

Abstract
Non-nucleoside inhibitors of HIV reverse transcriptase (NNRTIs) are being pursued with guidance from molecular modeling including free energy perturbation (FEP) calculations for protein-inhibitor binding affinities. The previously reported pyrimidinylphenylamine 1 and its chloro analog 2 are potent anti-HIV agents; they inhibit replication of wild-type HIV-1 in infected human T-cells with EC50 values of 2 and 10 nM. However, they show no activity against viral strains containing the Tyr181Cys (Y181C) mutation in HIV-RT. Modeling indicates that the problem is likely associated with extensive interaction between the dimethylallyloxy substituent and Tyr181. As an alternative, a phenoxy group is computed to be oriented in a manner diminishing the contact with Tyr181. However, this replacement leads to a roughly 1000-fold loss of activity for 3 (2.5 μM). The present report details the efficient, computationally-driven evolution of 3 to novel NNRTIs with sub-10 nM potency towards both wild-type HIV-1 and Y181C-containing variants. The critical contributors were FEP substituent scans for the phenoxy and pyrimidine rings and recognition of potential benefits of addition of a cyanovinyl group to the phenoxy ring.
Introduction
Non-nucleoside inhibitors of HIV reverse transcriptase (NNRTIs) are an important component of highly active antiretroviral therapy (HAART).1 They are known to function by binding to an allosteric pocket in the vicinity of RT’s polymerase active site.2 Though there are five FDA-approved drugs in the class (nevirapine, delavirdine, evafirenz, etravirine, and rilpivirine), there remain needs for alternatives for first-line therapy owing to issues of safety, resistance, and ease of administration.1,3 Common side effects associated with the currently approved drugs include severe skin reactions, liver toxicity, and sleep disorders. In view of the rapid mutation of the virus, resistance is problematic and the NNRTIs are given in combination therapies. Though the class has demonstrated significant utility and promise, further improvements are undoubtedly possible. There is also need to be positioned to respond to the emergence of pan-resistant viral variants and unknown effects of long-term treatment.4
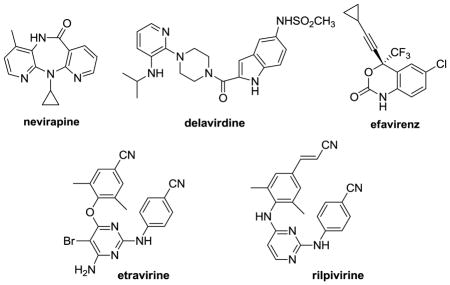
In our own pursuit of new NNRTIs, we have simultaneously sought improved computational methods of general utility to streamline the discovery process.5 The basic goal is to minimize the number of compounds that have to be synthesized and assayed to yield a drug candidate. Our computational approach is multi-faceted including virtual screening, de novo design, prediction of pharmacological properties and metabolites, and lead optimization guided by molecular modeling including free-energy perturbation (FEP) calculations.5 Though success has been demonstrated in the discovery and optimization of NNRTIs in several structural series,6 improvement in the performance against HIV variants with clinically relevant RT mutations is desired. Salient examples are provided by 1 and 2. They yield EC50 values of 2 and 10 nM,6b respectively, for inhibition of replication of wild-type (WT) HIV-1 in infected human T-cells. The activity for 1 is essentially the same as for efavirenz and etravirine in this assay and far greater than for nevirapine (110 nM). However, similar to nevirapine, 1 and 2 show no activity against virus encoding the Tyr181Cys (Y181C) mutation in the reverse transcriptase enzyme. This mutation often arises early in patients treated with NNRTIs.1 Most first-generation NNRTIs including nevirapine and delavirdine do not have useful activity towards Y181C containing variants, while efavirenz, etravirine, and rilpivirine retain 1–10 nM potency.7

A computed structure for 2 bound to WT HIV-RT is illustrated in Figure 1. Confidence in the structure comes from past experience with complexes for other NNRTIs including numerous crystal structures.2–6 Key features are the placement of the O-dimethylallyl (ODMA) group in the π-box formed by Tyr181, Tyr188, Phe227, and Trp229, and the hydrogen bond between the oxygen of Lys101 and the amino group of 2. The expectation has been that the inactivity towards the Y181C variant stems largely from loss of the favorable contact between Tyr181 and the ODMA group. Thus, ca. 15 alternatives to the ODMA group were tested, but they all led to much less potent or inactive inhibitors for replication of the WT virus.6b,6c
Figure 1.

Two orientations of NNRTI 2 bound to WT HIV-RT, as created using BOMB5 and optimized using MCPRO.8 The full model contains ca. 175 protein residues and the inhibitor. Carbon atoms of the NNRTI are colored gold. The image on the right is rotated 90° from the one on the left.
Nevertheless, in model building with the molecular growing program BOMB,5 the phenoxy substituted analog 3 is intriguing. It appears to deemphasize the interaction with Tyr181 in favor of an edge-to-face interaction between the phenyl rings of the phenoxy group and Tyr188, as illustrated in Figure 2. The problem is that the EC50 value for 3 deteriorates roughly 1000-fold to 2.5 μM.6c However, with experience gained with FEP-guided lead optimization,6 a challenge deemed worth pursuing was possible optimization of the substituents in the phenoxy and pyrimidine rings of 3 to seek analogs that would be very potent, e.g., with ca. 10 nM EC50 values, against both WT HIV-1 and variants containing the Y181C RT mutation.
Figure 2.

