

Abstract
Obesity is a low grade chronic inflammatory disease associated with an increased number of macrophages (ATM) in adipose tissue. Within the adipose tissue, ATM are the major source of visfatin/PBEF/NAMPT. The nuclear receptor Peroxisome Proliferator-Activated Receptor (PPAR)γ exerts anti-inflammatory effects in macrophages by inhibiting cytokine production and enhancing alternative differentiation. In this study, we investigated whether PPARγ modulates visfatin expression in murine (BMDM) and human (RM, M1, M2, ATM) macrophage models and preadipocyte-derived adipocytes. We show that synthetic PPARγ ligands increased visfatin gene expression in a PPARγ-dependent manner in primary human macrophages (RM) and ATM, but not in adipocytes. The increase of visfatin mRNA (3-fold) was paralleled by an increase of protein expression (30%) and secretion (30%). Electrophoretic Mobility Shift Assay (EMSA) experiments and transient transfection assays indicated that PPARγ induces visfatin promoter activity in human macrophages by binding to a DR1-PPARγ response element. Finally, we show that PPARγ ligands increase NAD+ production in primary human macrophages and this regulation is dampened in the presence of visfatin siRNA or by the visfatin-specific inhibitor FK866. Taken together, our results suggest that PPARγ regulates the expression of visfatin in macrophages leading to increased NAD+ levels.
Keywords: Animals; Base Sequence; Blotting, Western; Cells, Cultured; Enzyme Inhibitors; pharmacology; Enzyme-Linked Immunosorbent Assay; Gene Expression Regulation, Enzymologic; drug effects; Humans; Leukocytes, Mononuclear; enzymology; Mice; Mice, Inbred C57BL; Molecular Sequence Data; NAD; chemistry; Nicotinamide Phosphoribosyltransferase; genetics; metabolism; PPAR gamma; agonists; metabolism; RNA, Messenger; metabolism
Keywords: nuclear receptors, adipocytokines, visfatin, inflammation, macrophages
INTRODUCTION
Originally discovered in liver, skeletal muscle and bone marrow, also known as pre-B cell colony-enhancing factor (PBEF), a cytokine acting in B cell differentiation [1], visfatin is nicotinamide phosphoribosyl transferase (NAMPT) [2, 3], a rate-limiting enzyme in the synthesis of nicotinamide adenine dinucleotide (NAD+) from nicotinamide. Visfatin/PBEF/NAMPT is synthesized and secreted in adipose tissue by adipocytes and mostly by macrophages and circulates in plasma of humans and mice [4]. Plasma visfatin concentrations are positively associated with cytokines such as interleukin-6 (IL-6) and increase in morbidly obese subjects. Elevated circulating levels of visfatin have been observed in many inflammatory diseases such as rheumatoid arthritis, obesity, insulin resistance and type 2 diabetes [5–7]. Visfatin is secreted by neutrophils in response to inflammatory stimuli and is regulated in monocytes by pro-inflammatory factors such as interleukin-1beta (IL-1β), tumor necrosis factor alpha (TNFα), IL-6 via NFκB and AP-1-dependent mechanisms [8–10]. Visfatin activates pro-inflammatory signalling pathways in human endothelial and vascular smooth muscle cells through ROS-dependent NFκB activation or NAMPT activity, respectively, and therefore could provide a link between obesity and atherothrombotic diseases [11, 12]. Visfatin functions as an extra- and intracellular NAD biosynthetic enzyme that converts, in mammals, nicotinamide (NAM, a form of vitamin B3) to nicotinamide mononucleotide (NMN), a NAD precursor. Thus, the NAD pool is maintained at least in part by visfatin, which is for instance important for β-cell insulin secretion [2]. Although still controversial, visfatin is thought to have insulin mimetic effects and similarly as insulin, visfatin enhanced glucose uptake by myocytes and adipocytes and inhibited hepatocyte glucose release in vitro [13, 14]. Altogether, the pleiotropic role of visfatin suggests that the regulation of the NAD+ synthesis is critical for several aspects of cell physiology [15].
Macrophages, crucial cells in the development of inflammatory and metabolic disorders such as atherosclerosis and obesity, are a heterogeneous cell population that adapts and responds to a large variety of microenvironmental signals [16]. The activation states and functions of macrophages are regulated by several cytokines and microbial products. Th1 cytokines, such as interferon gamma (IFNγ), IL-1β or LPS, induce a classical proinflammatory M1 activation, while Th2 cytokines, such as IL-4 and IL-13, induce an alternative anti-inflammatory M2 phenotype [17]. In macrophages, many genes are regulated by transcription factors such as the nuclear receptors (NRs) which translate physiological signals into gene regulation. The Peroxisome Proliferator-Activated Receptor gamma (PPARγ) is a NR which regulates genes controlling lipid, glucose metabolism and inflammation. After activation by its ligands, PPARγ forms a heterodimer with the Retinoic X Receptor (RXR) [18]. The binding of this heterodimer to specific DNA sequences, called PPAR response elements (PPRE), results in the regulation of its target genes [18]. In this way PPARγ modulates crucial pathways of adipocyte differentiation and lipid metabolism, thus impacting on glucose metabolism and insulin sensitivity. Furthermore, activated PPARγ inhibits inflammatory response genes by negatively interfering with the NF-κB, STAT and AP-1 signaling pathways in a DNA-binding independent manner [19]. This trans-repression activity is likely the basis for the anti-inflammatory properties of PPARγ.
PPARγ is activated by natural or synthetic ligands such as GW1929 and the antidiabetic thiazolidinediones rosiglitazone and pioglitazone [20]. PPARγ expression is very low in human monocytes, but is induced upon differentiation into macrophages and is present in foam cells of atherosclerotic lesions [21–23]. More recently, PPARγ has been shown to enhance the differentiation of monocytes into alternative anti-inflammatory M2 macrophages [24, 25] and to promote infiltration of M2 macrophages into adipose tissue [26]. Consistent with these results, selective inactivation of macrophage PPARγ in BALB/C mice results in an impairment in the maturation of alternatively activated M2 macrophages and exacerbation of diet-induced obesity, insulin resistance, glucose intolerance and expression of inflammatory mediators [24, 27]. All these studies provide evidence that macrophage PPARγ is a central regulator of inflammation and insulin resistance.
Here, we identify visfatin as a novel PPARγ regulated gene in human macrophages. Interestingly, PPARγ activation enhanced visfatin gene expression both in classical as well as in alternative human macrophages but not in murine macrophages or human adipocytes. Finally, we show that intracellular NAD+ concentrations correlate with visfatin protein expression upon PPARγ ligand activation. Reduction of visfatin expression and activity by siRNA or a specific inhibitor abolished the PPARγ-mediated increase of NAD+.
