

Abstract
Protein-based liquid chromatography stationary phases are used in bioaffinity chromatography for studying drug-protein interactions, the determination of binding affinities, competitive and allosteric interactions, as well as for studying protein-protein interactions. This review addresses the development and characterization of protein-based stationary phase, and the application of these phases using frontal and zonal chromatography techniques. The approach will be illustrated using immobilized heat shock protein 90 and the immobilized estrogen related receptor stationary phases. In addition, the review discusses the use of the protein-coated magnetic beads for ligand and protein fishing as well as for the identification of unknown ligands from cellular or botanical extracts.
Keywords: Hsp90, ERR, HSA, protein fishing, frontal affinity chromatography
1. Introduction
The measurement of protein-protein interactions has become a key factor in drug discovery. The methods currently used for these measurements include but are not limited to static light scattering (SLS) [1,2], ultracentrifugation [3], X-ray scattering [4], self-interaction chromatography (SIC) [5, 6] and size-exclusion chromatography (SEC) [7, 8]. The characterization of the protein-protein interactions using these methods is typically carried out by determining the second virial coefficient parameter. A positive coefficient implies that repulsion interactions dominate the protein-protein interaction, while a negative number is indicative of a net attraction for the protein-protein interactions [9]. Dumetz et al, for example, carried out SIC to measure the effect of salt concentration on the protein-protein interactions, [6] and determined that the protein interactions show very low salt dependence for sodium chloride solutions, with a very pronounced effect when ammonium sulfate is employed, for their proteins tested. A limitation of SIC is that it requires prior immobilization of protein, which can affect protein structure and thus protein-protein interaction. In addition, SIC uses the same protein as ligand and ligate and thus is not useful to study multiprotein complexes, and several other methods are limited to protein-protein interactions between two different proteins. Bloustine et. al. used SEC technique to determine the solute distribution coefficient from the retention times measured by refractive index detector and diode array detector [7]. They also compared the result obtained by SEC technique to the results obtained by frontal chromatography [10, 11] and from light-scattering measurements [12, 13, 14]. Therefore, SIC and SEC techniques have the advantage of a shorter duration time relative to the other techniques. Although, these methods are very useful, they still use larger amounts of protein, albeit, Garcia et al., have recently shown that SIC technique can be miniaturized to a microchip, thus significantly reducing the amount of protein required for the determination of the second virial coefficient parameter [9]. SLS technique also requires large amounts of protein in order to determine the coefficient parameter and therefore is seldom used [1, 2].
Protein-protein and protein-ligand interactions have also been explored through protein immobilization on solid supports of chromatographic and non-chromatographic experimental techniques such as microarrays [15, 16], biosensors [17] and nanotechnology [18]. In protein microarrays, functionally active proteins were arrayed using microfabricated polyacrylamide gel pads to capture their samples [19], while more recently, Macbeath et al immobilized the protein onto the surface of the plates in order to probe thousands of protein-protein and ligand-protein interactions [15, 16]. This method was developed to take advantage of existing instrumentation. This was accomplished by immobilizing the protein onto a solid support while preserving its proper conformation. More specifically, the protein was immobilized covalently onto the surface of smooth flat surfaces of microscope slides. Although, several chemically derivatized slides were used the majority of the studies were carried out using the aldehyde-containing slides. This takes advantage of immobilizing the protein through primary amine containing residues, specifically the N-terminus lysine residue as they are a more reactive -amine. For details on the fabicrtaion of protein microarrays c.f. ref macbeth et al. Briefly, nanoliter volumes of protein samples is delivered on the slides in PBS with 40% glycerol, which prevents evaporation of the nanodroplets, thus keeping the proteins hydrated. The unreactive functional groups are capped using BSA. These slides have been used to study protein-protein interactions as well as small molecule protein interactions. Protein interaction at specific sites can also be studied using protein-domain microarrays, Espejo et al., used a microarray approach to study signal transduction issues and interactions that are sensitive to arginine methylation. This was accomplished using a glutathione S-transferase (GST)-fusion protein that contained a peptide-specific binding motif, allowing for the immobilization onto a support to study the specific protein interaction at a specific site [20]. Several other methods have been reported for protein microarrays and have been previously reviewed [21]. For example, Zhu et al. constructed yeast proteome microarrays containing 5800 yeast proteins and screened it for various biochemical activities [16]. Many known kinases and calcineurins were identified along with 33 new calmodulin binding partners. Knezevic et. al. showed the alteration in specific levels of more than ten cancer related proteins expression due to ionizing radiation treatment [22]. Miller et. al. also successfully showed the antibody microarrays containing 184 antibodies to profile the serum of patient diagnosed with prostate cancer to identify potential biomarkers [23]. The further development and characterization of this technology can lead to its application in a personalized medicine, where a treatment can be tailored to specific individual for an increased efficacy. .
An alternative approach is to use the immobilized protein as a stationary phase in bioaffinity chromatography. This technique is based on specific reversible interaction between the ligand and the immobilized protein.. A widely used method for the synthesis of protein-based stationary phases is the immobilization of the protein on the solid support using adsorption or covalent immobilization. The resulting protein-based LC phases (SPs) can be used to determine and characterize ligand-protein interactions [24, 25]. The theory and applicability of using immobilized proteins to explore the interactions between ligands/substrates and an immobilized cytosolic protein or enzyme were initially described by Chaiken [26] and Carr [27] and expanded to the study of transmembrane proteins by Lundahl, et al. [24, 25]. It was further expanded to the immobilization of cellular membrane fragments and their use in cellular membrane affinity chromatography (CMAC) [28]. Recent data using frontal and zonal chromatographic techniques have demonstrated that ligand binding affinities (Kd values) obtained using protein based stationary phases are comparable to affinities obtained using standard membrane binding techniques [29].