The same views as in Figure 1, but with NNRTI 3 bound to HIV-RT. Modeling suggested substitutions at the 3- and 5-positions in the phenoxy ring with possible passage of a substituent between Tyr181 and Tyr188 or towards Pro95.
Computational Details
The key computations were conjugate-gradient energy minimizations for protein-inhibitor complexes and FEP calculations to estimate differences in free energies of binding for series of inhibitors. The energetics for the systems were represented by classical force fields: the OPLS-AA force field for the protein, OPLS/CM1A for the inhibitors, and TIP4P for water molecules.8 The conjugate gradient optimizations use a dielectric constant of two to dampen the Coulombic interactions and 9-Å residue-based cutoffs. The configurational sampling for the FEP calculations was performed via Monte Carlo (MC) statistical mechanics simulations. In the MC/FEP calculations, small changes are made to convert one inhibitor to another, both unbound in water and bound to the protein; the difference in the two resultant free energy changes gives the relative free energy of binding, ΔΔGb.9,10 Standard protocols were followed.5,6 Briefly, all initial structures were generated with the molecule growing program BOMB starting from the 1s9e PDB file;11 the ligand was removed and replaced by cores such as aniline that are used by BOMB to grow the desired inhibitors in the binding site.5 For the Y181C variant, the 1s9e structure was also used as the starting point and the tyrosine was manually modified to cysteine. A reduced model of the protein was utilized that consisted of the ca. 175 amino acid residues closest to the NNRTI binding site; a few remote side chains were neutralized so that there was no net charge for the protein. The energy minimizations and MC/FEP calculations were executed with MCPRO.8 For the MC simulations, 1250 and 2000 water molecules in 25-Å caps were included for the complexes and unbound inhibitors, respectively. All degrees of freedom were sampled for the inhibitors, while the TIP4P water molecules only translated and rotated, as usual. Bond angles and dihedral angles for protein side chains were also sampled, while the backbone was kept fixed after conjugate-gradient relaxation.
The FEP calculations typically utilized 11 windows of simple overlap sampling for each modification of a non-hydrogen atom, e.g., CH3 → H or Cl → F (should be commas).10b,12 For the simulations of the protein-inhibitor complexes, each window covered at least 15 million (M) configurations of equilibration and 10 M configurations of averaging. A window refers to a MC simulation at one point along the mutation coordinate λ, which interconverts two inhibitors as λ goes from 0 to 1; two free-energy changes are computed at each window corresponding to a forward and backward increment. The spacing between windows, Δλ, is 0.1. The ligand and side chains within ca. 10 Å of the ligand were fully flexible, while the protein backbone was kept fixed during the MC runs. For the perturbations of the unbound inhibitors in water, equilibration covered 30 M configurations for the first window and 15 M for subsequent ones, while the averaging periods were extended to 40 M configurations. With current 3.0-GHz processors (Intel Q9650 Core2), the FEP calculations take approximately four days for the calculation on the complex and one day for the inhibitor alone in water on a single processor. It is easy to run the FEP windows simultaneously such that with 12 processors, one ΔΔGb result can be obtained in one day.
All MC simulations were run at 298 K. The reported uncertainties (±1σ) for the free energy changes were obtained from the fluctuation in separate averages over batches of 2 M configurations.13 Eq 1 is used where m is the number of batches, θi is the average of property θ for the i-th batch, and <θ> is the overall average for θ. Convergence is also checked through the hystereses in closed FEP cycles, as illustrated below.
| (1) |
Experimental Details
The syntheses of analogs of 3 and the corresponding 1,3,5-triazine benefited from previously described procedures.6b,6c Typically, the desired compounds arose from SNAr reaction of phenoxy or thiophenoxy anilines with heteroaryl chlorides (Scheme 1). The required anilines were also mostly prepared by SNAr chemistry from substituted phenols or thiophenols and 2-fluoro-4-nitro-benzonitrile (Scheme 2).14 However, when Y was chlorine, substitution of the chlorine was circumvented by copper- catalyzed selective O-arylation of 3-aminophenols,15 while the corresponding thioethers were prepared using cuprous thiophenolates (Scheme 2).16 Subsequently, when 3-cyanovinyl-substituted phenols were required, Heck coupling of aryl iodides with acrylonitrile using PdCl2(PPh3)2 as catalyst was effective (Scheme 3). This reaction afforded separable mixtures of E:Z (80:20) stereoisomers in 80–90% yield.17 Finally, some of the most potent compounds were cyclopropyltriazine analogs, which were prepared according to Scheme 4. Reaction between 2,4-dichloro-6-cyclopropyl-1,3,5-triazine and 4-amino-2-fluorobenzonitrile gave the corresponding intermediate, which was dechlorinated and coupled with 3-cyanovinyl-substituted phenols to yield the target compounds. Complete synthetic details are provided in the Supplementary Information. The identity of all assayed compounds was confirmed by 1H and 13C NMR and high-resolution mass spectrometry; purity was normally >95% as judged by high-performance liquid chromatography.
Scheme 1.

General procedure for synthesis of heteroarylaminodiphenyl ethers
Scheme 2.

General procedures for synthesis of diphenyl ethers and thioethers
Scheme 3.

Synthesis of cyanovinyl-substituted phenols
Scheme 4.

Synthesis of cyclopropyltriazine analogs
For the biology, the primary assay determined activities against the wild-type IIIB strain of HIV-118 using MT-2 human T-cells19 at an MOI of 0.1; EC50 values are obtained as the dose required for 50% protection of the infected cells using the MTT colorimetric method. The CC50 for inhibition of MT-2 cell growth in the absence of virus by 50% is obtained simultaneously.20,21 Analogous assays were performed using a variant HIV strain that encodes the Tyr181Cys (Y181C) mutation of HIV-RT.22 All assays were run in triplicate at inhibitor concentrations up to 100 μM.
Results
Alternative Conformers
The initially predicted structure of the parent 3 bound to RT is illustrated in Figure 2; it was built using BOMB and then subjected to conjugate gradient optimization using MCPRO. An alternative conformer for the bound ligand was also found as illustrated in Figure 3. The conformer from Figure 2 has the faces of the two phenyl rings roughly perpendicular, while the alternative is more reminiscent of a clamshell. The phenoxy groups are nearly orthogonal in the two complexes, though in both cases the interaction with Tyr181 appears to be reduced from what occurs with the ODMA group (Figure 1). The computed protein-ligand interaction energies from optimization of the two complexes are essentially identical at -65.5 kcal/mol. The conformational energetics were then investigated to try to better assess the relative likelihood of the two alternatives.
Figure 3.

