

Abstract
Objective
Conventional chemotherapy and radiotherapy produce marginal survival benefits in pancreatic cancer, underscoring the need for novel therapies. The aim of this study is to develop an adoptive T cell transfer approach to target tumours expressing prostate stem cell antigen (PSCA), a tumour-associated antigen that is frequently expressed by pancreatic cancer cells.
Methods
Expression of PSCA on cell lines and primary tumour samples was confirmed by immunohistochemistry. Healthy donor- and patient-derived T cells were isolated, activated in vitro using CD3/CD28, and transduced with a retroviral vector encoding a chimeric antigen receptor (CAR) targeting PSCA. The ability of these cells to kill tumour cells was analysed by chromium-51 (Cr51) release.
Results
Prostate stem cell antigen was expressed on >70% of the primary tumour samples screened. Activated, CAR-modified T cells could be readily generated in clinically relevant numbers and were specifically able to kill PSCA-expressing pancreatic cancer cell lines with no non-specific killing of PSCA-negative target cells, thus indicating the potential efficacy and safety of this approach.
Conclusions
Prostate stem cell antigen is frequently expressed on pancreatic cancer cells and can be targeted for immune-mediated destruction using CAR-modified, adoptively transferred T cells. The safety and efficacy of this approach indicate that it deserves further study and may represent a promising novel treatment for patients with pancreatic cancer.
Keywords: immunotherapy, chimeric antigen receptor, pancreatic cancer, prostate stem cell antigen
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related mortality in the USA with a 5-year survival rate of <5% (http://seer.cancer.gov/csr/1975_2007/index.html). Although the incidence of pancreatic cancer is lower than those of breast, prostate and colon cancer, no cancer is associated with a more dismal prognosis. Because its symptoms in the early stages of disease are vague, and effective screening tests are lacking, about 80% of patients are diagnosed with locally advanced or metastatic pancreatic cancer. Even among the minority of patients with localized disease that is eligible for surgery, 5-year survival is only 20%, which is much inferior to outcomes of complete resection in all other cancers. It is an understatement to say that pancreatic cancer is resistant to surgery and conventional chemoradiation. After resection with negative margins and adjuvant chemoradiation, most patients have recurrent disease in little over a year after surgery and die within 2 years of surgery. Treatment of pancreatic cancer remains an enormous clinical challenge. Therefore, novel therapeutic strategies are urgently needed and adoptive T cell transfer represents a promising approach.1
Since the targeting of chemoresistant tumours using T cell therapy was demonstrated, interest has increased in combining these strategies and recruiting the host immune system to help eradicate disease that remains after conventional therapy.2,3 We and others have shown that the infusion of T cells engineered with tumour specificity through the transgenic expression of a chimeric antigen receptor (CAR) can effectively treat even disseminated malignancies including neuroblastomas and B cell tumors.4–8 Chimeric antigen receptors combine the antigen-binding properties of a monoclonal antibody with the lytic capacity of T cells, thus allowing transgenic cells to recognize both protein and non-protein antigens on tumour cells and to kill target cells in a major histocompatibility complex (MHC)-independent fashion.9,10
In the current study, we sought to extend T cell therapy to the treatment of pancreatic cancer. However, a prerequisite step involved the identification of a target antigen which is present on the cell surface and is expressed or overexpressed on malignant cells, but has limited expression on vital organs. Prostate stem cell antigen (PSCA) meets all these criteria. This antigen is a glycosylphosphatidylinositol (GPI)-anchored cell surface protein thought to be involved in intracellular signalling, although much remains unknown about its physiological function.11 It is upregulated in several major cancers, including prostate, bladder and pancreatic cancers, but in normal cells its expression is restricted, making it an ideal target antigen.11,12
We now show, using immunohistochemistry (IHC), that PSCA is overexpressed in both pancreatic cancer cell lines and in the majority of pancreatic ductal adenocarcinomas. To target this antigen, we constructed a retroviral vector encoding a CAR-targeting PSCA and demonstrated that both normal donor- as well as patient-derived T lymphocytes can be genetically modified to express this CAR. Transgenic cells are able to specifically kill PSCA-positive tumour cell lines without impacting non-antigen expressing targets and can be produced in large numbers,13 thus demonstrating the feasibility of this approach in a clinical setting.