In addition, Belanger (2009) et al. immobilized Mex67-Nep1 onto sepharose beads and determined protein-protein interactions by LC/MS and western blotting techniques [30]. Magnetic beads have gained a significant amount of interest as an alternative method for fishing experiments for both ligand and protein binders. It has been demonstrated and will be discussed in greater detail below that the formation of protein-protein complexes remain intact on the surface of the protein coated magnetic beads [31, 32, 33, 34]. The protein coated magnetic beads, were successfully used to fish out binders from a mixture of binders and non-binders for HSA [35], and the identification of ligands that modulate protein-protein interactions [31]. Further, Jonker et al, have shown that the magnetic nano-particles can be used as a novel high throughput screening methodology, to determine whether a compound has an affinity for an immobilized target in a ‘yes’ or ‘no’ method [32]. The formation of a multiprotein complex was also carried out on the surface of the protein coated magnetic beads, and it has been demonstrated that the resulting protein coated magnetic bead was able to fish out a binding partner in a complex matrix, the KU-812 cellular matrix [34].
The development of protein-based stationary phase and their characterization using frontal and zonal chromatographic techniques will be demonstrated using Hsp90a and the estrogen related receptors. The use of HSA and Hsp90α-coated magnetic beads for ligand and protein fishing will also be reviewed.
2. Experimental Approach
The protein immobilized stationary phases utilized in biochromatography can be created using a variety of experimental approaches and the resulting columns used in a variety of chromatographic techniques. These various approaches are described below in section 2.1.
2.1. Synthesis of protein-immobilized LC phases
Protein immobilization onto a stationary phases can be carried out using adsorption, covalent immobilization and directional immobilization using his-tag or GST fusion proteins. Apart from these widely used approaches, it can also be carried out on the surface of an open tubular capillary or magnetic beads. The various advantages and disadvantages of these approaches have been discussed below.
The major advantage of an adsorption technique is that modification of the protein is not required. Conversely, this technique forms a weak and mainly reversible interaction between the stationary phase and protein. As a result, a slow leakage of immobilized protein from the stationary phase can be observed, which would eventually result in loss of activity [36].
The most frequently used method of protein immobilization is covalent immobilization, although commonly used, this occasionally prevents the protein from immobilizing in its proper conformation. The immobilizations are typically carried out using the N-terminal or C-terminal of the protein with the intent of preventing steric interaction between the solid support and the active sites of the protein, which are not usually present at the terminals. however, this method has the disadvantage of potentially disrupting the proper protein conformation. These methods have been extensively reported and will not be discussed here [34, 37–44].
Directional immobilization can be carried out using various techniques, including the use of fusion proteins with either a His-tag [45] or glutathione S-transferase (GST) tagged fusion proteins [46]. In addition, the proteins can be immobilized with their corresponding binder, nickel and glutathione, respectively. This approach (His-tag) has been used to immobilize the His-tagged estrogen receptor ligand binding domain, ER-LBD [47] and the His-tagged DNA unwinding element binding (DUE-B) [45] using Ni+2 as the coordinating metal ion. In addition Jonker et al., used a Cobalt (II) coated magnetic bead to immobilize the His-tagged protein [32], while McFadden et al., used a Nickel coated magnetic bead to isolate a His-tagged protein from a mixture of proteins and small molecules [31].
Open tubular capillaries [28,37,48], are a promising format for which to immobilize proteins as a significant reduction in run time is seen, thus increasing throughput. However, due to the decrease in surface area, a significant reduction in the active binding site will also be observed. The immobilization of proteins onto the surface of magnetic beads in order to analyze protein-protein or drug-protein interaction by western-blot or by LC/MS system has also been carried out. Several articles have been recently published on the use of magnetic beads for ligand and protein fishing [31, 32, 34, 35]. The major advantage of protein-coated magnetic beads is the ease in isolating an active compound from a mixture of compounds, since it does not require any additional columns or fraction collectors. Using this approach, human serum albumin was immobilized onto the surface of silica coated magnetic beads and was able to differentiate between ligands and nonligands. This screening process was also automated [25]. This application can be extended to immobilize other proteins to determine protein-protein and ligand-protein interactions and potential ligand fishing from plant extracts.
2.2. Chromatographic techniques
2.2.1. Frontal affinity chromatography
Frontal affinity Chromatography (FAC) is used in bioaffinity chromatography to study ligand-protein interactions. Schreimer et al. and Calleri et. al. have reviewed the application of FAC by immobilizing various proteins onto the columns and using it to determine binding affinities of potential therapeutic compounds [49, 50]. Briefly, a marker ligand is placed in the mobile phase and passed through the column in the presence or absence of serial concentrations of a displacing ligand under dynamic equilibrium conditions in FAC [51]. The marker ligand can be monitored using a radioflow detector (radiolabeled ligand), mass spectrometer, uv or fluorescence (for a fluorescence ligand). The resulting chromatographic traces contains initially a relatively flat initial portion, which represents the nonspecific and specific binding of the marker ligand to the stationary phase, followed by a vertical breakthrough, which reflects the saturation of the specific binding sites on the immobilized protein, and ending in a plateau, which corresponds to the complete saturation of these sites, Figure 1.
Figure 1.