RESULTS
PPARγ agonists induce visfatin gene expression in human macrophages in a PPARγ-dependent manner
To investigate whether PPARγ regulates visfatin gene expression, Q-PCR analysis was performed in primary human resting macrophages (RM) upon PPARγ activation. Time course experiments showed that visfatin induction was already observed after 9 hours of stimulation with GW1929 (600 nM) or rosiglitazone (RSG, 100 nM) and became maximal at 24 hours (Figure 1A), with no significant further increase after 48 hours (data not shown). Treatment of RM with increasing concentrations of the PPARγ ligands GW1929 (300, 600 and 3000 nM) or RSG (50, 100 and 1000 nM) for 24 hours significantly increased visfatin mRNA levels in a concentration-dependent manner (Figure 1B). Expression of CD36, a known PPARγ target gene [23], was also induced to a similar extent in a dose-dependent manner (data not shown). Interestingly, visfatin regulation by PPARγ was also observed in macrophage foam cells, obtained by AcLDL-loading (Figure 1C). Moreover, GW1929 (600 nM) also regulated visfatin expression in infiltrated adipose tissue macrophages (ATM) derived from visceral fat depots (Figure 1D). To determine whether PPARγ agonists up-regulate visfatin expression in a PPARγ-dependent manner, the effect of GW1929 (600 nM) was analysed in the presence or in the absence of the PPARγ inhibitor T0070907 (1 μM) [28]. T0070907 abolished GW1929-induced visfatin mRNA expression (Figure 1E). Furthermore, infection of RM with PPARγ-expressing adenovirus resulted in a significant further increase of visfatin expression in the presence of the agonist (Figure 1F). Expression of two PPARγ target genes, CD36 and FABP4 (aP2), measured as positive control, was also increased (Figure 1G and 1H). Taken together, these data demonstrate that PPARγ ligands induce visfatin gene expression in human macrophages through a PPARγ-dependent mechanism.
Figure 1. PPARγ agonists regulate visfatin gene expression in human macrophages and foam cells in a PPARγ dependent manner.
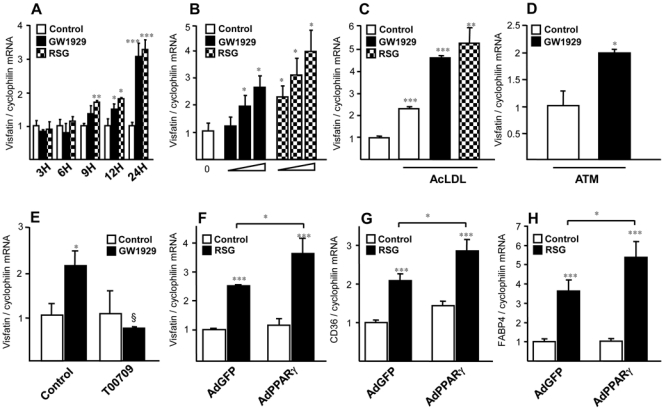
Primary human macrophages were incubated or not (control) with (A) GW1929 (600 nM) RSG (100 nM), for indicated time points, or (B) with GW1929 (300, 600 and 3000 nM) or RSG (50, 100 and 1000 nM) for 24 hours, or (C) transformed into foam cells by AcLDL (50μg/mL) loading before treatment with PPARγ ligands. (D), Human visceral ATM where treated with GW1929 (600 nM) during 24 hours. (E), Primary human monocytes were differentiated in macrophages in the presence or absence of GW1929 (600 nM), T0070907 (1μM) or both, added at the beginning of the differentiation. Primary human macrophages were infected with recombinant adenovirus AdGFP or AdPPARγ and treated with RSG (100 nM) for 24 hours. Visfatin (F), CD36 (G) and FABP4 (H) mRNA were analyzed by quantitative PCR and normalized to cyclophilin mRNA. Results are representative of those obtained from 3 independent macrophage preparations and are expressed relative to the levels in untreated cells set as 1. Each bar is the mean value ± SD of triplicate determinations. Statistically significant differences between treatments and controls are indicated (t test; *P<0.05; **P<0.01; ***P<0.001).
PPARγ agonists do not regulate visfatin gene expression in murine macrophages or human adipocytes
To determine whether regulation of visfatin also occurs in mouse macrophages, experiments were performed in murine bone marrow-derived macrophages, treated with GW1929 (1200 nM) or RSG (1000 nM) for 24 hours. PPARγ activation did not increase visfatin gene expression, although expression of CD36 was induced (Figure 2A and B). Similar results were observed with murine macrophage cell line RAW264.7 incubated with increasing concentrations of GW1929 and rosiglitazone (data not shown). Furthermore, PPARγ activation with GW1929 (600nM) during 24 hours did not lead to an increased visfatin expression in human mature adipocytes derived from the differentiation of primary preadipocytes in vitro, while the expression of CD36 was strongly induced (Figure 2C and D). Similar results were obtained in the murine preadipocyte cell line 3T3L1 after treatment with RSG or pioglitazone (data not shown), in line with a previous report [29].
Figure 2. PPARγ agonists do not regulate visfatin gene expression in murine macrophages or human adipocytes.
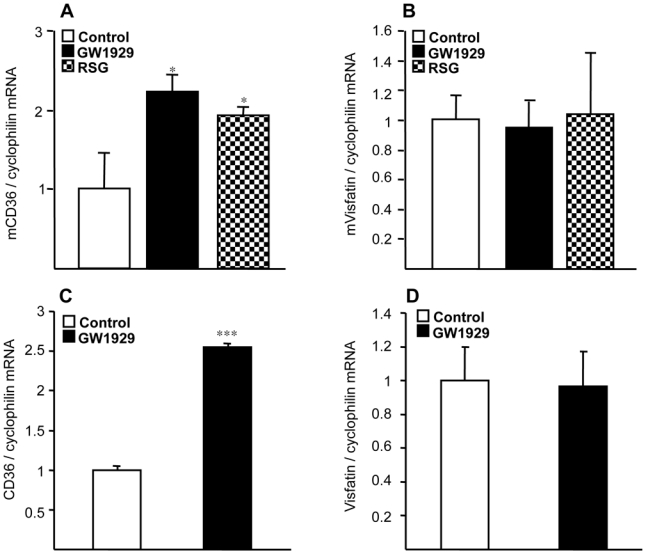
(A, B), Murine bone marrow-derived macrophages (BMDM) were incubated or not (control) in the presence of PPARγ ligands GW1929 (1.2 μM) or rosiglitazone (1μM). (C,D), human mature adipocytes derived from the differentiation of preadipocytes in vitro were incubated or not (control) in the presence of PPARγ ligands GW1929 (600 nM). CD36 (A, B) and visfatin (C, D) mRNA was analyzed by quantitative PCR and normalized to cyclophilin mRNA. Results are representative of at least 3 independent macrophage preparations and are expressed relative to the levels in untreated cells set as 1. Each bar is the mean value ± SD of triplicate determinations. Statistically significant differences between treatments and controls are indicated (t test; *P<0.05; ***P<0.001).
PPARγ regulates visfatin gene expression at the transcriptional level
To determine whether visfatin is a direct PPARγ target gene, the human visfatin promoter was examined by bio-informatic analysis. Three putative DR1-like PPRE motifs were identified in the 2150 bp sequence upstream of the ATG start site of the visfatin gene [30]. Among these sites, only the putative PPRE identified at position -1501/-1513 (AGGGCA A AGATCA) was found to be functional in EMSA experiments (Figure 3A). Incubation of the labeled -1501/-1513 visfatin-PPRE oligonucleotide with in vitro translated PPARγ and RXRα resulted in the formation of a retarded complex (Figure 3A, lane 6). The binding specificity of PPARγ to this DR1-visfatin-PPRE site was demonstrated by the competitive inhibition with excess cold unlabeled wild type (Figure 3A, lanes 7–11), but not mutated (Figure 3A, lanes 12–17) visfatin-PPRE oligonucleotide, as well as by the supershift with a specific anti-human PPARγ antibody (Figure 3A, lane 18). Binding of RXRα and PPARγ to labelled DR1-consensus PPRE was assayed as positive control (Figure 3A, lane 2).