(Left) Alternative conformer of 3 bound to HIV-RT. (Right) Comparison of the two ligand conformations. In the top one, the faces of the phenyl rings are roughly perpendicular, while the geometry is more like a clamshell in the alternative conformer.
The optimized dihedral angles for the C-C-O-C-C fragment are 16° and 43° for the perpendicular conformer and 71° and 37° for the clamshell in the complexes. For isolated diphenyl ether there is only one unique energy minimum with both dihedral angles near 40°, though significant deviations are observed in numerous crystal structures for molecules containing diphenyl ether substructures.23 Energy minimizations for diphenyl ether and 2-chlorodiphenyl ether with the OPLS/CM1A force field yield optimal dihedral angles of 40 ± 4° in both cases.
In order to attempt to assess the difference in strain energy for the ligand in the two complexes (Figures 2 and 3), single-point OPLS/CM1A calculations were performed on the extracted conformers. The results favor the perpendicular conformer by 3.4 kcal/mol. For the clamshell conformer, a contact between two ortho-hydrogens is short at 2.18 Å. Optimization of the extracted structure of the clamshell conformer causes the CCOCC dihedral angles to relax to 71° and 20°, the short H-H contact lengthens to 3.31 Å, and the energy declines by 7.8 kcal/mol; the terminal phenyl ring rotates to a more perpendicular arrangement. Optimization of the extracted, perpendicular conformer changes the dihedral angles to 36° and 45°, and the energy declines by 4.4 kcal/mol. In view of the similar protein-ligand interaction energies, this analysis favors the complex in Figure 2 with the perpendicular conformer for the inhibitor. However, some motional freedom can be expected for the phenoxy group in the binding site, which is desirable for binding and response to mutational variations.24,25 It is also likely that different substitution patterns for the phenoxy ring can change the conformational preference. It should be noted that only the ligand strain and protein-ligand interaction energy have been considered here. The total energies of the complexes are −3385 (perpendicular) and −3376 kcal/mol (clamshell); however, the separation is sensitive to small differences in the convergence of the optimizations.
Chlorine and Methyl Scans
Starting from 3, placement of substituents on the phenoxy ring was first considered. FEP calculations were performed for the conversion of each hydrogen in the phenoxy ring of 3 to a chlorine or methyl group. The perpendicular conformer (Figure 2) was used as a starting point for the FEP calculations. The chloro and methyl derivatives of 3 were also built using BOMB and each was converted to 3 in the FEP calculations. The FEP calculations include 1250 water molecules for the complexes and extensive configurational sampling, which could interconvert the ligand conformers. The FEP results are listed in Table 1 with ΔΔGb representing the predicted change in free energy of binding for the chlorine or methyl to hydrogen conversion. A positive value indicates that chlorine or a methyl group is preferred over hydrogen. The structure in Table 1 also corresponds to the structure in Figure 2 with the 2- and 3-positions being proximal to Y188; in the FEP calculations C2 and C6 are not equivalent as they do not interconvert during the MC simulations. The barrier for rotation of the substituted phenoxy ring in the complexes is sufficiently high that with normal MC sampling these equivalent positions do not interconvert. The normal range of such dihedral angle changes for a MC move is ±15º. Though larger moves could be attempted, which would allow interconversion of the equivalent positions, knowing the exact preferences at each position provides added design information.
Table 1.
FEP Results for Replacements of Chlorine and CH3 by Hydrogen (kcal/mol).

The results indicated that gains in binding should be achievable by substitutions only at C3 and C5. The preferences are not obvious by visual inspection of the built complex structures; often, one cannot tell if a non-bonded contact is attractive or somewhat too short. The convergence of the calculations for the chlorine analogs was checked using the 3,5-dichloro derivative, as summarized in Figure 4. The hysteresis for the cycle of four FEP calculations, 0.50 kcal/mol, was consistent with the individual uncertainties in Table 1. The results also indicated that 3,5-disubstitution was viable. During the MC simulations for the complexes, the conformation of the ligand did not convert from perpendicular to clamshell. In the simulations for the isolated ligands in water, display of numerous configurations also did not reveal any evidence for sampling of clamshell conformers. The dominant conformation is perpendicular with the methyl group of the methoxy substituent packed well against the phenoxy ring.
Figure 4.