Cartoon showing the basic principal of Frontal Chromatography {Reprinted from Reference 50}
Frontal chromatography allows the determination of the binding affinity of the competing ligand (Kd) and the number of active binding sites (Bmax) for the immobilized protein. This is determined using Eqn 1.
| (1) |
Where [D] is the concentration of displacer ligand, V is the retention volume corresponding to 50% maximal response; Vmin is the retention volume of displacer ligand when the specific interaction is completely suppressed and P is the product of the Bmax (the number of active binding sites) and (Kd/KdM). From the plot of [D] (V – Vmin) versus [D], dissociation constant values (Kd), for displacer ligand can be obtained. For the results to be quantitative, Eqn 1 assumes that the concentration of the Marker ligand is at least one order of magnitude less than its Kd for the immobilized protein.
FAC can be used to screen mixture of compounds, for example, a library of 356 β-galactopyranoside compounds were divided in a group of 25–40 compounds which were screened using an immobilized β-galactosidase enzyme on a LC stationary phase column [52].
2.2.2 Non linear chromatography
Non-linear Chromatograph (NLC) is also used in bioaffinity chromatography to study ligand-protein interactions. The zonal injections of ligands result in asymmetric peak profiles with tailing that is proportional to the injected concentration The shape of this chromatographic peak represents specific and nonspecific interactions between the solute and the stationary phase. When the stationary phase contains an immobilized protein, the mass transfer process defined by the association and dissociation of a ligand–protein complex usually is slow, producing broad, non-Gaussian chromatographic peaks with significant tailing. The degree of deviation from a Gaussian distribution is a function of applied ligand concentration, and the concentration-dependent asymmetry can be used with NLC techniques to characterize the separation processes occurring on the column, including the kinetics involved in the formation and dissociation of the solute–stationary phase complex, the association (ka) and dissociation (kd) rate constants, and the equilibrium constant (Ka, calculated as ka/kd) [37, 53].
The observed peak asymmetries were analyzed using Impulse Input Solution (see Eqn 3 below as described previously [53–55].
| (3) |
Where y is intensity of signal, x is reduced retention time, I0 and I1 are modified Bessel functions, a0 is area parameter and a1 is center parameter (which determines the true thermodynamic capacity factor k’), a2 is width parameter, and a3 is distortion parameter. Kinetic parameters can be calculated as follows: kd = 1/a2/t0, Ka = a3/C0, and ka = Ka/kd, where t0 is the dead time of the column and C0 is the concentration of ligand injected multiplied by the width of the injection pulse [55, 56].
3. Application of Biochromatography
Various membrane and cytosolic proteins have been immobilized onto silica-based stationary phases by several groups, allowing them to study the binding interactions of single or multiple compounds using biochromatography techniques. These applications are exemplified here by the synthesis and application of stationary phases containing immobilized estrogen related receptors α and γ (ERRα, ERRγ), heat shock protein 90α (HSP90 α) and human serum albumin (HSA) proetin. These phases have been used in high-throughput screening methods including ligand and protein fishing using protein coated magnetic beads, microaffinity columns etc. [34, 35].
3.1. Estrogen Related Receptor (ERRαand ERRβ)
ERRs are a subfamily of nuclear receptors that is currently composed of three isoform (ERRα, ERRβ, and ERRγ) [57, 58]. All three proteins possess the typical functional domains of nuclear receptors and were orphan receptors identified based on their high level of sequence identity with estrogen receptors (ER) α and β[36].
There are a number of characterized ERR agonists which include daidzein, biochanin A, genistein and 6,3′,4′-trihydroxyflavone [59] and DY131. DY131, in particular, has been shown to increase ERR transcriptional activity by ~3–4 fold in CV-1 cells [60]. ERR antagonists include diethylstilbestrol (DES) and 4-hydroxytamoxifen (4-OHT) [59]. 4-OHT is selective for the ERRβ and γ while DES is an antagonists for all three isoforms, ERRα ERRβ and ERRγ [59, 61-63]. Greshil et. al. reported the mechanism of ERR deactivation upon binding of antagonists like DES and 4-OHT [63].
The ERR proteins (Genscript, Piscataway, NJ) were initially immobilized onto the surface of an amine-coated silica stationary phase. The ERRα and ERRγ columns were characterized by FAC and the obtained binding affinities correlated with previously reported data [36].
Although the results of the initial studies with the ERRα and ERRγ LC column demonstrated that the phases could be used to characterize binding to the immobilized receptor, the application of these columns to compound screening was limited by the required 3–6 h wash time. In order to increase throughput, ERRα and ERRγ was immobilized on the surface of open tubular capillaries resulting in ERRα-OT and ERRγ-OT columns. DES was used as the marker ligand to characterize the ERRα-OT and ERRγ-OT columns using FAC techniques and obtained chromatograph using various concentrations of DES is shown in Figure 2. The calculated Kd value for ERRγ–OT, 237 nM [37], was the same as the value obtained on the ERRγ-silica column, which clearly indicates that the protein interactions with DES were similar on both columns. However, the calculated maximum binding capacity value was 40-fold lower relative to the ERRγ-silica column (4 nmol) [37]. In contrast to ERRγ-silica column, the maximum binding capacity of ERRα on the silica support did not differ significantly between the open tubular format and the silica column and the calculated Kd value was 929 nM [37].
Figure 2.

A. The effect of increasing concentration of diethylstilbesterol on its chromatographic retention on the ERRγ-OT column from right to left (0.05, 0.1, 0.25 and 0.5 μM). B. The effect of increasing concentration of diethylstilbesterol on its chromatographic retention on the ERRγ-column from right to left (0.05, 0.1, 0.25, 0.5, 1, 2μM) (Reprinted from reference 37).