Figure 3. PPARγ binds to and activates a PPRE in the human visfatin gene promoter.
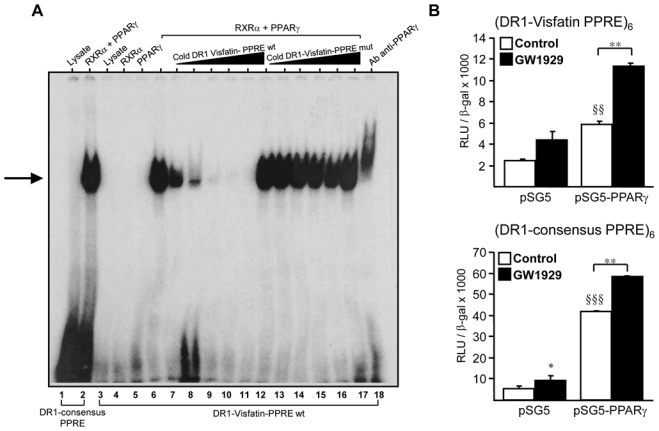
(A) EMSA were performed using the end-labeled DR1-consensus-PPRE (lanes 1–2) or DR1- visfatin-PPREwt oligonucleotide in the presence of unprogrammed reticulocyte lysate or in vitro translated hPPARγ and hRXRα (lanes 3–5). Competition experiments were performed in the presence of excess of cold unlabeled wild type (wt) (lanes 6–11) or mutated (mut) DR1-visfatin-PPRE oligonucleotides (lanes 12–17). Supershift assays were performed using a anti-human PPARγ antibody (lane 18). (B) Primary human macrophages were transfected with the indicated reporter constructs (DR1-visfatin PPRE)6 or (DR1-consensus PPRE)6, in the presence of pSG5 empty vector or pSG5-PPARγ. Cells were treated or not (Control) with GW1929 (600nM) and luciferase activity was measured. Statistically significant differences are indicated (pSG5 vs pSG5-PPARγ §§ p< 0.01, §§§ p< 0.001; control vs GW1929 *p< 0.05, **p< 0.01).
To determine whether PPARγ activates transcription from the (-1501/-1513) PPRE site, 6 copies of this element were cloned in front of the heterologous herpes simplex virus thymidine kinase promoter to obtain the (DR1-visfatin-PPRE)6x-Tk-Luc luciferase reporter vector. Co-transfection of the pSG5-PPARγ expression vector with the (DR1-visfatin PPRE)6 reporter vector in primary human RM, led to a significant induction of transcriptional activity compared with the pSG5 empty vector, an effect enhanced in the presence of GW1929 (600 nM) (Figure 3B). The consensus DR1-PPRE site cloned in 6 copies (DR1-consensus PPRE)6, used as positive control, was strongly induced by PPARγ (Figure 3B). Taken together these results indicate that visfatin is a direct PPARγ target gene in human macrophages.
PPARγ activation induces visfatin gene expression in M1 and M2 macrophages
Since macrophages are heterogeneous cells [16, 17], we decided to investigate whether induction of visfatin also occurs after PPARγ activation in classical (M1) or alternative (M2) macrophages. Human monocytes were differentiated in vitro into RM macrophages and activated into inflammatory M1 macrophages with recombinant human TNFα (5 ng/ml), IL-1β (5 ng/ml) or LPS (100 ng/ml). As expected [8], expression of visfatin was strongly induced by pro-inflammatory stimuli (Figure 4A). Interestingly, the effects of TNFα and LPS treatment were amplified in the presence of the PPARγ agonist GW1929 (Figure 4A). Under the same experimental conditions, PPARγ inhibited the induction of TNFα or IL-1β induced by inflammatory stimuli, indicative of its anti-inflammatory activity (data not shown).
Figure 4. PPARγ agonists induce visfatin gene expression in M1, M2 and adipose tissue macrophages.
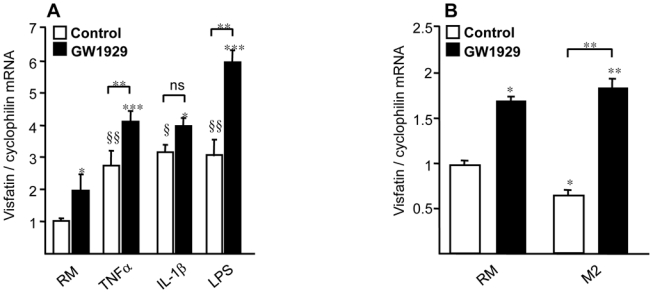
(A), Primary human monocytes were differentiated to resting macrophages (RM), treated for 24 hours with GW1929 (600 nM). Where indicated, RM were activated to M1 macrophages with recombinant human TNFα (5 ng/ml), recombinant human IL-1β (5 ng/ml) for 4 hours or LPS (100 ng/ml) for 1 hour after GW1929 treatment. (B), Primary human monocytes were differentiated in RM or M2 macrophages in the presence of IL-4 (15 ng/ml) and the PPARγ agonist GW1929 (600 nM) was added or not during differentiation process. Visfatin mRNA was analyzed by quantitative PCR and normalized to cyclophilin mRNA. Results are representative of those obtained from 5 independent macrophage preparations and are expressed relative to the levels in untreated cells set as 1. Each bar is the mean value ± SD of triplicate determinations. Statistically significant differences between treatments and controls are indicated (control vs PPARγ agonists *p< 0.05, ***p< 0.001; control vs cytokines §p<0.05, §§p<0.01).
In parallel experiments, human monocytes were differentiated in vitro into M2 macrophages with recombinant IL-4 (15 ng/ml) in the absence or in the presence of the PPARγ agonist GW1929 added at the beginning of the differentiation process [25]. As shown in Figure 4B, the expression of visfatin was significantly decreased by IL-4 stimulation. However, the PPARγ agonist GW1929 enhanced visfatin gene expression in M2 macrophages similar as in RM. A similar regulation was observed in monocytes differentiated into M2 macrophages in the presence of IL-13 (data not shown).
PPARγ activation regulates visfatin protein expression and secretion in human macrophages
To determine whether visfatin gene induction by PPARγ agonists leads to an increase of protein level, western blot analysis was performed on human RM treated with GW1929 (600 nM) or DMSO for 24 hours. Activation of PPARγ caused a significant increase (approximately 30%) of visfatin protein expression (Figure 5A). To examine whether this induction was followed by an increased secretion, we examined the ability of PPARγ to stimulate visfatin release. As shown in Figure 5B, GW1929 markedly increased (approximately 30%) visfatin concentration in macrophage supernatants after 24 hours of treatment.
Figure 5. PPARγ regulates visfatin protein expression and secretion in primary human macrophages.
