RJ, Droogmans G, Nilius B. Modulation of the epithelial Ca2+ channel ECaC by extracellular pH. Pflugers Arch. 2001;442:237–42. doi: 10.1007/s004240100517. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chang Q, Tang W, Sun Y, Zhou B, Li H, Lin X. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem Biophys Res Commun. 2009;385:33–7. doi: 10.1016/j.bbrc.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P, Liu J, Marcus DC. Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear Res. 1995;84:19–29. doi: 10.1016/0378-5955(95)00009-s. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Shen Z, Liu J. K(+)-induced stimulation of K+ secretion involves activation of the IsK channel in vestibular dark cells. Hear Res. 1996;100:201–10. doi: 10.1016/0378-5955(96)00127-x. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Nakaya K, Wu T, Maganti RJ, Itza EM, Sanneman JD, Harbidge DG, Billings S, Marcus DC. Loss of cochlear HCO3- secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2007;292:F1345–53. doi: 10.1152/ajprenal.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P, Kim HM, Billings S, Nakaya K, Li X, Singh R, Sharlin DS, Forrest D, Marcus DC, Fong P. Developmental delays consistent with cochlear hypothyroidism contribute to failure to develop hearing in mice lacking Slc26a4/pendrin expression. Am J Physiol Renal Physiol. 2009;297:F1435–47. doi: 10.1152/ajprenal.00011.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004;2:30. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DF, Hodgson RS, Talbot JM. Endolymphatic sac obliteration for large vestibular aqueduct syndrome. Am J Otol. 1997;18:101–6. discussion 106–7. [PubMed] [Google Scholar]
- Wu CC, Lu YC, Chen PJ, Yeh PL, Su YN, Hwu WL, Hsu CJ. Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol Neurootol. 2010;15:57–66. doi: 10.1159/000231567. [DOI] [PubMed] [Google Scholar]
- Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4) Am J Hum Genet. 2007;80:1055–63. doi: 10.1086/518314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Gurrola JG, 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–7. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, You Y, Huang D, Cui J, Wang Y, Wang Q, Yu F, Kang D, Yuan H, Han D, Dai P. Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. J Transl Med. 2009;7:79. doi: 10.1186/1479-5876-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci U S A. 2005;102:15201–6. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]