3.2. Human Serum Albumin (HSA)
Ligand fishing using protein coated magnetic beads (MB) has been reported for the first time by our laboratory [35]. HSA coated magnetic beads were used to fish out known binders from a mixture of nonbinders. The protocol was optimized by studying incubation time and elution conditions. A mixture of known binders (Warfarin, Azidothymidine and Naproxen) and nonbinders (Nicotine, Fenoterol and Labetolol) were incubated with magnetic beads for 15 minutes. The supernatant was collected using Dynal magnetic separator followed by wash and elution. Load, wash and elution containing individual or mixture of binders were detected and quantified by Agilent LC-MSD. As shown in Figure 3, HSA ligands were retained until the final elution (A4), whereas the nonbinders were not retained and were present in the loading solution (A1) (Nicotine, fenoterol, labetolol). This method was then automated using the Magtration system 12GC PSS Bio Instruments Inc., which can run upto 12 samples in parallel. The known binder ligands were successfully separated from nonbinders using the magtration system (Figure 3). This demonstrates the versatility of the use of protein coated magnetic beads to isolate ligands/proteins from a complex matrix, for example cellular or botanical extracts.
Figure 3.

HSA coated magnetic beads were incubated with warfarin, Azidothymidine, naproxen, nicotine, fenoterol and labetolol. Nonabsorbed and eluted material were analyzed. A direct comparison of the manual ligand fishing results [A1 (Non-absorbed) Vs A4 (Eluted)] and the automated ligand fishing results [B1 (Non-absorbed) Vs B4 (Eluted)]. (Reprinted from Reference 35)
3.3. Heat Shock Protein α Hsp90α)
Hsp90αis the only inducible form in a family of molecular chaperones (Hsp90α, Hsp90β Grp94, Trap1), which are involved in the folding, intracellular disposition and proteolytic turnover of many key regulators for cell growth and survival [65]. Hsp90α exists in multiple conformations including, an ATP-free state, ATP-bound state or in a protein complex with other co-chaperones and client proteins. It has been recently demonstrated that Hsp90α is significantly elevated in glioma cells compare to normal astrocytes and this disparity indicates that Hsp90α is a potential therapeutic target for the treatment of astrocytomas [66]. Hsp90α is also viewed as a promising target for the treatment of many forms of cancer and a number of drug development programs are aimed at the discovery of compounds that will selectively inhibit the proteins intrinsic ATPase activity [67].
HSP90α usually exists as a homodimer made up of monomers that consist of three main domains, each of which have important functional interactions, Figure 4 [65]. Atomic resolution of crystal structures have only been solved for individual monomers due to intrinsic conformational mobility of the intact protein. The protein contains highly conserved N-, middle-, and C-terminal domains [65]. The N-terminal domain contains an adeninenucleotide-binding pocket, which has been associated with the intrinsic ATPase activity. There are some reports on extensive structural alterations driven by the hydrolysis of ATP to ADP in the adenine nucleotide-binding pocket, which have an essential role in the chaperoning activity of the HSP90 dimer [68, 69]. The chaperone function of Hsp90α is sensitive to oxidative stress and it has been confirmed by using chromatographic approach (See section 3.3.2.).
Figure 4.

The numbering 1–732 indicates the approximate positions in the amino acid sequence of the human protein that define its functional domains. ‘CR’ refers to a charged region which serves as a flexible linker between the N-terminal and middle domains. The locations where various small molecules bind HSP90 (heat-shock protein of 90 kDa) and modulate its function are indicated. The biochemical functions of each domain are also shown. 17AAG, 17-allylaminogeldanamycin; GA, geldanamycin {Reprinted from Ref. 65}.
Hsp90α was immobilized at N-terminus (Figure 5a) and C-terminus (Figure 5b) onto the silica stationary phase and the resulting columns were characterized using C-terminal ligands and N-terminal ligands respectively [38]. Coumermycin A1 (CA1) and novobiocin (NOVO), C-terminal ligands, were used to characterize binding to the exposed C terminus on the Hsp90α-NT column, while geldanamycin (GM), 17-allylamino-17-demethoxygeldanamycine (17-AAG) and radicicol (RAD) were used to characterize the exposed N-terminus on the HSP90α-CT column. The addition of GM, an N-terminus ligand, to the mobile phase did not displace NOVO on the HSP90α-NT, indicating that they are not competing for the same site. These results indicate that GM did not specifically bind to the Hsp90α-NT column, nor did it competitively or allosterically displace the C-terminus ligands. Similar results were obtained with the Hsp90α-CT column and C-terminus ligand binders.
Figure 5.

The synthetic approaches used in the covalent immobilization of Hsp90α on an aminopropyl silica liquid chromatography stationary phase via the a) amino terminus and b) carboxyl terminus (Reprinted from Reference - 38).
The relative affinities were determined using the Hsp90α-CT column, RAD > GM > 17-AAG and are consistent with the previously reported relative IC50 values for the inhibition of Hsp90 ATPase activity [70].
3.3.1. ATPase activity of Hsp90α-CT column
Since the inhibition of the ATPase activity of Hsp90α is one of the properties used to screen for new drug candidates, the effect of immobilization of Hsp90α on ATPase activity and the sensitivity to inhibition of this activity were determined. The ATPase activity of Hsp90α has been associated with an adenine nucleotide binding pocket in the N-terminus [65, 71], and, therefore, the Hsp90α-CT column was used in these studies. The initial studies demonstrated that the ATPase mediated conversion of ATP to ADP occurred on the Hsp90α CT-column [38], and that this hydrolysis could be inhibited by GM.