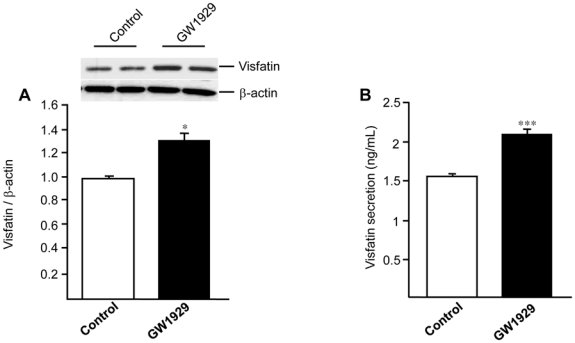
Primary human macrophages were treated or not (control) with GW1929 (600 nM) for 24 hours. (A), Intracellular visfatin and β-actin protein expression was analyzed by western blot and relative signal intensities were quantified using Quantity One Software. Results are representative of 4 independent macrophage preparations and are expressed relative to the levels in untreated cells set as 1. (B), Visfatin protein secretion was quantified in macrophage supernatant by ELISA. Results are representative of 3 independent macrophage preparations. Each bar is the mean value ± SD of triplicate determinations. Statistically significant differences between treatments and controls are indicated (t test; *P<0.01; ***P<0.001).
PPARγ activation increases the intracellular NAD+ concentration in human macrophages
Since visfatin is known as a nicotinamide phosphoribosyl transferase [2], we investigated whether the induction of visfatin by PPARγ affects NAD+ concentrations. Human RM were treated or not with GW1929 (600 nM) for 24 hours and intracellular NAD+ levels were determined by an enzymatic assay. Our results show that PPARγ activation significantly enhances cellular NAD concentration (Figure 6), an effect in line with the observed induction of visfatin expression (Figures 1 and 5).
Figure 6. PPARγ activation affects intracellular NAD concentrations in primary human macrophages.
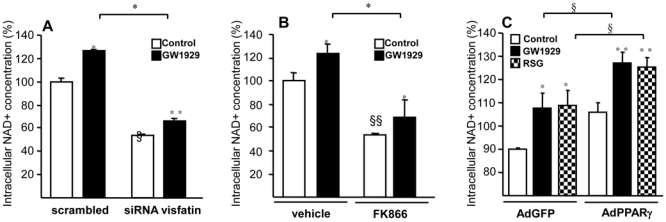
Primary human macrophages were transfected or not with non-silencing control or silencing siRNA against human visfatin (A) or treated or not with the visfatin inhibitor FK866 (100nM) (B) or infected or not with PPARγ-expressing (AdPPARγ) or GFP (AdGFP) adenovirus (C) and subsequently treated with GW1929 (600 nM), RSG (100 nM) or DMSO during 24 hours. Cells were lysed in NAD extraction buffer and NAD+ concentrations were measured by an enzymatic cycling reaction assay and normalized to protein levels and are expressed in percentage, the control non-stimulated cells being expressed as 100%. Results are representative of those obtained from 3 independent macrophage preparations. Values are means ± SD of triplicates. Statistically significant differences are indicated (t test; control vs PPARγ agonists *p< 0.05, **p< 0.01; scrambled vs siRNA visfatin or vehicle vs FK866 §p<0.05, §§p<0.05; AdGFP +PPARγ agonists vs AdPPARγ +PPARγ agonists §p<0.05).
To determine whether the NAD+ enhancement by PPARγ was dependent on visfatin induction, experiments were performed in RM macrophages in the absence or in the presence of a specific visfatin siRNA. Q-PCR analysis showed a significant decrease in visfatin gene expression after siRNA (scrambled = 1±0.019 vs siRNA visfatin = 0.27±0.01), whereas PPARγ activation increased visfatin gene (scrambled + GW1929 = 2.04±0.4 and siRNA visfatin + GW1929 = 0.51±0.022). siRNA-mediated visfatin knock-down resulted in a reduction of the basal as well as of GW1929-induced NAD+ concentration (Figure 6A). Moreover, experiments performed in the presence of a specific non-competitive inhibitor of visfatin (FK866) in the presence or absence of GW1929 demonstrated that the induction of NAD+ by GW1929 was inhibited in the presence of FK866 (Figure 6B). Finally, PPARγ over-expression increased NAD+ levels, an effect enhanced by its synthetic ligands GW1929 and RSG (Figure 6C).
DISCUSSION
Visfatin has been suggested to act as an inflammatory mediator, expressed in blood monocytes and foam cell macrophages within unstable atherosclerotic lesions where it potentially plays a role in plaque destabilization [8, 31]. Visfatin induces leukocyte adhesion to endothelial cells by inducing the expression of the cell adhesion molecules ICAM-1 and VCAM-1, thus potentially contributing to endothelial dysfunction [11]. Moreover, visfatin increases matrix metalloprotein-9 activity, TNFα and IL-8 in THP-1 monocytes [8]. These effects of visfatin were abolished when insulin receptor signalling was blocked [8], in line with the report that visfatin could bind and activate insulin receptors [14]. However, the insulin mimetic actions of visfatin are still debated [13]. All these data suggest that visfatin might be a player linking several inflammatory pathologies including obesity-associated insulin resistance, diabetes mellitus and vascular wall dysfunctions [9, 32].
In this study we show that PPARγ activation up-regulates the expression of visfatin in human monocyte-derived macrophages and ATM. This induction is concentration dependent and does not occur at a short incubation time generally required for macrophage activation, but requires more than 9 hours. The maximum effect was obtained at 24 hours with no significant further increase at 48 hours (data not shown). In addition, treatment with AcLDL induced visfatin mRNA levels and PPARγ activation further increases visfatin expression in these AcLDL-loaded macrophages.
By over-expressing PPARγ with adenovirus constructs or by inhibiting PPARγ with a specific antagonist, we demonstrate that PPARγ agonists induce visfatin gene expression in a PPARγ-dependent manner [33]. By bio-informatics analysis, we detected the presence of three DR1-like motifs which might serve as PPREs in the 2150 bp sequence upstream of the ATG codon of the human visfatin gene [30]. By EMSA analysis and transient transfection experiments in primary human macrophages a functional PPRE was identified at position - 1501/-1513 within the promoter. This PPRE is distinct from the described AP-1 or NFκB-RE like elements (located at the position -1757/-1767) within the human visfatin promoter [30]. This can explain our observation that inflammatory cytokines and PPARγ agonists have an additive effect on visfatin mRNA expression, an effect apparently in contrast to the known anti-inflammatory actions of PPARγ in macrophages due to its ability to interfere with NFκB and AP-1 signaling pathway [19]. This is similar as already reported for other nuclear receptors such as Liver X Receptor (LXR), of which short term pre-treatment with LXR agonists significantly reduced the LPS induced-inflammatory response, whereas 24 hour pre-treatment of macrophages with agonists resulted in an enhanced inflammatory response [34].