Computed differences in free energies of binding, ΔΔGb (kcal/mol), for chlorophenyl analogs. The hysteresis of 0.50 kcal/mol for the cycle provides a measure of the precision of the calculations.
As summarized in Table 2, the 2-, 3-, and 4-methyl analogs, 6 – 8, were prepared and confirmed the preference for the 3-methyl compound; 7 provided a significant activity gain to 0.54 μM compared to 3 at 2.5 μM. Results for 4 are included to demonstrate the benefits of the methoxy group in 3. The 3-Cl and 3,5-dichloro analogs, 9 and 10, were then prepared. Though they are also more potent than 3, greater activity gains might have been expected based on the results in Table 1. An issue for the activities is that the computed results are for free energies of binding and the EC50 is for a cell-based assay. It may be noted that the correlation between observed cell-based and enzymatic activities for NNRTIs is high with r2 values of 07.–0.9.27 The FEP results normally provide valuable qualitative guidance, while quantitative accord with the cell assay results is not expected.5 For example, chlorine has a higher Hansch π value (0.71) than a methyl group (0.56) or hydrogen (0.0),26 so addition of chlorine to a phenyl ring is expected to make a molecule more lipophilic than addition of a methyl group. The more lipophilic compound may then partition more into cell membranes and also be lost to non-specific complexation with other cellular proteins. Another small point is that the computed results have not been corrected for possible symmetry effects. For example, the parent 3 or the 3,5-dichloro analog 10 have a factor of two advantage for binding over the unsymmetrical 9, if there is strong preference for 9 binding with the chlorine in just the 3-position or the 5-position.
Table 2.
WT Inhibitory Activity (EC50) and Cytotoxicity (CC50) in μM for Pyrimidines with HIV-1
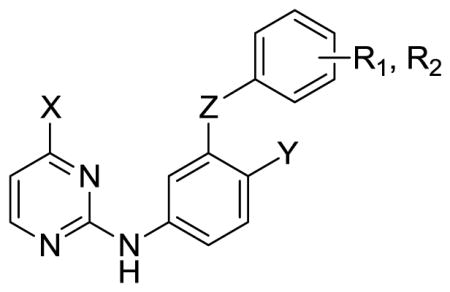 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | X | Y | Z | R1a | R2 | EC50b | CC50c |
| 3 | OCH3 | Cl | O | - | - | 2.5 | 38 |
| 4 | H | Cl | O | - | - | 13.0 | 30 |
| 5 | OCH3 | H | O | 3-Cl | - | 9.1 | 33 |
| 6 | OCH3 | Cl | O | 2-CH3 | - | NA | 13 |
| 7 | OCH3 | Cl | O | 3-CH3 | - | 0.54 | 18 |
| 8 | OCH3 | Cl | O | 4-CH3 | - | 10 | >100 |
| 9 | OCH3 | Cl | O | 3-Cl | - | 1.1 | 23 |
| 10 | OCH3 | Cl | O | 3-Cl | 5-Cl | 0.80 | 4.3 |
| 11 | OCH3 | CN | O | 3-Cl | - | 0.64 | >100 |
| 12 | OCH3 | CN | O | 3-Cl | 5-Cl | NA | 0.27 |
| 13 | OCH3 | CN | O | 3-CH3 | 5-CH3 | 0.49 | 1.3 |
| 14 | OCH3 | CN | O | 3-CN | - | 0.31 | 8.5 |
| 15 | OCH3 | CN | O | 3-CV | - | 0.26 | 6.9 |
| 16 | CH3CH2 | CN | O | 3-CV | - | 0.048 | 0.36 |
| 17 | SCH3 | CN | O | 3-CV | - | 0.048 | 1.2 |
| 18 | CH(CH3)2 | CN | O | 3-CV | - | 0.046 | 4.1 |
| 19 | c-C3H5 | CN | O | 3-CV | - | 0.032 | 1.1 |
| 20 | c-C3H5 | CN | O | 3-CV | 5-Cl | 0.012 | 1.1 |
| 21 | c-C3H5 | CN | O | 3-CV | 5-F | 0.0085 | 0.26 |
| 22 | c-C3H5 | CN | O | 3-F | 5-F | 0.78 | 4.3 |
| 23 | c-C3H5 | CN | O | 3-Cl | 5-CN | 0.015 | 0.15 |
| 24 | c-C3H5 | CN | O | 3-ClV | 0.49 | 1.9 | |
| 25 | OCH3 | Cl | S | - | - | 0.32 | 21 |
| 26 | OCH3 | Cl | S | 3-Cl | - | 0.31 | 4.9 |
| 27 | OCH3 | Cl | S | 3-CH3 | - | 0.31 | 8.9 |
| 28 | OCH3 | CN | S | 3-Cl | - | 0.091 | 0.29 |
| 29 | OCH3 | CN | S | 3-CH3 | 0.062 | 0.47 | |
| 30 | c-C3H5 | CN | S | - | - | NA | 0.039 |
| d4T | 1.4 | >100 | |||||
| nevirapine | 0.11 | >100 | |||||
| efavirenz | 0.002 | 15 | |||||
| etravirine | 0.001 | 11 | |||||
| rilpivirined | 0.0004 | 8 | |||||
CV = E-cyanovinyl. ClV = E-chlorovinyl.
For 50% protection in MT-2 cells; antiviral curves used triplicate samples at each concentration. NA for EC50 > CC50.
For 50% inhibition of MT-2 cell growth; toxicity curves also used triplicate samples.
Ref. 7.
Continuing, a Cl to CN change in the anilinyl ring of 9 did provide an expected6b boost to 0.64 μM for 11. Based on the EC50 values for 9 – 11, ca. 0.4 μM might have been anticipated for the 3,5-dichloro analog 12, but this was masked by the sudden enhancement of the cytotoxicity to 0.27 μM. The 3,5-dimethyl analog 13 was also prepared and yielded an EC50 of 0.49 μM, but it too was somewhat cytotoxic with a CC50 of 1.3 μM. The sensitivity of the cytotoxicity to structural changes is often high, as demonstrated here for 11 and 12 and considered further below. The origin of the cytotoxicity is unclear, but it is expected to arise from multiple mechanisms. In any event, it is desirable to have a safety margin (CC50/EC50) of ca. 1000 or more in the WT assay, as in the cases of the approved drugs in Table 2. The optimization strategy has been to drive down the EC50 values to the low nM range and hopefully achieve CC50 values at μM levels.
Phenoxy versus Thiophenyl
An excursion at this point was to consider replacing the oxygen linker with sulfur, which might insert the terminal phenyl ring farther into the π-box. Thus, FEP calculations were executed for the interconversion of 3 and 25; the outcome is summarized in Figure 5. The calculations were preformed in both directions and they consistently found that the sulfur analog should be better bound to WT HIV-RT by 0.9 kcal/mol. Compound 25 was synthesized and the assay results did yield a good activity gain to 0.32 μM. It was natural to then prepare the 3-Cl and 3-CH3 analogs with both chlorine and CN at the 4-position in the anilinyl ring. The assay results for 26 – 29 were curious in that activity boosts were not found for 26 and 27, while for the CN analogs nice EC50 gains were obtained to 91 and 62 nM for 28 and 29. However, cytotoxicity again intervened with CC50 values near 0.4 μM. Coupled with the tendency of thioethers to undergo metabolic oxidation, interest in their further pursuit was dampened. Later, one additional pyrimidinylthioether, 30, which differs from 25 by Cl for CN and OCH3 for cyclopropyl replacements, was synthesized and was found to be highly cytotoxic with a CC50 of 39 nM.
Figure 5.