Materials and methods
Donors and cell lines
Pancreatic cancer cell lines CAPAN-1, CFPAC, PL45 and ASPC-1 were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were maintained in a humidified atmosphere containing 5% carbon dioxide (CO2) at 37 °C. Peripheral blood and tumour samples were collected from pancreatic cancer patients and healthy volunteers with their informed consent under Baylor College of Medicine Institutional Review Board-approved protocols. Tumour arrays (human cancers B IMH-327) were purchased from Imgenex Corp. (San Diego, CA, USA).
OKT3 blasts generation
Peripheral blood mononuclear cells (PBMCs) obtained from healthy donors and patients with pancreatic cancer were activated with OKT3 (1 mg/ml) (Ortho Biotech, Inc., Bridgewater, NJ, USA) and CD28 (Becton Dickinson & Co., Mountain View, CA, USA) antibodies (1 mg/ml) and plated in a non-tissue culture-treated 24-well plate at 1 × 106 PBMCs/2 ml complete media (RPMI 1640; Gibco BRL Life Technologies, Inc., Gaithersburg, MD, USA) containing 45% Clicks medium (Irvine Scientific, Inc., Santa Ana, CA, USA), 10% foetal calf serum (FCS) (HyClone Laboratories, Inc., Logan, UT, USA) and 2 mM L-glutamine (Gibco BRL Life Technologies, Inc.). On day 1 after activation, the cells were supplemented with recombinant human interleukin-2 (rhIL-2, 100 U/ml, Proleukin; Chiron Corp., Emeryville, CA, USA) and subsequently split and fed with fresh media plus IL-2 (50 U/ml).
Generation of retroviral constructs and retroviral transduction
We synthesized (DNA 2.0) a human codon-optimized single-chain variable fragment (scFv) of PSCA based on a published sequence.14,15 The scFv fragment was cloned in frame with the human IgG1-CH2CH3 domain and with the ζ-chain of the TCR/CD3 complex in the SFG retroviral backbone, previously established in our laboratory.4,5 To produce the retroviral supernatant, 293T cells were co-transfected with the CAR-PSCA vector, the Peg-Pam-e plasmid containing the sequence for MoMLV gag-pol, and the DRF plasmid containing the sequence for the RD114 envelope, using the Fugene6 transfection reagent (Roche Diagnostics Corp., Indianapolis, IN, USA), according to the manufacturer's instructions. Supernatant containing the retrovirus was collected at 48 h and 72 h post-transfection, filtered (using a 0.45-µm filter) and stored at −80 °C.5,16,17
T lymphocyte transduction
For transduction, 0.2 × 106/ml OKT3/CD28-activated PBMCs were plated in complete media in a non-tissue culture-treated 24-well plate (1 ml/well) pre-coated with a recombinant fibronectin fragment (FN CH-296; Retronectin; Takara Shuzo Co. Ltd, Otsu, Japan). After the addition of viral supernatant (1 ml/well), the cells were spun at 1000 g for 30 min, and then transferred to the 37 °C, 5% CO2 incubator. Expression of CARs on T lymphocytes was measured 72 h post-transduction by flow cytometry. To detect CAR-transduced cells, T lymphocytes were stained with a monoclonal antibody Fc-specific cyanine-Cy5-conjugated (Fc-γCy5) provided by Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA), which recognizes the IgG1-CH2CH3 component of the receptor. Cells were analysed using a FACScan (Becton Dickinson & Co.) equipped with the filter set for four fluorescence signals. Cells were maintained or expanded in complete media supplemented with rhIL-2 (50 U/ml) every 3 days.