PPARγ agonists induce visfatin protein expression and secretion in human primary macrophages. Visfatin is a secreted cytokine-like protein [35], although it has been speculated that the release of visfatin may be due to either cell lysis or cell death [36, 37]. However, it has been demonstrated in adipocytes and CHO cells that visfatin is actively secreted through a nonclassical (non Golgi-endoplasmic reticulum system) secretory pathway [2]. In our experiments we did not observe any cellular toxicity after PPARγ agonist treatment, thus suggesting that the secretion of visfatin in human macrophages may be an active process.
Since visfatin is the rate-limiting enzyme for the conversion of nicotinamide to NAD+ in mammals, the increase of intracellular NAD+ concentration by PPARγ agonists is likely the consequence of visfatin induction. NAD+ modulates various signalling pathways. For instance it regulates the transcription and function of NAD+-dependent SIRTs and increased expression of visfatin upregulates SIRT1 activity [2]. The observed variation of intracellular NAD+ concentrations after visfatin modulation (by siRNA or PPARγ activation) are in the same order of magnitude as previously reported in murine NIH-3T3 fibroblasts transduced with visfatin-specific shRNAs. The reduction of intracellular visfatin protein in these cells led to a reduction of NAD+ levels ranging from 20 to 40%, whereas cells over-expressing visfatin displayed a 15–25% increase in total intracellular NAD+ levels [38]. By using the pharmacological visfatin inhibitor, FK866, a significant decrease in intracellular NAD+ concentration was observed even in the presence of PPARγ ligand, confirming the role of the enzymatic activity of visfatin and the possibility that PPARγ can modulate intracellular NAD+ levels via an increase of visfatin expression. Indeed, a small increase of NAD+ concentrations in response to GW1929 in siRNA visfatin treated-macrophages was observed, suggesting that additional PPARγ related pathway might modulate NAD+ levels.
Moreover, we have shown that PPARγ agonists increase expression of visfatin in macrophages irrespectively of their M1 or M2 polarization. Visfatin-dependent recycling of nicotinamide to NAD+ may represent a physiologically important homeostatic mechanism to avoid depletion of the intracellular NAD+ pool during its active use as a substrate by sirtuins, cADP-ribose synthases or PARPs [15]. It has been recently shown that pharmacological SIRT1 activators exert broad anti-inflammatory effects in macrophages [39]. Conversely, SIRT1 knockdown leads to an increase in the basal expression of TNFα, MCP-1 and KC. The activity of SIRT1 requires an increase of visfatin expression to compensate the consumption of NAD+. Van Gool et al. have identified SIRT6, another member of the sirtuin family, as the NAD-dependent enzyme able to increase TNFα production in macrophages by acting at a post-transcriptional level [40]. Taken together these observations suggest that NAD+ can exert pro- and/or anti-inflammatory properties depending on the activated sirtuins.
It is also possible that macrophage-produced visfatin has a local paracrine effect on surrounding cells, such as smooth muscle cells (SMC) within atherosclerotic plaques since in vascular SMC, overexpression of visfatin promotes cell maturation by regulating NAD+-dependent SIRT deacetylase activity [41]. Visfatin has been reported as a longevity protein that extends the lifespan of human SMC, suggesting that visfatin allows vascular cells to resist to stress and senescence, a hallmark of atherosclerotic lesions [42]. The ability of visfatin to prolong cellular longevity of vascular SMC might contribute to the stabilization efficiency of a developing atherosclerotic lesion by SMC. Treatment of humans with PPARγ ligands does not alter both adipose visfatin gene expression and circulating visfatin levels, as reported by several papers [43–45]. However, other authors reported that in lean as well as in lean-HIV infected patients, rosiglitazone treatment increased the amounts of circulating visfatin [46, 47]. It thus appears that the effect of PPARγ ligand treatment on circulating visfatin levels is highly dependent on the patient phenotype. However, in such studies the net contribution of visfatin coming from adipocytes or macrophages cannot be evaluated and cell-specific PPARγ regulation of visfatin may have a local effect.
Adipose tissue is not only composed by adipocytes, but several other types of cells including macrophages, lymphocytes and endothelial cells are also present. It has been shown that PPARγ agonists induce the expression of visfatin in visceral fat of OLETF rats [48]. The authors analyzed whole adipose tissue, thus it can not be determined whether PPARγ regulation of visfatin occurred in macrophages or in adipocytes. Here we show that PPARγ activation leads to an increased visfatin expression in ATM. However, this regulation does not occur in human primary mature adipocytes derived from preadipocyte differentiation in vitro. It has been shown recently that PPARγ binding in macrophages occurs at genomic locations different from those in adipocytes, showing that PPARγ binding sites are cell type-specific [49]. These results are in agreement with a previous report showing that in human, PPARγ has distinct functions in different cell types since treatment with pioglitazone induces apoptotic cell death specifically in macrophages, whereas differentiated adipocytes did not show any significant increase in apoptosis [50]. Furthermore, treatment with pioglitazone for 3 weeks did not alter visfatin gene expression in adipose cells both in non-diabetic and diabetic individuals [43]. Altogether, these results may allow to shed some light on regulation of visfatin expression by PPARγ in human adipose tissue, an effect limited to ATM.
In conclusion, our results identify visfatin as a novel PPARγ target gene in human macrophages and demonstrate that PPARγ activation induces visfatin gene and protein secretion in different types of human macrophages. This induction of visfatin by PPARγ in macrophages contributes to enhanced intracellular NAD+ concentrations.
MATERIALS AND METHODS
Cell culture
Mononuclear cells were isolated from blood (buffy coats; thrombopheresis residues) of human healthy normolipidemic donors by Ficoll gradient centrifugation [21]. Briefly, after Ficoll gradient centrifugation, peripheral blood mononuclear cells (PBMC) were suspended in RPMI 1640 medium (Gibco, Invitrogen) containing gentamycin (40 μg/ml), glutamine (0,05%) (both from Gibco, Invitrogen). Cells were cultured depending on the experiment at a density of 1 or 2.106 cells/well in six-well plastic culture dishes (Primaria, Becton Dickinson Labware). Selection of pure monocyte population occurred spontaneously after two hours of cell adhesion to the culture dish. After 2 washing steps with PBS, cells were cultured in RPMI 1640 medium, containing gentamycin (40 μg/ml), glutamine (0,05%), supplemented with 10% pooled human serum (Biowest). Differentiation of monocytes into macrophages is completed after 7 days as characterized by immunocytochemistry or flow cytometry analysis using macrophage marker anti-CD68 antibody [21]. These primary human macrophages, also called resting macrophages (RM), were used for experiments after 7 days of differentiation. RM were incubated for 3, 6, 9, 12 or 24 hours in the presence of PPARγ ligands GW1929 (300, 600, 3000 nM), rosiglitazone (RSG 50, 100, 1000 nM) or DMSO (Control). Where indicated, RM were transformed to foam cells by 48 hour loading with acetylated LDL (AcLDL) (50μg/mL) and treated with PPARγ ligands GW1929 (600 nM), RSG (100 nM) or DMSO (Control). Where indicated, the PPARγ antagonist T0070907 (1 μM) (Tocris Bioscience) or the NAMPT inhibitor FK866 (100nM) (Cayman Chemical) were added. In other experiments, RM were treated with GW1929 (600 nM) or DMSO for 24 hours and then activated into M1 macrophages by recombinant human TNFα (5 ng/ml), human IL-1β (5 ng/ml) (Promokines) during 4 hours or LPS (lipopolysaccharide, 100 ng/ml) (Sigma) during 1 hour. M2 macrophages were obtained by differentiating monocytes in the presence of recombinant human IL-4 (15 ng/ml) (Promokines).