Computed differences in free energies of binding, ΔΔGb (kcal/mol), with HIV-RT and observed anti-HIV activities for phenoxy and thiophenoxy analogs. The FEP calculations were run in both directions yielding the indicated results in kcal/mol.
Cyanovinyl Introduction and OCH3 Replacement
So far, 11 was the best phenoxy compound, though the gain in activity over 3 was modest. However, it was clear that two chlorines could fit at the 3- and 5-positions of the phenoxy ring. This knowledge and extensive viewing of structures of 3 bound to HIV-RT, as in Figure 2, proposed something bolder. The hydrogen on C3 in the phenoxy ring is pointing directly between Tyr181 and Tyr188. Though unprecedented in NNRTI design, this suggested that it might be possible to thread a substituent, no wider than chlorine, between the two tyrosine rings. A thin, relatively rigid substituent seemed advisable and cyanovinyl (CV) came to mind. Structure building with BOMB, followed by energy minimization, showed that complexes with compounds such as 15 should be possible with no significant protein-ligand strain. A computed structure for a complex illustrating the possible positioning of the CV group is provided in Figure 6. The CV extension is seen to fill the space well between Tyr181, Tyr188, and Trp229. It also fills the space much better in this region for the Y181C variant (Figure 6, right) than for 1 or 2.
Figure 6.

Optimized structures for 19 bound to wild-type (left) HIV-RT and the Tyr181Cys mutant (right). Highlighted residues are labeled in Figure 1.
However, given the discussion of alternative conformations above, it was prudent to attempt again to explore alternative structures. In fact, conjugate-gradient optimizations for complexes with the cyanovinyl analog of 3 revealed four possible placements for the CV group corresponding to attachment at either meta position in both the perpendicular and clamshell conformations (Figure 7). For the perpendicular conformer, it is possible for the CV group to project between Tyr181 and Tyr188 as in Figure 6, and it is also possible to place it between Pro95 and Pro97 (Figure 7, top). For the clamshell conformation, the CV group can project between Tyr188 and Phe227 or below Pro95 (Figure 7, bottom). Options B and D differ primarily in the conformation of the CV group, i.e., the dihedral angle about the bond to the phenoxy ring. The computed protein-ligand interaction energies are again similar, −68.5, −73.5, −69.8, and −72.6 kcal/mol for the structures in Figures 7A – 7D, respectively. The corresponding relative energies for the extracted ligands are 0.0, 0.9, 4.3, and 2.8 kcal/mol, and the total energies are −3394, −3381, −3382, and −3394 kcal/mol. Thus, there is uncertainty as to the preferred conformer with a single CV group on the phenoxy ring. However, addition of a second meta substituent would disfavor conformer B owing to steric conflict with Tyr188.
Figure 7.

Computed structures illustrating alternative placement of the cyanovinyl group for analogs of 15 bound to wild-type HIV-RT: projecting (A) between Tyr181 and Tyr188; (B) towards Pro95 and Pro97; (C) between Tyr188 and Phe227; and (D) below Pro95.
Given the likelihood that a CV group could be accommodated, 15 was synthesized. It was found to improve the inhibitory activity to 0.26 μM for WT HIV-1 (Table 2). With this success, attention turned to the substitution of the pyrimidine ring. The methoxy group had been inherited from 1;6b however, the change from ODMA to the cyanovinylphenoxy group is so large that it seemed likely that there might be more optimal substituents to fill the space near the Cβ’s of Tyr181 and Tyr188. Consequently, FEP calculations were executed to optimize the substituent on the pyrimidine ring in 15. The calculations used the conformation of the inhibitors with the CV group pointing between Tyr181 and Tyr188. The results are summarized in Table 3 with the fluorine-substituted case as the reference point. It was very encouraging to find multiple possibilities that were predicted to yield better binding than the methoxy analog. Among the options in Table 3, the ethyl, methylthio, cyclopropyl, and isopropyl analogs, 16 – 19, were synthesized and assayed. They are indeed much more active than 15 with the cyclopropyl analog 19 being the most potent at 32 nM (Table 2; Figures 5 and 6). The qualitative accord between the EC50 values and the FEP results is not perfect, perhaps owing to the conformational uncertainty; however, the FEP results indicated that there were better alternatives than methoxy and provided the motivation to pursue this course. The payoff was a factor of 8 activity boost in progressing from 15 to 19. For comparison, Table 3 also contains the computed protein-inhibitor interaction energies, EP-L, from energy minimizations in vacuum. In this case, the EP-L values correlate well with the observed activities with ethyl and methylthio similarly better than methoxy, and with isopropyl and cyclopropyl better still.
Table 3.
FEP and Energy-Minimization Results for Side-Chain Variations (kcal/mol).
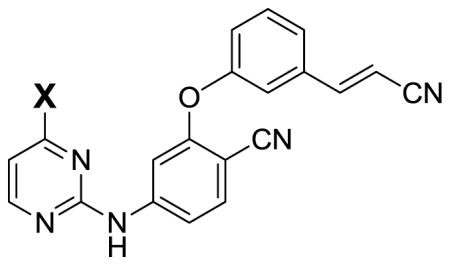 | |||||||
|---|---|---|---|---|---|---|---|
| X | ΔΔGba | σb | EP-Lc | X | ΔΔGba | σb | EP-Lc |
| H | 0.67 | 0.07 | −67.6 | OCF3 | −4.52 | 0.40 | −71.7 |
| F | 0 | 0 | −68.1 | c-C3H5 | −4.74 | 0.47 | −75.6 |
| CH3 | −0.36 | 0.12 | −70.8 | SCH3 | −4.96 | 0.43 | −71.9 |
| CH2CH3 | −2.68 | 0.19 | −73.4 | SCHF2 | −6.16 | 0.44 | −72.5 |
| OCH3 | −3.00 | 0.39 | −69.9 | CH(CH3)2 | −6.55 | 0.23 | −74.9 |
| OCHF2 | −4.46 | 0.40 | −71.5 | ||||
Computed change in free energy of binding from FEP calculations in water relative to X = F for WT HIV-RT.
Statistical uncertainties computed using Eq 1.
Protein-inhibitor interaction energy from energy minimization of the complex in vacuum with ε = 2.0.
The next step was to add again a halogen at the other meta-position in the phenoxy ring giving the chloro and fluoro analogs, 20 and 21; they provided further activity gains to 12 and 8.5 nM. Thus, starting with 3 or 4, it was possible to improve the WT activity roughly 1000-fold in the course of synthesis and assaying of ca. 20 compounds. The key advances hinged on (1) the FEP results for the optimal substitution pattern of the phenoxy ring, (2) FEP guidance on the substituent in the pyrimidine ring, and (3) the recognition from the visualization and modeling that a cyanovinyl substituent should be viable in the phenoxy ring.
At this point, improvement in the CC50 values and the activity towards the Y181C variant need to be addressed. However, first, results are noted for several additional compounds, which were synthesized to test alternative substitution patterns in the phenoxy ring. The EC50 of 0.78 μM for the 3,5-difluoro analog 22 confirms the importance of the cyanovinyl group, while the 3-Cl, 5-CN analog 23 is potent at 15 nM, but too cytotoxic (CC50 = 150 nM). In addition, replacing the CV group in 19 by chlorovinyl in 24 was found to cause a 15-fold activity loss. As for CV, the model building indicates that there are multiple alternatives for orientation of the chlorovinyl group in 24, analogous to Figure 7. However, for the 3,5-disubstituted cases, 20 and 21, the calculations favor the conformation with the CV group projecting between Tyr181 and Tyr188 or between Tyr188 and Phe227.
In closing this section, it is noted that the presence of a CV group in NNRTIs has precedent, most notably in the recently approved drug rilpivirine. However, the topology of rilpivirine and the present NNRTIs is very different such that there is no option for the CV group of rilpivirine except to be positioned between Tyr188 and Phe227 (Figure 8).24 For rilpivirine, the CV group is inserted into the π-box from the left-side near Tyr181, while it is inserted from the right-side for the present CV-containing NNRTIs.
Figure 8.