Cytotoxicity assay
The cytotoxic specificity of control and CAR+ T lymphocytes was evaluated in a standard 4–6-h chromium-51 (Cr51) release assay using effector : target (E : T) ratios of 40 : 1, 20 : 1, 10 : 1 and 5 : 1, as previously described.5,18 The targets tested included CAPAN-1, CFPAC, PL45 and 293T. Target cells incubated in media alone or in 1% Triton X-100 (Sigma-Aldrich, Inc., St Louis, MO, USA) were used to determine spontaneous and maximum Cr51 release, respectively. The mean percentage of specific lysis of triplicate wells was calculated as follows: ([test counts – spontaneous counts]/[maximum counts – spontaneous counts]) × 100%.
Immunohistochemistry
Pancreatic cancer cell lines and samples were resuspended in phosphate-buffered saline (PBS), mounted onto glass slides (Shandon Cytospin 4 Cytocentrifuge; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and then placed in a steamer for 10 min (high pressure) in Target Retrieval solution (Dako). Slides were incubated in pre-block/diluent for 30 min, after which they were incubated with rabbit anti-human PSCA antibody (AbCam, Inc., Cambridge, MA, USA) diluted 1 : 100 in PBS/1% bovine serum albumin (BSA) for 1 h at room temperature. Anti-rabbit horseradish peroxidase (HRP) as Biotin was used to detect positive cells, followed by enzymatic conversion using the chromogenic substrate 3,3 diaminobenzidine (DAB).
Results
Expression of PSCA in pancreatic cancer cell lines and tumour samples
We assessed the expression of tumour-associated antigen (TAA) PSCA in the pancreatic cancer cell lines ASPC-1, PL45, CAPAN-1 and CFPAC by IHC, using 293T cells as a negative control cell line. All four of the cell lines strongly overexpressed PSCA (Fig. 1) as assessed by both IHC and quantitative real-time polymerase chain reaction (qRT-PCR; data not shown).
Figure 1.
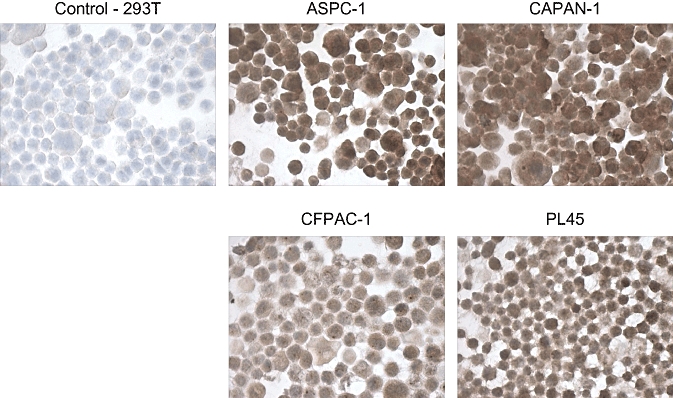
Prostate stem cell antigen (PSCA) expression in pancreatic cancer cell lines. The pancreatic cancer cell lines ASPC-1, CAPAN-1, CFPAC and PL45 were analysed for PSCA expression by immunohistochemistry. Expression of PSCA was strong in all four lines tested, but not in the 293T cells used as a control
To confirm that the same was true using primary tissue isolated from pancreatic adenocarcinoma patients, we performed IHC on paraffin-embedded tissue samples from 14 patients. We graded the tissue samples according to the intensity of PSCA staining (Fig. 2A). In total, eight of 14 samples (57%) were strongly PSCA-positive, five (36%) were intermediate and one (7%) was PSCA-negative. Figure 2B shows the staining in six individual samples that were strongly PSCA positive. This analysis confirms that PSCA is frequently expressed in pancreatic adenocarcinoma and validates its usefulness as a potential target for immunotherapy.19
Figure 2.