3.3.2. Thiol Oxidation of Hsp90α
The chaperone function of Hsp90α is sensitive to oxidative stress [72], therefore the Hsp90α CT column was used to ascertain whether a reversible S-thiolation reaction plays a role in regulating the intrinsic ATPase function of Hsp90α by carrying out the S-Glutathionylation of the immobilized Hsp90α This reaction was performed according to the protocol described in Caplan et. al. [73]. Briefly, Hsp90α CT was incubated with 2 mM GSH and 1mM diamide in degassed buffer A [40 mM Tris-HCl (pH 8.0), 140 mM NaCl, and 0.2 mM EDTA] for 1 h at room temperature in the dark, followed by extensive washing to remove excess reagents. 3 mM dithiothreitol was included in buffer A as a control to monitor the reversibility of diamide-induced S-thiolation of immobilized Hsp90α. ATPase activity was carried out in the presence or absence of 10 μM GM in the mobile phase as discussed previously [38]. Further, the thiol oxidation of Hsp90α-column by diamide plus GSH mediated functional impairment in the chaperone s ATPase activity, which was reversed by subsequent incubation of the column with the reducing agent dithiothreitol, as shown in Figure 6. This data demonstrated that the immobilized Hsp90α could undergo S-thiolation, which interfered in the intrinsic ATPase actiivity and thus resulted in a disruption of the chaperone function toward client proteins.
Figure 6.
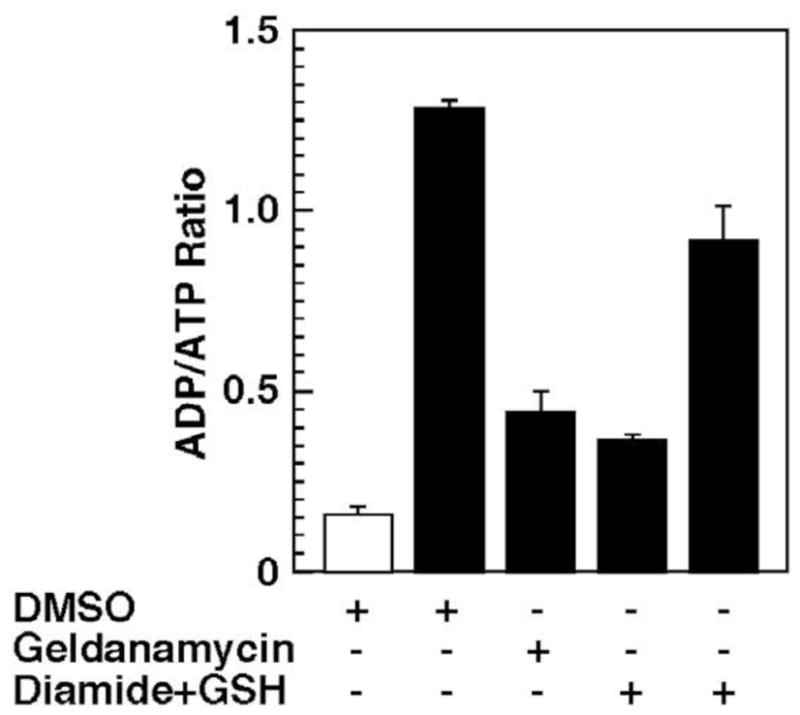
Effect on the ATPase activity of an Hsp90α(CT) column of the addition of either the Hsp90-binding drug, geldanamycin, to the mobile phase or from diamide-mediated S-glutathionylation of the immobilized Hsp90. Calculation of the ADP/ATP ratio represents the AUC (expressed as ion abundance) of the peak produced by mass spectral analysis of ATP and ADP. Bars represent the mean ± S.D. of three independently conducted experiments.
3.4 Ligand fishing using Hsp90 coated magnetic beads (Hsp90α–MB)
3.4.1. Preparation of Hsp90α–MB
Hsp90α was also immobilized through the N-terminus and C-terminus onto the surface of magnetic beads resulting in the Hsp90α(CT)-MB and Hsp90α(NT)-MB, respectively [34]. The detailed procedure for the immobilization of Hsp90α at N-terminus and C-terminus onto magnetic beads has been previously reported [34]. Briefly, N-terminus of Hsp90α protein was immobilized onto an amine coated magnetic beads by reductive amination using gluteraldehyde and sodium cyanoborohydride. In order to immobilize Hsp90α at C-terminus, amine coated magnetic beads were incubated by Hsp90α protein and EDAC for the conjugation reaction in presence of Sulfo-NHS.
3.4.2. Application of Hsp90α(CT)-MB and Hsp90α(NT)-MB
Hsp90α(CT)-MB and Hsp90α(NT)-MB were incubated with individual compound solutions of two C-terminus binders (CA1, NOVO), two N-terminus binders (17-AAG and GM), and two non-binders (nicotine, propranolol). The magnetic beads were isolated using a Dynal Magnetic Separator, and the supernatant, which contains the unbound ligands, was then analyzed by mass spectrometry. The results indicated that the Hsp90α (CT)-MB bound the N-terminus ligands from the mixture at greater than 80%, with less than 30% of the non-binders and C-terminus ligands [34]. The Hsp90α(NT)-MB captured the C-terminus binders at levels greater than 75%, with less than 35% of the nonbinders and the N-terminus ligands [34].