Visceral adipose tissue (AT) biopsies were obtained from consenting obese patients undergoing bariatric surgery. This study was approved by the ethics committee of the University Hospital of Lille, France. After removing all fibrous materials and visible blood vessels, AT was cut into small pieces and digested in Krebs buffer pH 7.4 containing collagenase (1.5 mg/ml, Roche Diagnostic). The cell suspension was filtered through a 200 μm filter and centrifuged at 300g for 15 minutes to separate floating adipocytes. The stromal vascular fraction (SVF) was pelleted, treated with erytrocyte lysing buffer (NH4Cl 131mM, NH4CO3 9mM, EDTA 1mM, pH 7.4) for 10 minutes and filtered through meshes with pore size of 70 μm. The SVF was then subjected to magnetic-activated cell sorting of CD14+ cells (MACS, Miltenyi Biotec) using CD14-labelled magnetic beads and MS columns (Miltenyi) according to the manufacturer’s instructions to yielding ATM. Purity of CD14+ cells was assessed by flow cytometry analysis. ATM where cultured for 24 hours in Endothelial Cell Basal Medium (ECBM) supplemented with 0.1% BSA before treatment with GW1929 (600 nM) or DMSO for 24 hours.
The CD14− fraction was cultured in Preadipocyte Basal Medium (Promocell) for 24 hours, washed with PBS to remove floating cells. Adherent preadipocytes were then cultured in Preadipocyte Growth Medium (Promocell) according to the manufacturer’s instructions until confluence. After confluence, preadipocyte were cultured in Preadipocyte Differentiation Medium (Promocell) for 72 hours. To complete the differentiation process into mature adipocytes, cells were fed every 2–3 days during 12 days with Adipocyte Nutrition Medium (Promocell). At the end of the differentiation, mature adipocytes were treated with the PPARγ ligand GW1929 (600nM).
Murine BMDM were prepared from C57BL/6J mice. Bone marrow cell suspensions were isolated by flushing the femurs and tibias with PBS and cells were cultured as previously described [51]. BMDM were treated with PPARγ ligands GW1929 (1.2 μM) and rosiglitazone (1 μM) for 24 hours.
RNA extraction and analysis
Total cellular RNA was extracted from human macrophages using Trizol (Invitrogen, France) for RM or RNeasy micro kit (Qiagen) for ATM. For quantitative PCR, total RNA was reverse transcribed and cDNAs were quantified by quantitative polymerase chain reaction (Q-PCR) on a MX 4000 apparatus (Stratagene) using specific primers for human visfatin (5′-GCC AGC AGG GAA TTT TGT TA-3′ forward and 5′-TGA TGT GCT GCT TCC AGT TC-3′ reverse), mouse visfatin (5′-TCCGGCCCGAGATGAAT-3′ forward and 5′-GTGGGTATTGTTTATAGTGAGTAACCTTGT-3′ reverse), human CD36 (5′-TCAGCAAATGCAAAGAAGGGAGAC-3′ forward and 5′-GGTTGACCTGCAGCCGTTTTG-3′ reverse), mouse CD36 (5′-GGATCTGAAATCGACCTTAAAG-3′ forward and 5′-TAGCTGGCTTGACCAATATGTT-3′ reverse ), human FABP4 (5′-TACTGGGCCAGGAATTTGAC-3′ forward 5′-GTGGAAGTGACGCCTTTCAT-3′ reverse) and human/mouse cyclophilin (5′-GCA TAC GGG TCC TGG CAT CTT GTC C-3′ forward and 5′-ATG GTG ATC TTC TTG CTG GTC TTG C-3′ reverse). Visfatin mRNA levels were subsequently normalized to those of cyclophilin.
Adenovirus preparation and cell infection
The recombinant adenovirus AdGFP and AdPPARγ were obtained by homologous recombination in Escherichia coli after insertion of the cDNAs into the pAdCMV2 vector (Q.BIOgene, Illkirch, France). Viral stocks were created as previously described (Meisner F, et al, 2006). Viral titers were determined by plaque assay on HEK 293 cells and defined as plaque-forming units/ml. For the infection experiments, primary human macrophages were seeded in 6-well Prim aria plates at a density of 106 cells/well and viral particles were added at a multiplicity of infection of 100 for 12 hours. Cells were subsequently incubated for 24 hours with rosiglitazone (100 nM) or DMSO.
In vitro translation and Electrophoretic Mobility Shift Assay (EMSA)
PPARγ and RXRα were in vitro transcribed from the pSG5-hPPARγ and pSG5-hRXRα plasmids, respectively, using T7 polymerase and subsequently translated using the TNT coupled transcription/translation system (Promega, Madison, WI). Proteins were then incubated for 10 minutes at room temperature in a binding buffer (Hepes 10mM pH 7.8, NaCl 100mM, EDTA 0.1mM, 10% glycerol, 1mg/ml BSA) containing 1μg of poly(dI-dC) and 1μg of herring sperm DNA in a total volume of 20μl. Double stranded oligonucleotides containing the wild type DR1-PPARγ response element (PPRE) present at position -1501/-1513 of the human visfatin promoter, end-labeled using T4 polynucleotide kinase and γ32P-ATP, was added as probe to the binding reaction. For competition experiments, increasing amounts (5, 10, 50, 100 and 200-fold excess) of unlabeled visfatin-PPREwt (5′-CAATACAGGGCAAAGATCATGGAAG-3′) or visfatin-PPREmut (5′-CAATACAGGAAAAAGAAAATGGAAG-3′) oligonucleotides were added to the mixture 10 minutes before the DR1-visfatin-PPRE wt. The binding reaction was incubated for a further 15 minutes at room temperature. For supershift assays, 2μl of monoclonal mouse antihuman PPARγ antibody (Sc-7273, Santacruz Biotechnology) were added to the binding reaction. DNA/protein complexes were resolved by 6% non-denaturing polyacrylamide gel electrophoresis in 0.25X Tris-Borate-EDTA.
Plasmid cloning and transient transfection experiments
The reporter plasmid (DR1-visfatin-PPREwt)6-TK-pGL3 was generated by inserting 6 copies of the double-strand oligonucleotides (for 5′-CAATACAGGGCAAAGATCATGGAAG-3′; rev 5′-CTTCCATGATCTTTGCCCTGTATTG-3) into the pTK-pGL3 plasmid. Primary human macrophages were transfected overnight in a RPMI medium containing 10% of human serum with reporter plasmids and expression vectors (pSG5-empty or pSG5-hPPARγ) using jetPEI (Polyplus transfection, France). β-galactosidase expression vectors were used as internal control of transfection efficiency. Subsequently, cells were incubated for additional 24 hours in RPMI medium containing 2% of human serum in the presence of GW1929 (600 nM) or DMSO. At the end, cells were lysed and luciferase and β-galactosidase activities measured on cell extracts using a luciferase buffer (Promega, Madison, WI).