Depiction from the 2zd1 crystal structure24 of rilpivirine bound to WT HIV-RT illustrating the positioning of the cyanovinyl group between Tyr188 and Phe227. Carbon atoms of rilpivirine are in yellow.
Pyrimidine versus Triazine
It was previously found that the 1,3,5-triazine 31 is more than 100-fold less cytotoxic (CC50 = 42 μM) than the analogous pyrimidine 1 (0.23 μM).6c The reduction in WT activity from 2 nM for 1 to 11 nM for 31 is comparatively small. The 4,6-dimethoxy analog of 31 was also potent (EC50 = 22 nM) and non-toxic (CC50 > 100 μM).6c Thus, it was natural to consider switching to triazines for the cyanovinylphenoxy series in order to improve the cytotoxicity from the level for 21 and hopefully from that of 20. To gauge the potential impact on WT potency, FEP calculations were executed for the pyrimidine to triazine conversions with a methoxy or cyclopropyl group on the heterocycle. As summarized in Figure 9, the results were encouraging in indicating that there would be a small penalty with the methoxy substituent, but that the cyclopropyl-containing inhibitors should have similar potency in either azine series.
Figure 9.

Computed differences in free energies of binding, ΔΔGb (kcal/mol), with HIV-RT for conversion of pyrimidines to 1,3,5-triazines. The computed statistical uncertainties in both cases are ± 0.33 kcal/mol.