3.4.3. Protein Fishing with Protein Coated Magnetic Beads
Besides ligand fishing, Hsp90α monomer coated magnetic beads were also used to isolate proteins from a mixture of proteins, as well as a cellular extract [34]. Hsp90α coated magnetic beads were incubated with individual protein (eNOS, p60 HOP and Hsp70) and both non-absorbed and eluted material were analyzed by western blotting techniques (Figure 7). Considering the fact that p60 HOP binds at the C-terminal domain of Hsp90α, Hsp90αNT-MB extracted more protein then Hsp90αCT MB. The result showed that only 8% and 14% remained in the supernatant [34]. In contrast, 70% Hsp70 was retained in the supernatant [34]. These results were consistent with the previously reported data that eNOS and p60 HOP bind directly to Hsp90, whereas Hsp70 binds indirectly through the co-chaperone p60 HOP (Figure 7a; 7b). When Hsp90α coated magnetic beads were incubated with a mixture of these proteins, Hsp70 was retained in the presences of p60 HOP (Figure 7b). This demonstrates that a multiprotein complex was formed in the presences of p60 HOP and Hsp70. It was also demonstrated that the Hsp90 coated magnetic beads could also be used to ‘fish’ out client proteins from a cellular matrix. Hsp90α coated magnetic beads were incubated with lysates from KU-812 basophiles containing p60 HOP. The non-absorbed and eluted material was analyzed by western blot experiment with antibodies against known proteins. The Hsp90α coated magnetic beads succesfully “fished” out p60 HOP from the cellular matrix [34], indicating that protein coated magnetic beads can be used to identify or isolate new drug candidates or client proteins in complex chemical and biological mixtures. In addition, Hsp90α dimer coated magnetic beads were used to attempt ligand and protein fishing experiments. However, we did not have any success due to high non-specific interaction. Different approach needs to be performed in order to decrease non-specific interaction.
Figure 7.

Hsp90αNT- MBs were incubated with eNOS, p60 HOP and Hsp70 proteins individually (a) or in mixture (b). The nonabsorbed and eluted material was collected and analyzed using western blot analysis. Lane 1, control; Lane 2, Hsp90αNT supernatant (non-absorbed); Lane 3, eluted material. (Reprinted from reference 34).
McFadden et al. also demonstrated that ‘protein complex fishing’ using a His-tagged calmodulin as the primary protein to be isolated from a cellular matrix onto a Nickel coated magnetic beads [31]. It has been previously demonstrated that His-tagged fusion protein could be immobilized using a Nickel coated stationary phase and used for characterization of the fusion protein [45, 47]. In the current study, they demonstrated that the calmodulin-melittin complex was screened against 50 mixtures of 20 compounds and correctly identified three known antagonists as well as 2 previously unknown antagonist. Several other studies have also reported on the versatility of the magnetic beads for the study of protein-protein interactions [31, 32, 33].
4. Conclusions
Bioaffinity columns, as well as protein-coated magnetic beads can be used to study ligand–protein and protein-protein interactions including the determination of binding affinities and thermodynamic properties of the ligand protein interaction. In addition to the characterization of the agonist binding site, this technique can also be used to study immobilized proteins in distinct conformations [74], as well as at different allosteric sites. The development of allosteric modifiers as therapeutic agents is a growing area in drug discovery and bioaffinity chromatography is an excellent method for the determination and measurement of this property [75]. Bioaffinity chromatography has been extensively applied to the characterization of an immobilized protein using one ligand at a time. However, this approach can also be used to simultaneously characterize individual compounds contained within complex chemical mixtures [34, 76] and to isolate and identify active components from complex biological matrices [34, 77]. The extraction of active components from complex biological matrices is historically and currently a major component of drug discovery. While advances in chromatographic separations and spectroscopic identification have reduced the effort required to identify new lead drugs, the process is still time consuming and problematic. Bioaffinity chromatography has a promising role to play in this process as the versatility of the protein based stationary phases can be adapted to potentially isolate new, previously unknown, therapeutic drugs. In addition, to the columns, our initial work with the protein-coated magnetic beads demonstrates that this technique may have a greater advantage than the protein-based stationary phase in this regard.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging. There are no financial conflicts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Curtis R, Ulrich AJ, Montaser A, Prausnitz JM, Blanch HW. Biotech Bioeng. 2002;79:367. doi: 10.1002/bit.10342. [DOI] [PubMed] [Google Scholar]