Short-Interfering RNA
Short-interfering (si) RNA specific for human PBEF1 (Visfatin NAMPT) and non-silencing control siRNA (siScrambled) were purchased from Dharmacon. 7-day-old human macrophages were transfected with siRNA using the transfection reagent DharmaFECT Reagent 4. 16 hours after transfection, cells were incubated in the presence of GW1929 (600 nM) or vehicle (DMSO) and harvested 24 hours later.
Protein extraction and Western blot analysis
Cells were washed twice with ice-cold PBS and harvested in ice-cold protein lysis buffer (RIPA). Cell homogenates were collected by centrifugation at 13,000 rpm at 4 °C and protein concentrations were determined using the BCA assay (Pierce Interchim). 10 μg of protein lysate was separated by 10% SDS-PAGE and transferred to nitrocellulose membranes (Amersham). Equal loading of proteins was verified by Ponceau red staining. Membranes were then subjected to immunodetection using rabbit polyclonal antibodies against visfatin (Abcam, ab24149) or against β-actin (Santacruz Biotechnology, I-19). After incubation with a secondary peroxidase-conjugated antibody (Cell Signaling Technology), immunoreactive bands were revealed using a chemiluminescence ECL detection kit (Amersham) and intensity of signals was subsequently analyzed by densitometry and quantified using Quantity One software.
Measure of visfatin protein secretion by ELISA
Human RM were treated with the PPARγ ligand GW1929 (600 nM) or DMSO for 24 hours. Supernatants were collected and extracellular visfatin concentrations measured using a commercially available ELISA kit with a human visfatin (COOH-terminal) enzyme immunometric assay (Phoenix Pharmaceuticals, Karlsruhe, Germany), according to the manufacturer’s instructions.
Measure of cellular NAD content
Total nicotinamide adenine dinucleotide (NADt = NAD+NADH) levels were determined in cell lysates, using the NADH/NAD quantification kit according to the manufacturer’s instructions (Biovision research products). Briefly, human RM treated or not with FK866 (100nM), infected or not with adenovirus (AdGFP, AdPPARγ), transfected or not with siRNA (siScrambled, siVisfatin) were treated with the PPARγ ligand GW1929 (600 nM), Rosiglitazone (100 nM) or DMSO during 24 hours. Cells were lysed in NAD+ extraction buffer after three times washing with ice-cold PBS. The NAD/NADH ratio was calculated as (NADt-NADH)/NADH. NAD levels were normalized to protein content. Results are expressed as a percentage, the control non-stimulated cells being expressed as 100%. All assays were done in triplicate in at least 3 independent experiments.
Statistical analysis
Statistically differences between groups were analysed by Student’s t tests and were considered significant when p≤0.05.
Acknowledgments
We thank R. Dievart, B. Derudas, A. Blondy, C. Eberle, MF. Six and Dr. L. Arnalsteen for their contribution. We acknowledge grant support from the “Nouvelle Societé Française d’Athérosclérose” (to T.H. Mayi) and Fondation Coeur et Artères. The research leading to these results has received funding from the European Community’s 7th Framework Programme (FP7/2007-2013) under grant agreement n° 201608.
References
- 1.Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–7. doi: 10.1128/mcb.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, Imai S. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–75. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–34. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 4.Curat CA, Wegner V, Sengenes C, Miranville A, Tonus C, Busse R, Bouloumie A. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia. 2006;49:744–7. doi: 10.1007/s00125-006-0173-z. [DOI] [PubMed] [Google Scholar]
- 5.Seo JA, Jang ES, Kim BG, Ryu OH, Kim HY, Lee KW, Kim SG, Choi KM, Baik SH, Choi DS, Kim NH. Plasma visfatin levels are positively associated with circulating interleukin-6 in apparently healthy Korean women. Diabetes Res Clin Pract. 2008;79:108–11. doi: 10.1016/j.diabres.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 6.Haider DG, Holzer G, Schaller G, Weghuber D, Widhalm K, Wagner O, Kapiotis S, Wolzt M. The adipokine visfatin is markedly elevated in obese children. J Pediatr Gastroenterol Nutr. 2006;43:548–9. doi: 10.1097/01.mpg.0000235749.50820.b3. [DOI] [PubMed] [Google Scholar]
- 7.Brentano F, Schorr O, Ospelt C, Stanczyk J, Gay RE, Gay S, Kyburz D. Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 2007;56:2829–39. doi: 10.1002/art.22833. [DOI] [PubMed] [Google Scholar]
- 8.Dahl TB, Yndestad A, Skjelland M, Oie E, Dahl A, Michelsen A, Damas JK, Tunheim SH, Ueland T, Smith C, Bendz B, Tonstad S, Gullestad L, Froland SS, Krohg-Sorensen K, Russell D, Aukrust P, Halvorsen B. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation. 2007;115:972–80. doi: 10.1161/CIRCULATIONAHA.106.665893. [DOI] [PubMed] [Google Scholar]
- 9.Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748–58. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- 10.Kendal CE, Bryant-Greenwood GD. Pre-B-cell colony-enhancing factor (PBEF/Visfatin) gene expression is modulated by NF-kappaB and AP-1 in human amniotic epithelial cells. Placenta. 2007;28:305–14. doi: 10.1016/j.placenta.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH, Koo TH, Jang HO, Yun I, Kim KW, Kwon YG, Yoo MA, Bae MK. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim Biophys Acta. 2008;1783:886–95. doi: 10.1016/j.bbamcr.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Romacho T, Azcutia V, Vazquez-Bella M, Matesanz N, Cercas E, Nevado J, Carraro R, Rodriguez-Manas L, Sanchez-Ferrer CF, Peiro C. Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia. 2009;52:2455–63. doi: 10.1007/s00125-009-1509-2. [DOI] [PubMed] [Google Scholar]
- 13.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Retraction. Science. 2007;318:565. doi: 10.1126/science.318.5850.565b. [DOI] [PubMed] [Google Scholar]
- 14.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–30. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 15.Galli M, Van Gool F, Rongvaux A, Andris F, Leo O. The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res. 70:8–11. doi: 10.1158/0008-5472.CAN-09-2465. [DOI] [PubMed] [Google Scholar]
- 16.Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, Meerschaut S, Beschin A, Raes G, De Baetselier P. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211:487–501. doi: 10.1016/j.imbio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 18.Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116:581–9. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- 20.Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. 2008;28:1050–9. doi: 10.1161/ATVBAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- 21.Chinetti G, Griglio S, Antonucci M, Pineda Torra I, Delerive P, Majd Z, Fruchart JC, Chapman J, Najib J, Staels B. Activation of peroxisome proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J Biol Chem. 1998;273:25573–25580. doi: 10.1074/jbc.273.40.25573. [DOI] [PubMed] [Google Scholar]