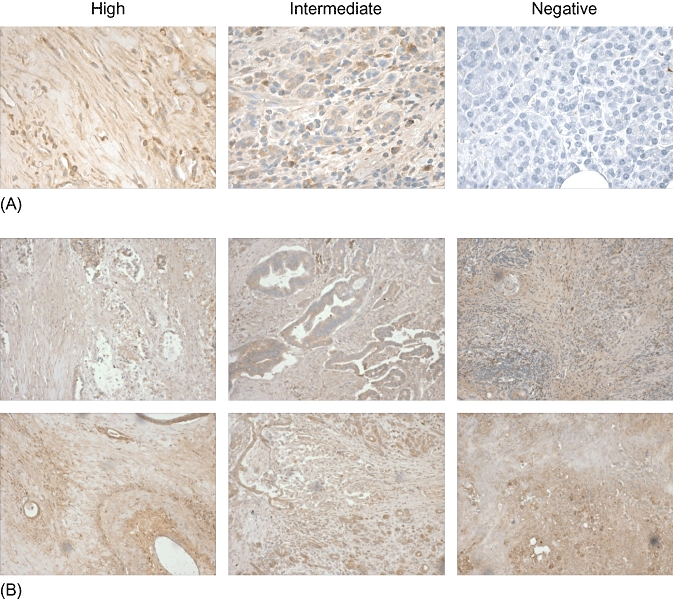
Prostate stem cell antigen (PSCA) expression in primary tumour samples. To assess whether PSCA was also frequently expressed in primary tissue samples obtained from patients with pancreatic cancer, 14 samples were stained and antigen expression evaluated by immunohistochemistry. (A) The grading strategy used to assess expression based on the intensity of PSCA staining. (B) Examples of tumour isolates from six patients deemed to be highly PSCA-positive
Generation of CAR-PSCA-modified T cells
T lymphocytes obtained from four normal donors and four patients with pancreatic carcinoma were activated using anti-CD3/CD28, expanded in the presence of IL-2 (Fig. 3A) and a fraction of the cells were transduced with a retroviral vector encoding PSCA. Figure 3B shows the CAR-PSCA vector map. After a single transduction, 53% (range: 20–92%) of healthy donor and 50% (range: 37–70%) of patient T lymphocytes expressed CAR-PSCA. Both CD4+ and CD8+ T cells were transduced (data not shown). Figure 3C shows a representative example of transgenic T cells and Fig. 3D summarizes the results in all donors.
Figure 3.
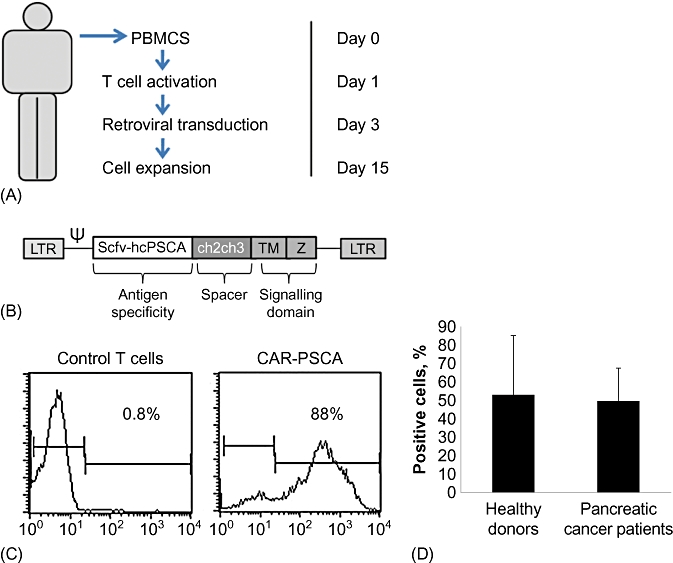
Generation of CAR-PSCA T cells. (A) Schematic of the protocol used to activate and transduce CAR-PSCA T cells in vitro. PBMCs, peripheral blood mononuclear cells. (B) Retroviral vector map of the CAR-PSCA construct. (C) Transduction efficiency of CAR-PSCA transduced T cells from a representative donor as evaluated by flow cytometry using an antibody directed against the CH2CH3 region of the retroviral construct. (D) T cells generated from both normal donors (n = 7) and pancreatic cancer patients (n = 3) were efficiently transduced
CAR-PSCA-modified T cells kill PSCA-expressing tumour cells
To determine whether the CAR-PSCA T cells were cytolytic, we incubated transgenic (69% transduced; data not shown) or control (non-transduced) T cells with the PSCA-positive cell lines PL45, CFPAC and CAPAN-1, as well as the PSCA-negative cell line 293T as a negative control. Specific Cr51 release was measured 4–6 h after co-incubation. At E : T ratios ranging from 40 : 1 to 5 : 1, CAR-PSCA T cells killed PL45, CFPAC and CAPAN-1 cell lines (Cr51 release of 46%, 55% and 76%, respectively, at E : T 40 : 1), but achieved minimal recognition of control 293T cells (Cr51 release: 29%). By contrast, control, non-transduced T cells showed minimal or no specific recognition of the target PSCA-positive cell lines (Cr51 release of 22%, 17% and 42%, respectively, at E : T 40 : 1) (Fig. 4A). These results were confirmed in four additional T cell lines generated from healthy donors. Figure 4B shows that CAR-PSCA T cells from four additional donors efficiently lysed the CFPAC and CAPAN-1 cell lines (Cr51 release of 30 ± 9% and 68 ± 9%, respectively, at an E : T ratio of 40 : 1 ± standard deviation [SD]) with no recognition of control 293T cells (16 ± 3%).
Figure 4.
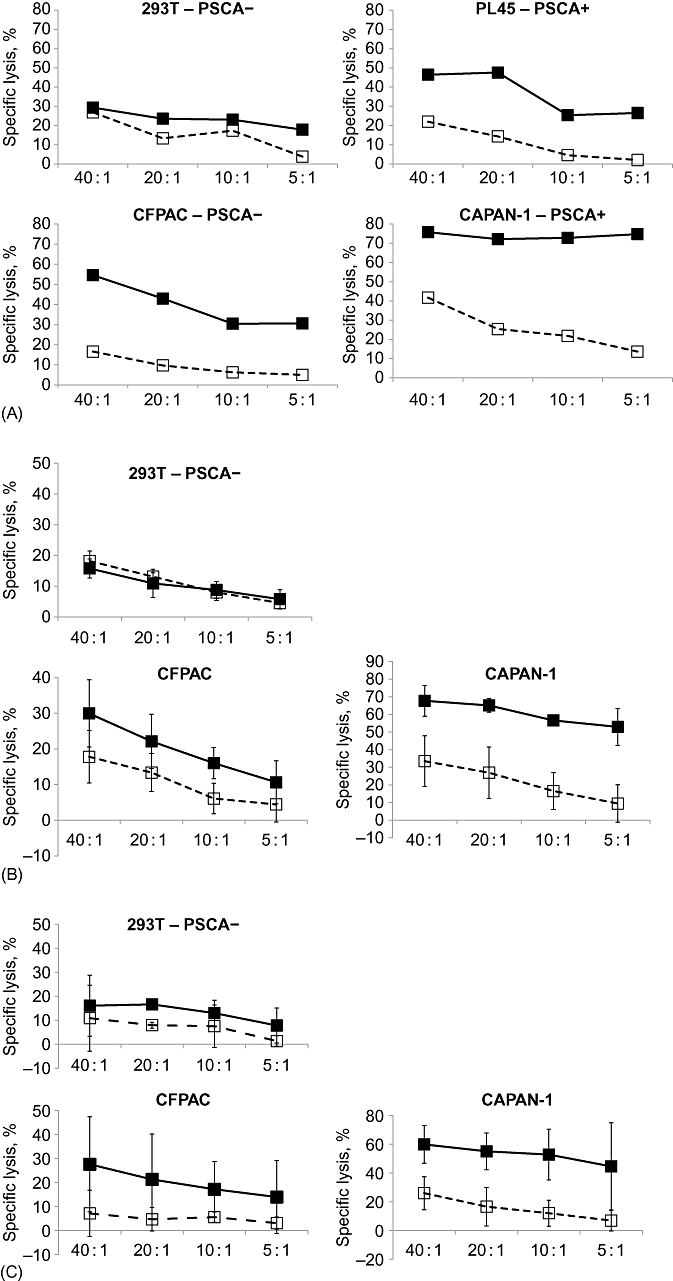
Chimeric antigen receptor (CAR) prostate stem cell antigen (PSCA) T cells are cytotoxic in vitro. (A) To determine whether the CAR-PSCA T cells could lyse tumour antigen-expressing target cells, we tested the ability of a line generated from a representative donor to kill PSCA-expressing tumour cell lines PL45, CAPAN-1 and CFPAC in a standard 6-h Cr51 release assay. The PSCA negative cell line 293T served as a control. Effector : target (E : T) ratios of 40 : 1, 20 : 1, 10 : 1 and 5 : 1 were tested. (B) Summary of results using CAR-PSCA T cells from four healthy donors. (C) Summary of results using CAR-PSCA T cells from four pancreatic cancer patients.  , CAR-PSCA cells;
, CAR-PSCA cells;  , non-treated T cells
, non-treated T cells
As the clinical use of CAR-PSCA T cells for the treatment of pancreatic cancer relies on the generation of T cell lines from autologous PBMCs, we next determined whether our generation protocol would be effective in material from patients who had been heavily treated with lymphocyte-directed chemotherapy and irradiation. Figure 4C shows that CAR-PSCA T cells from three patients with pancreatic adenocarcinoma efficiently lysed the CFPAC and CAPAN-1 cell lines (achieving lysis of 28 ± 20% and 60 ± 11%, respectively, at E : T 40 : 1 ± SD) with no recognition of control 293T cells (16 ± 15% lysis).
Discussion
Pancreatic cancer is an aggressive and lethal disease which is highly resistant to conventional treatments. As a result of its rapid progression, absence of symptoms and lack of early screening methods, most patients are diagnosed when the disease is already advanced and when conventional treatments such as surgery, chemotherapy and radiation therapy are mostly ineffective. Adoptive transfer of T lymphocytes genetically modified to express CARs targeting TAAs is an attractive treatment alternative and hence, to assess the feasibility of this approach, we cloned a novel CAR targeting the frequently expressed TAA PSCA, which is commonly expressed