Consequently, several triazine analogs were synthesized and assayed as summarized in Table 4. The trisubstituted triazines 32 – 34 were prepared first owing to their relative ease of synthesis. A nice boost in potency was again found by replacing the methoxy group in 32 with cyclopropyl in 33. Then, the greater activity of 34 (43 nM) than 33 (160 nM) is expected to stem from hydrogen bonding of the amino group of 34 with the carboxylate group of the proximal Glu138. However, further enhanced activity is obtained by removal of the substituent at the 6-position yielding 35 (21 nM), which is the 1,3,5-triazine analog of the pyrimidine 19 (32 nM). Subsequent addition of the chlorine in the phenoxy ring gave the extremely potent 36 (2.5 nM), which can be compared to the pyrimidine 20 (12 nM). Thus, as anticipated by the FEP results in Figure 9, the switch to the triazine core was not deleterious for WT activity of the cyclopropyl-containing analogs.
Table 4.
Assay Results in μM for Triazines with WT HIV-1a
 | |||||
|---|---|---|---|---|---|
| Compd | X | Y | R | EC50 | CC50 |
| 32 | OCH3 | OCH3 | H | 0.85 | 6.3 |
| 33 | c-C3H5 | OCH3 | H | 0.16 | 10 |
| 34 | c-C3H5 | NH2 | H | 0.043 | 1.7 |
| 35 | c-C3H5 | H | H | 0.021 | 1.8 |
| 36 | c-C3H5 | H | Cl | 0.0025 | 0.12 |
| 37 | c-C3H5 | H | CH3 | 0.0017 | 1.5 |
Details as in Table 2.
Concerning the toxicity towards the MT-2 cells, 35 (1.8 μM) showed only slight improvement over 19, and the much greater cytotoxicity for the potent triazine 36 (0.12 μM) versus the corresponding pyrimidine 20 (1.1 μM) was very surprising. This illustrates well that the structure-activity data for cytotoxicity is complex and difficult to predict. Addition of the 5-chlorine had no effect for 19 and 20, while there is a 15 or 35-fold deficit for 35 and 36. Another example is the greater than 4-fold benefit for cyano over chlorine in the anilinyl ring for 9 and 11, while the opposite ordering is found by 17-fold for 26 and 28, and by 39-fold for 2 and 1.6c For the pyrimidines 12, 13, and 26 - 29, it appears that cytotoxicity diminishes for replacement of chlorine by a methyl group in the phenoxy ring. So, 37 was the final compound to be prepared. The idea paid off. 37 is a very potent NNRTI (1.7 nM) and with a CC50 of 1.5 μM, it has a safety margin of about 1000.
Viral Mutations
The remaining issue is the performance of the designs incorporating the 3-cyanovinylphenoxy group on the troublesome Tyr181Cys-containing variant form of HIV-1. The strategy was to reduce contact with Tyr181 and to better fill the space that is vacated by the Tyr181 to Cys change. The present NNRTIs also incorporate several rotatable bonds, which is viewed as beneficial for responding to structural changes in the binding site.24,25 The results are summarized for some of the more potent compounds in Table 5, which includes reference data for five drugs.
Table 5.
Activity (EC50) and Cytotoxicity (CC50) in μM for Wild-Type and the Tyr181Cys Variant Strain of HIV-1a
| WT | Y181C | |||
|---|---|---|---|---|
| Compd | EC50 | CC50 | EC50 | CC50 |
| 1 | 0.002 | 0.23 | NA | 0.11 |
| 2 | 0.010 | 9.0 | NA | 11.0 |
| 17 | 0.048 | 1.2 | 0.25 | 0.98 |
| 19 | 0.032 | 1.1 | 0.16 | 0.98 |
| 20 | 0.012 | 1.1 | 0.032 | 0.82 |
| 31 | 0.011 | 42.0 | 12.5 | 20.0 |
| 35 | 0.021 | 1.8 | 0.25 | 2.2 |
| 36 | 0.0025 | 0.12 | 0.0049 | 0.073 |
| 37 | 0.0017 | 1.5 | 0.015 | 2.2 |
| d4T | 1.4 | >100 | 0.64 | >100 |
| nevirapine | 0.11 | >100 | NA | >100 |
| efavirenz | 0.002 | 15.0 | 0.010 | >0.1 |
| etravirine | 0.001 | 11.0 | 0.008 | >0.1 |
| rilpivirine | 0.0004b | 8.0b | 0.0013b | |
As noted above, 1 and 2 show no activity against the Y181C strain, though the CC50 for 1 is low (100–200 nM), which would mask any activity above that level. The triazine 31 corresponding to 1 does show weak antiviral activity towards the Y181C variant (12.5 μM). When new NNRTIs were obtained with EC50 values below 100 nM in the WT assay, the mutant assay was initiated. The first Y181C results for the thiomethylpyrimidine 17 and the cyclopropyl analog 19 were very encouraging with EC50 values of 250 and 160 nM. Then, addition of the 5-Cl group in 20 achieved a significant milestone with EC50 values below 50 nM towards both the WT virus and the Y181C variant. The cyclopropylpyrimidine 35 was found to perform similarly to its pyridine analog 19, while 6-fold gains in potency were obtained with 36 over 20 for both the WT virus and Y181C variant. The resultant EC50 values for 36 of 2.5 nM (WT) and 4.9 nM (Y181C) are striking given the starting point of the original phenoxy compound 3. In addition, the fold-change (ratio of Y181C to WT activities) of only a factor of 2 for 36 and its greater potency towards the Y181C variant than for the reference NNRTIs, efavirenz and etravirine, are notable. Finally, the potency results for the methyl analog 37 are also excellent with EC50 values of 1.7 nM and 15 nM towards the WT and mutant viruses, which are similar to those for efavirenz and etravirine. The CC50 values for 37 are also good at 2 μM. The improved performance of the 3,5-disubstituted compounds, 20, 36 and 37, can be attributed to more complete filling of space in the region spanning between Cys181, Pro95, and Pro97.
Conclusion
The present study demonstrates the benefit of extensive molecular modeling for efficient discovery of enzyme inhibitors. The calculations featured structure building with the molecule growing program BOMB and free energy perturbation calculations for estimation of relative free energies of binding. The key goal was to obtain anti-viral agents with high potency towards variant forms of HIV-1 that incorporate the clinically important Tyr181Cys mutation in HIV reverse transcriptase. With guidance from the computations, it was possible to progress from the parent phenoxy-containing 3 with micromolar activity to the extremely potent analogs 36 and 37 with low-nanomolar potency against both wild-type HIV-1 and the Tyr181Cys variant. The transformation only required the synthesis and assaying of roughly 30 compounds. The FEP calculations were essential for predicting optimal substitution patterns for the phenoxy and pyrimidinyl rings of 3 and for investigating the change of the heterocycle from pyrimidine to triazine. A key outcome is the structurally novel NNRTI 37, which has both high potency and a large safety margin.
An issue that emerged is the need for better understanding of the origins and structure-activity relationships for the T-cell cytotoxicity, which provided sudden roadblocks at several points, e.g., for 21, 28, and 36. The extreme cytotoxicity of the structurally simple compound 30 is particularly intriguing. Interesting complexity also arose concerning the preferred conformations of the new NNRTIs when bound to HIV-RT, as illustrated in Figures 3 and 7. On-going computational and experimental investigations are addressing further these structural questions and activity towards additional viral variants.