- 22.Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1998;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tontonoz P, Nagy L, Alvarez J, Thomazy V, Evans R. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 24.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Eagle AR, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (London) 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadski C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARg activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metabolism. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Stienstra R, Duval C, Keshtkar S, van der Laak J, Kersten S, Muller M. Peroxisome proliferator-activated receptor gamma activation promotes infiltration of alternatively activated macrophages into adipose tissue. J Biol Chem. 2008;283:22620–7. doi: 10.1074/jbc.M710314200. [DOI] [PubMed] [Google Scholar]
- 27.Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, Watt MJ, Benner C, Febbraio MA, Nguyen AK, Folian B, Subramaniam S, Gonzalez FJ, Glass CK, Ricote M. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117:1658–69. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi H, Fujita K, Fujisawa T, Yonemitsu K, Tomimoto A, Ikeda I, Yoneda M, Masuda T, Schaefer K, Saubermann LJ, Shimamura T, Saitoh S, Tachibana M, Wada K, Nakagama H, Nakajima A. Inhibition of peroxisome proliferator-activated receptor gamma activity in esophageal carcinoma cells results in a drastic decrease of invasive properties. Cancer Sci. 2006;97:854–60. doi: 10.1111/j.1349-7006.2006.00250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lv Q, Wang Y, Wang W, Wang L, Zhou X. Effect of pioglitazone on visfatin expression in 3T3-L1 adipocytes and SD rats. Endocr Res. 2009;34:130–41. doi: 10.3109/07435800903287061. [DOI] [PubMed] [Google Scholar]
- 30.Ognjanovic S, Bao S, Yamamoto SY, Garibay-Tupas J, Samal B, Bryant-Greenwood GD. Genomic organization of the gene coding for human pre-B-cell colony enhancing factor and expression in human fetal membranes. J Mol Endocrinol. 2001;26:107–17. doi: 10.1677/jme.0.0260107. [DOI] [PubMed] [Google Scholar]
- 31.Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, Marshall JC. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest. 2004;113:1318–27. doi: 10.1172/JCI19930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luk T, Malam Z, Marshall JC. Pre-B cell colony-enhancing factor (PBEF)/visfatin: a novel mediator of innate immunity. J Leukoc Biol. 2008;83:804–16. doi: 10.1189/jlb.0807581. [DOI] [PubMed] [Google Scholar]
- 33.Zhou H, Yang X, Wang NL, Zhang YO, Cai GP. Macrostemonoside A promotes visfatin expression in 3T3-L1 cells. Biol Pharm Bull. 2007;30:279–83. doi: 10.1248/bpb.30.279. [DOI] [PubMed] [Google Scholar]
- 34.Fontaine C, Rigamonti E, Nohara A, Gervois P, Teissier E, Fruchart JC, Staels B, Chinetti-Gbaguidi G. Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ Res. 2007;101:40–9. doi: 10.1161/CIRCRESAHA.106.135814. [DOI] [PubMed] [Google Scholar]
- 35.Ognjanovic S, Ku TL, Bryant-Greenwood GD. Pre-B-cell colony-enhancing factor is a secreted cytokine-like protein from the human amniotic epithelium. Am J Obstet Gynecol. 2005;193:273–82. doi: 10.1016/j.ajog.2004.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hug C, Lodish HF. The role of the adipocyte hormone adiponectin in cardiovascular disease. Curr Opin Pharmacol. 2005;5:129–34. doi: 10.1016/j.coph.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Stephens JM, Vidal-Puig AJ. An update on visfatin/pre-B cell colony-enhancing factor, an ubiquitously expressed, illusive cytokine that is regulated in obesity. Curr Opin Lipidol. 2006;17:128–31. doi: 10.1097/01.mol.0000217893.77746.4b. [DOI] [PubMed] [Google Scholar]
- 38.Rongvaux A, Galli M, Denanglaire S, Van Gool F, Dreze PL, Szpirer C, Bureau F, Andris F, Leo O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J Immunol. 2008;181:4685–95. doi: 10.4049/jimmunol.181.7.4685. [DOI] [PubMed] [Google Scholar]
- 39.Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, Oh da Y, Lu M, Milne JC, Westphal C, Bandyopadhyay G, Olefsky JM. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298:E419–28. doi: 10.1152/ajpendo.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, Bedalov A, Mostoslavsky R, Alt FW, De Smedt T, Leo O. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009;15:206–10. doi: 10.1038/nm.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Veer E, Nong Z, O’Neil C, Urquhart B, Freeman D, Pickering JG. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ Res. 2005;97:25–34. doi: 10.1161/01.RES.0000173298.38808.27. [DOI] [PubMed] [Google Scholar]
- 42.van der Veer E, Ho C, O’Neil C, Barbosa N, Scott R, Cregan SP, Pickering JG. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem. 2007;282:10841–5. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 43.Hammarstedt A, Pihlajamaki J, Rotter Sopasakis V, Gogg S, Jansson PA, Laakso M, Smith U. Visfatin is an adipokine, but it is not regulated by thiazolidinediones. J Clin Endocrinol Metab. 2006;91:1181–4. doi: 10.1210/jc.2005-1395. [DOI] [PubMed] [Google Scholar]
- 44.Pfutzner A, Marx N, Walcher D, Lobig M, Seidel D, Forst T. Impact of rosiglitazone on visfatin and adiponectin plasma concentrations in patients with type 2 diabetes and coronary artery disease. Clin Lab. 2008;54:237–41. [PubMed] [Google Scholar]
- 45.Varma V, Yao-Borengasser A, Rasouli N, Bodles AM, Phanavanh B, Lee MJ, Starks T, Kern LM, Spencer HJ, 3rd, McGehee RE, Jr, Fried SK, Kern PA. Human visfatin expression: relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J Clin Endocrinol Metab. 2007;92:666–72. doi: 10.1210/jc.2006-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haider DG, Schindler K, Mittermayer F, Muller M, Nowotny P, Rieger A, Luger A, Ludvik B, Wolzt M. Effect of rosiglitazone on visfatin and retinol-binding protein-4 plasma concentrations in HIV-positive patients. Clin Pharmacol Ther. 2007;81:580–5. doi: 10.1038/sj.clpt.6100047. [DOI] [PubMed] [Google Scholar]
- 47.Haider DG, Mittermayer F, Schaller G, Artwohl M, Baumgartner-Parzer SM, Prager G, Roden M, Wolzt M. Free fatty acids normalize a rosiglitazone-induced visfatin release. Am J Physiol Endocrinol Metab. 2006;291:E885–90. doi: 10.1152/ajpendo.00109.2006. [DOI] [PubMed] [Google Scholar]
- 48.Choi KC, Ryu OH, Lee KW, Kim HY, Seo JA, Kim SG, Kim NH, Choi DS, Baik SH, Choi KM. Effect of PPAR-alpha and -gamma agonist on the expression of visfatin, adiponectin, and TNF-alpha in visceral fat of OLETF rats. Biochem Biophys Res Commun. 2005;336:747–53. doi: 10.1016/j.bbrc.2005.08.203. [DOI] [PubMed] [Google Scholar]
- 49.Lefterova MI, Steger DJ, Zhuo D, Qatanani M, Mullican SE, Tuteja G, Manduchi E, Grant GR, Lazar MA. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol Cell Biol. 2010;30:2078–89. doi: 10.1128/MCB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bodles AM, Varma V, Yao-Borengasser A, Phanavanh B, Peterson CA, McGehee RE, Jr, Rasouli N, Wabitsch M, Kern PA. Pioglitazone induces apoptosis of macrophages in human adipose tissue. J Lipid Res. 2006;47:2080–8. doi: 10.1194/jlr.M600235-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci USA. 2004:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]