in the majority of pancreatic adenocarcinomas. We demonstrated in vitro that activated T lymphocytes, derived from both healthy donors and patients, can be readily prepared in large numbers and can be efficiently gene-modified to express this receptor. Finally, we showed that these CAR-PSCA-expressing T cells efficiently kill PSCA-positive pancreatic cancer cell lines in vitro.
Adoptive T cell transfer offers a real therapeutic alternative to patients with pancreatic cancer, not only because this treatment has the ability to target chemoresistant tumours, but also because the infused T cells can target even disseminated disease with few side-effects compared with conventional therapies. However, the efficacy of any T cell-based therapy relies heavily on the nature of the antigen(s) targeted. The ideal target antigen should be expressed on the tumour cell surface, and uniquely or highly expressed by tumour cells compared with normal tissues in order to limit collateral damage. It should also be directly involved in maintaining the oncogenic tumour phenotype, thereby limiting the emergence of tumour escape mutants. In the current study, we focused on preparing T cells specific to the TAA PSCA. Cheever et al. recently identified PSCA as a high-priority target for cancer therapy based upon criteria including tumour specificity, oncogenicity, expression level and number of identified epitopes.20 The PSCA gene encodes a 123-amino acid glycoprotein, with 30% homology to stem cell antigen 2 (Sca 2), and, like Sca-2, PSCA belongs to a member of the Thy-1/Ly-6 family and is anchored by a GPI linkage. It was originally identified as a potential target for adoptive immunotherapy in the context of prostate cancer, in which its overexpression was found to correlate with disease progression and prognosis. However, it has subsequently been found to be overexpressed in a broad spectrum of tumours, including non-small cell lung carcinoma, bladder cancer and pancreatic cancer.11,19 The function of PSCA is unknown, but anti-PSCA antibodies and Fab fragments have been shown to inhibit tumour growth in pancreatic xenograft models,19,21 whereas vaccine studies targeting PSCA induced effective antitumour immune responses in vivo, resulting in the inhibition of tumour growth and metastasis in a prostate cancer xenograft model and no evidence of collateral autoimmune responses in normal tissues,22 thus establishing this antigen as an attractive target antigen for immunotherapy.