Supplementary Material
Acknowledgments
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, GM49551) for support and to Dr. Julian Tirado-Rives for computational assistance. Receipt of the following reagents through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH is also greatly appreciated: MT-2 cells, catalog #237, and Nevirapine-Resistant HIV-1 (N119), catalog #1392, from Dr. Douglas Richman; HTLV-IIIB/H9, #398, from Dr. Robert Gallo; and HIV-1IIIB (A17 Variant) from Dr. Emilio Emini.
Footnotes
Supporting Information Available: Synthetic details, NMR and HRMS spectral data for compounds in Tables 2 and 4. These materials are available free of charge via the Internet at http://pubs.acs.org .
References
- 1.(a) Prajapati DG, Ramajayam R, Yadav MR, Giridhar R. Bioorg Med Chem. 2009;17:5744–5762. doi: 10.1016/j.bmc.2009.06.060. [DOI] [PubMed] [Google Scholar]; (b) de Bethune MP. Antiviral Res. 2010;85:75–90. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]; (b) Smerdon SJ, Jäger J, Wang J, Kohlstaedt LA, Chirino AJ, Friedman JM, Rice PA, Steitz TA. Proc Natl Acad Sci U S A. 1994;91:3911–3915. doi: 10.1073/pnas.91.9.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Rossotti R, Rusconi S. HIV Therapy. 2009;3:63–77. [Google Scholar]; (b) Chiao SK, Romero DL, Johnson DE. Curr Opin Drug Disc Dev. 2009;12:53–60. [PubMed] [Google Scholar]; (c) Adams J, Patel N, Mankaryous N, Tadros M, Miller CD. Ann Pharmacotherapy. 2010;44:157–165. doi: 10.1345/aph.1M359. [DOI] [PubMed] [Google Scholar]
- 4.Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 5.Jorgensen WL. Acc Chem Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For example, see: Jorgensen WL, Ruiz-Caro J, Tirado-Rives J, Basavapathruni A, Anderson KS, Hamilton AD. Bioorg Med Chem Lett. 2006;16:663–667. doi: 10.1016/j.bmcl.2005.10.038.Ruiz-Caro J, Basavapathruni A, Kim JT, Wang L, Bailey CM, Anderson KS, Hamilton AD, Jorgensen WL. Bioorg Med Chem Lett. 2006;16:668–671. doi: 10.1016/j.bmcl.2005.10.037.Thakur VV, Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2006;16:5664–5667. doi: 10.1016/j.bmcl.2006.08.037.Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. J Am Chem Soc. 2006;128:15372–15373. doi: 10.1021/ja066472g.Zeevaart JG, Wang L, Thakur VV, Leung CS, Tirado-Rives J, Bailey CM, Domaoal RA, Anderson KS, Jorgensen WL. J Am Chem Soc. 2008;130:9492–9499. doi: 10.1021/ja8019214.Leung CS, Zeevaart JG, Domaoal RA, Bollini M, Thakur VV, Spasov K, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2010;20:2485–2488. doi: 10.1016/j.bmcl.2010.03.006.
- 7.Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune MP, Pauwels R, Das K, Clark AD, Jr, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. J Med Chem. 2005;48:1901–1919. [Google Scholar]
- 8.(a) Jorgensen WL, Tirado-Rives J. J Comput Chem. 2005;26:1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]; (b) Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]; (c) Jorgensen WL, Tirado-Rives J. Proc Nat Acad Sci USA. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 9.Jorgensen WL, Ravimohan C. J Chem Phys. 1985;83:3050–3054. [Google Scholar]
- 10.For recent reviews, see: Chipot C, Pohorille A. Springer Series in Chemical Physics. In: Chipot C, Pohorille A, editors. Free Energy Calculations: Theory and Applications in Chemistry and Biology. Vol. 86. Springer-Verlag; Berlin: 2007. pp. 33–75.Jorgensen WL, Thomas LT. J Chem Theory Comput. 2008;4:869–876. doi: 10.1021/ct800011m.Michel J, Essex JW. J Comput Aided Mol Des. 2010;24:639–658. doi: 10.1007/s10822-010-9363-3.
- 11.Das K, Clark AD, Jr, Lewi PJ, Heeres J, de Jonge MR, Koymans LMH, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, de Béthune MP, Boyer PL, Clark P, Hughes SH, Janssen PAJ, Arnold E. J Med Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 12.Lu N, Kofke DA, Woolf TB. J Comput Chem. 2004;25:28–39. doi: 10.1002/jcc.10369. [DOI] [PubMed] [Google Scholar]
- 13.Allen MP, Tildesley DJ. Computer Simulations of Liquids. Clarendon; Oxford: 1987. [Google Scholar]
- 14.Purstinger G, De Palma AM, Zimmerhofer G, Huber S, Ladurner S, Neyts J. Bioorg Med Chem Lett. 2008;18:5123–5125. doi: 10.1016/j.bmcl.2008.07.099. [DOI] [PubMed] [Google Scholar]
- 15.Maiti D, Buchwald SL. J Am Chem Soc. 2009;131:17423–17429. doi: 10.1021/ja9081815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong D, Li J, Yuan C. Syn Comm. 2005;35:55–66. [Google Scholar]
- 17.Reyes-Gutiérrez PE, Camacho JR, Ramírez-Apan MT, Osornio YM, Martínez R. Org Biomol Chem. 2010;8:4374–4382. doi: 10.1039/c004399k. [DOI] [PubMed] [Google Scholar]
- 18.(a) Popovic M, Read-Connole E, Gallo RC. Lancet. 1984;2:1472–1473. doi: 10.1016/s0140-6736(84)91666-0. [DOI] [PubMed] [Google Scholar]; (b) Popovic M, Sarngadharan MG, Read E, Gallo RC. Science. 1984;224:497–500. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]
- 19.(a) Haertle T, Carrera CJ, Wasson DB, Sowers LC, Richmann DD, Carson DA. J Biol Chem. 1988;263:5870–5875. [PubMed] [Google Scholar]; (b) Harada S, Koyanagi Y, Yamamoto N. Science. 1985;229:563–566. doi: 10.1126/science.2992081. [DOI] [PubMed] [Google Scholar]
- 20.Lin TS, Luo MZ, Liu MC, Pai SB, Dutschman GE, Cheng YC. Biochem Pharmacol. 1994;47:171–174. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 21.Ray AS, Yang Z, Chu CK, Anderson KS. Antimicrob Agents Chemother. 2002;46:887–891. doi: 10.1128/AAC.46.3.887-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richman D, Shih CK, Lowy I, Rose J, Prodanovic P, Goff S, Griffin J. Proc Natl Acad Sci USA. 1991;88:11241–11245. doi: 10.1073/pnas.88.24.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Hao MH, Haq O, Muegge I. J Chem Inf Model. 2007;47:2242–2252. doi: 10.1021/ci700189s. [DOI] [PubMed] [Google Scholar]; (b) Brameld KA, Kuhn B, Reuter DC, Stahl M. J Chem Inf Model. 2008;48:1–24. doi: 10.1021/ci7002494. [DOI] [PubMed] [Google Scholar]
- 24.Das K, Bauman JD, Clark AD, Jr, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. Proc Nat Acad Sci USA. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang DP, Rizzo RC, Tirado-Rives J, Jorgensen WL. Bioorg Med Chem Lett. 2001;11:2799–2802. doi: 10.1016/s0960-894x(01)00510-8. [DOI] [PubMed] [Google Scholar]
- 26.Hansch C, Leo L, Unger SH, Kim KH, Nikaitani D, Lien EJ. J Med Chem. 1973;16:1207–1216. doi: 10.1021/jm00269a003. [DOI] [PubMed] [Google Scholar]
- 27.Rizzo RC, Udier-Blagovic M, Wang DP, Watkins EK, Kroeger Smith MB, Smith RH, Jr, Tirado-Rives J, Jorgensen WL. J Med Chem. 2002;45:2970–2987. doi: 10.1021/jm010580q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.