- 2.Piazza R. J Crystal Growth. 1999;196:415. [Google Scholar]
- 3.Behlke J, Ristau O. Biophys Chem. 1999;76:13. doi: 10.1016/s0301-4622(98)00212-9. [DOI] [PubMed] [Google Scholar]
- 4.Vivares D, Bonnete F. Acta Crystallogr D. 2002;58:472. doi: 10.1107/s0907444902000124. [DOI] [PubMed] [Google Scholar]
- 5.Tessier PM, Lenhoff AM, Sandler SI. Biophys J. 2002;82:1620. doi: 10.1016/S0006-3495(02)75513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumetz AC, Snellinger-O’Brien AM, Kaler EW, Lenhoff AM. Protein Science. 2007;16:1867. doi: 10.1110/ps.072957907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bloustine J, Berejnov V, Fraden S. Biophys J. 2003;85:2619. doi: 10.1016/s0006-3495(03)74684-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bajaj H, Sharma VK, Kalonia DS. Biophys J. 2004;87:4048. doi: 10.1529/biophysj.104.048686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garica CD, Hadley DJ, Wilson WW, Henry CS. Biotechnology Progress. 2003;19:1006. doi: 10.1021/bp025788z. [DOI] [PubMed] [Google Scholar]
- 10.Nichol LW, Siezen RJ, Winzor DJ. Biophys Chem. 1978;9:47. doi: 10.1016/0301-4622(78)87014-8. [DOI] [PubMed] [Google Scholar]
- 11.Willis PR, Nichol LW, Siezen RJ. Biophys Chem. 1980;11:71. doi: 10.1016/0301-4622(80)85009-5. [DOI] [PubMed] [Google Scholar]
- 12.Gripon C, Legrand L, Rosenman I, Vidal O, Robert MC, Boue F. J Crystal Growth. 1997;178:575. [Google Scholar]
- 13.Kulkarni AM. Master’s thesis Uni of Illinois. Urbana-Champaign, IL: [Google Scholar]
- 14.Veleve OD, Kaler EW, Lenhoff AM. Biophys J. 1998;75:2682. doi: 10.1016/S0006-3495(98)77713-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacBeath G, Schreiber SL. Science. 2000;289:1760. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M. Science. 2001;293:2101. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 17.Wu G, Datar RH, Hansen KM, Thundat T, Cote RJ, Majumdar A. Nat Biotechnol. 2001;19:856. doi: 10.1038/nbt0901-856. [DOI] [PubMed] [Google Scholar]
- 18.Yadavalli VK, Forbes JG, Wang K. Langmuir. 2006;22:6969. doi: 10.1021/la060320h. [DOI] [PubMed] [Google Scholar]
- 19.Arenkov P, Kukhtin A, Gemmell A, Voloshchuk S, Chupeeva V, Mirzbekov A. Anal Biochem. 2000;278:123. doi: 10.1006/abio.1999.4363. [DOI] [PubMed] [Google Scholar]
- 20.Espejo A, Cote J, Bednarek A, Richard S, Bedford MT. Biochem J. 2002;367:697. doi: 10.1042/BJ20020860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoll D, Templin MF, Bachmann J, Joos TO. Curr Opin in Drug Discov and Dev. 2005;8:239. [PubMed] [Google Scholar]
- 22.Knezevic V, Leethanakul C, Bichsel VE, Worth JM, Prabhu VV, Gutkind JS, Liotta LA, Munson PJ, Petricoin EF. Proteomics. 2001;1:1271. doi: 10.1002/1615-9861(200110)1:10<1271::AID-PROT1271>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 23.Miller JC, Zhou H, Kwekel J, Cavallo R, Burke J, Butler EB, Teh BS, Haab BB. proteomics. 2003;3:56. doi: 10.1002/pmic.200390009. [DOI] [PubMed] [Google Scholar]
- 24.Brekkan E, Lundqvist A, Lundahl P. Biochemistry. 1996;35:12141. doi: 10.1021/bi9603231. [DOI] [PubMed] [Google Scholar]
- 25.Yang Q, Lundahl P. Biochemistry. 1995;34:7289. doi: 10.1021/bi00022a001. [DOI] [PubMed] [Google Scholar]
- 26.Chaiken IM. J Chromatogr. 1986;376:11. doi: 10.1016/s0378-4347(00)80821-x. [DOI] [PubMed] [Google Scholar]
- 27.Wade JL, Bergold AF, Carr PW. Anal Chem. 1987;59:1286. [Google Scholar]
- 28.Moaddel R, Wainer IW. Nat protocol. 2009;4:197. doi: 10.1038/nprot.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moaddel R, Jozwiak K, Wainer IW. Med Res Rev. 2007;27:723. doi: 10.1002/med.20091. [DOI] [PubMed] [Google Scholar]
- 30.Belanger KD. Life Sci Edu. 2009;8:214. doi: 10.1187/cbe.09-03-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mcfadden MJ, Junop MS, Brennan JD. Anal Chem. 2010;82:9850. doi: 10.1021/ac102164d. [DOI] [PubMed] [Google Scholar]
- 32.Jonker N, Krestchmer A, Kool J, Fernandez A, Kloos D, Krabbe JG, Lingeman H, Irth H. Anal Chem. 2009;81:4263. doi: 10.1021/ac9000755. [DOI] [PubMed] [Google Scholar]
- 33.Li JS, Ge JP, Yin YD, Zhong WW. Anal Chem. 2008;80:7068. doi: 10.1021/ac801251y. [DOI] [PubMed] [Google Scholar]
- 34.Marszall MP, Moaddel R, Kole S, Gandhari M, Bernier M, Wainer IW. Anal Chem. 2008;80:7571. doi: 10.1021/ac801153h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moaddel R, Marszall MP, Bighi F, Yang Q, Duan X, Wainer IW. Anal Chem. 2007;79:5414. doi: 10.1021/ac070268+. [DOI] [PubMed] [Google Scholar]
- 36.Wong LS, Khan F, Micklefield J. Chem Rev. 2009;109:4025. doi: 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- 37.Sanghvi M, Moaddel R, Frazier C, Wainer IW. J Pharm Biomed Anal. 2010;53:777. doi: 10.1016/j.jpba.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marszall MP, Moaddel R, Jozwiak K, Bernier M, Wainer IW. Anal Biochem. 2008;373:313. doi: 10.1016/j.ab.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larsson PO. Methods Enzymol. 1984;104:212. doi: 10.1016/s0076-6879(84)04091-x. [DOI] [PubMed] [Google Scholar]
- 40.Erler U, Heublein G. J Chromatogr A. 1991;588:340. [Google Scholar]
- 41.Zhang Q, Hunag R, Guo L. Chi Sci Bull. 2009;54:2620. [Google Scholar]
- 42.Axen R, Porath J, Ernback S. Nature. 1967;214:1302. doi: 10.1038/2141302a0. [DOI] [PubMed] [Google Scholar]
- 43.Kohn J, Wilchek M. Biochem Biophys Res Commun. 1982;197:878. doi: 10.1016/0006-291x(82)90604-0. [DOI] [PubMed] [Google Scholar]
- 44.Paul R, Anderson GW. J Chem Soc. 1960;82:4596. [Google Scholar]
- 45.Moaddel R, Price GB, Juteau JM, Leffak M, Wainer IW. J Chromatogr B. 2005;820:197. doi: 10.1016/j.jchromb.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 46.Smith DB, Johnson KS. Gene. 1988;67:31. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 47.Moaddel R, Lu L, Baynham M, Wainer IW. J Chromatogr B. 2002;768:41. doi: 10.1016/s0378-4347(01)00484-4. [DOI] [PubMed] [Google Scholar]
- 48.Moaddel R, Bullock IP, Wainer IW. J Chromatogr B. 2004;799:255. doi: 10.1016/j.jchromb.2003.10.054. [DOI] [PubMed] [Google Scholar]
- 49.Calleri E, Temporini C, Massolini G. J Pharm Biomed Anal. 2011;54:911. doi: 10.1016/j.jpba.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 50.Ng ESM, Chora NWC, Lewis DF, Hindsgaul O, Schriemer D. Nat Protocol. 2007;2:1907. doi: 10.1038/nprot.2007.262. [DOI] [PubMed] [Google Scholar]
- 51.Calleri E, Temporini C, Caccialanza G, Massolini G. Chem Med Chem. 2009;4:905. doi: 10.1002/cmdc.200800436. [DOI] [PubMed] [Google Scholar]
- 52.Chan NW, Lewis DF, Hewko S, Hindsgaul O, Schriemer DC. Comb Chem High Throughput Screen. 2002;5:395. doi: 10.2174/1386207023330192. [DOI] [PubMed] [Google Scholar]
- 53.Schiel JE, Joseph KS, Hage DS. Adv Chromatogr. 2010;48:145. [PubMed] [Google Scholar]
- 54.Jozwiak K, Haginaka J, Moaddel R, Wainer IW. Anal Chem. 2002;74:4618. doi: 10.1021/ac0202029. [DOI] [PubMed] [Google Scholar]
- 55.Wade J, Bergold AF, Carr PW. Anal Chem. 1987;59:1286. [Google Scholar]
- 56.Jaulmes A, Vidal-Madjar C. Advances in Chromatography. 1989;28:1. [PubMed] [Google Scholar]
- 57.Gigue’re V, Yang N, Segui P, Evans RM. Nature. 1988;331:91. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 58.Eudy JD, Yao S, Weston MD, Ma-Edmonds M, Talmage CB, Cheng JJ, Kimberling WJ, Sumegi J. Genomics. 1998;50:382. doi: 10.1006/geno.1998.5345. [DOI] [PubMed] [Google Scholar]
- 59.Suetsugi M, Su L, Karlsberg K, Yuan YC, Chen S. Mol Cancer Res. 2003;1:981. [PubMed] [Google Scholar]
- 60.Coward P, Lee D, Hull MV, Lehmann J. Proc Natl Acad Sci U S A. 2001;98:8880. doi: 10.1073/pnas.151244398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tremblay GB, Kunath T, Bergeron D, Lapointe L, Champigny C, Bader JA, Rossant J, Giguere V. Genes Dev. 2001;15:833. doi: 10.1101/gad.873401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tremblay GB, Bergeron D, Giguere V. Endocrinology. 2001;142:4572. doi: 10.1210/endo.142.10.8528. [DOI] [PubMed] [Google Scholar]
- 63.Greschik H, Flaig R, Renaud J, Moras D. The Journal of Biol Chem. 2004;279:33639. doi: 10.1074/jbc.M402195200. [DOI] [PubMed] [Google Scholar]
- 64.Yu DD, Forman BM. Bioorg Med Chem Lett. 2005;15:1311. doi: 10.1016/j.bmcl.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 65.Whitesell L, Lindquist SL. Nat Rev Cancer. 2005;5:671. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 66.Shervington A, Cruickshanks N, Lea R, Roberts G, Dawson T, Shervington L. Cancer Investigation. 2008;26:900. doi: 10.1080/07357900802087259. [DOI] [PubMed] [Google Scholar]
- 67.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. Nature. 2003;425:407. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 68.Meyer P. Mol Cell. 2003;1:647. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 69.McLaughlin SH, Ventouras LA, Lobbezoo B, Jackson SE. J Mol Biol. 2004;344:813. doi: 10.1016/j.jmb.2004.09.055. [DOI] [PubMed] [Google Scholar]
- 70.Rowlands MR, Newbatt YM, Prodromou C, Pearl LH, Workman P, Aherne W. Anal Biochem. 2004;327:176. doi: 10.1016/j.ab.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 71.Calderwood SK, Khaleque MD, Sawyer DB, Ciocca DR. Trends Biochem Sci. 2006;31:164. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 72.Song S, Kole S, Precht P, Pazin MJ, Bernier M. Int J Biochem Cell Biol. 2010;42:1856. doi: 10.1016/j.biocel.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Caplan JF, Filipenko NR, Fitzpatrick SL, Waisman DM. J Biol Chem. 2004;279:7740. doi: 10.1074/jbc.M313049200. [DOI] [PubMed] [Google Scholar]
- 74.Moaddel R, Wainer IW. Anal Chim Acta. 2006;564:97. doi: 10.1016/j.aca.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 75.Chen J, Hage DS. Nat Biotechnol. 2004;22:1445. doi: 10.1038/nbt1022. [DOI] [PubMed] [Google Scholar]
- 76.Calleri E, Ceruti S, Cristalli G, Martini C, Temporini C, Parravicini C, Volpini R, Daniele S, Caccialanza G, Lecca D, Lambertucci C, Trincavelli ML, Marucci G, Wainer IW, Ranghino G, Fantucci P, Abbracchio MP, Massolini G. J Med Chem. 2010;53:3489. doi: 10.1021/jm901691y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maciuk A, Moaddel R, Haginaka J, Wainer IW. J Pharm Biomed Anal. 2008;48:238. doi: 10.1016/j.jpba.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]