Chimeric antigen receptor-modified T cells have been adoptively transferred to a wide spectrum of patients as tumour immunotherapy. The infused cells have been modified with a variety of receptors. ‘First generation’ CARs consist of a tumour-directed scFV linked to the transmembrane and intracellular signalling domains of either CD3ζ or FcRγ. ‘Second and third generation’ constructs contain additional intracellular endodomains added to enhance in vivo T cell function by enabling cytokine production in the absence of co-stimulation, which is often lacking on tumour cells, and improving effector function.9 In the current study, we developed a first-generation ‘humanized’ CAR targeting PSCA for two main reasons.14,15 Firstly, the infusion of T cells modified with this CAR should not be inherently immunogenic, a problem which has been encountered in previous studies using T cells modified with receptors derived from murine antibodies.23 For example, Lamers and colleagues used a CAR targeting carbonic anhydrase as treatment for renal cell carcinoma in three patients, all of whom mounted anti-scFv antibody responses, directed against the murine portion of the CAR.24 Secondly, there have been recent reports of toxicity associated with the infusion of high numbers of T cells modified with second- and third-generation receptors that are easily triggered by low-avidity ‘off-target’ binding to produce a potent activation signal that leads to a lethal cytokine storm in vivo.8,25,26 We reasoned that our CAR-PSCA receptor would be safe for infusion because: (i) PSCA expression is highly restricted to tumours and prostate tissue,19 and (ii) this first-generation CAR lacks co-stimulatory domains which have previously been associated with toxicity. Fortunately, PSCA-directed T cells induced using DNA plasmids have revealed no major systemic toxicities22 and T cells directed against PSCA peptides have been isolated from the peripheral blood of patients.27,28
Our in vitro experiments showed that CAR-PSCA T cells generated from both healthy donors and pancreatic cancer patients were able to specifically target and efficiently and rapidly kill PSCA-positive tumour cells. Thus, this approach deserves further study and may represent a promising novel treatment for patients with pancreatic cancer. Ultimately, however, a strategy in which T cells are combined with conventional therapies may produce maximal benefit in vivo. Previous studies have shown that engraftment of transferred T cells can be facilitated by prior immunodepletion with chemotherapy or total body irradiation because this favours subsequent homeostatic T cell expansion post-infusion.10,29–31 The combination of chemotherapy with the CAR-PSCA T cells described here may be of considerable value in the treatment of patients with pancreatic cancer but can be extended essentially to any patient with a PSCA-positive tumour, including those with prostate and bladder cancer.
Conclusions
Prostate stem cell antigen is frequently expressed on the surface of pancreatic cancer cells and can be targeted in vitro for immune-mediated destruction using CAR-PSCA modified, adoptively transferred T cells. The safety and efficacy of this approach indicate that it deserves further study and may represent a promising novel treatment for patients with pancreatic cancer.
Acknowledgments
This work was supported by a pilot project grant awarded by the Dan L. Duncan Cancer Center, Baylor College of Medicine (Houston, TX, USA).
Conflicts of interest
None declared.
References
- 1.Sweeney AD, Wu MF, Hilsenbeck SG, Brunicardi FC, Fisher WE. Value of pancreatic resection for cancer metastatic to the pancreas. J Surg Res. 2009;156:189–198. doi: 10.1016/j.jss.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 2.Bollard CM, Gottschalk S, Leen AM, Weiss H, Straathof KC, Carrum G, et al. Complete responses of relapsed lymphoma following genetic modification of tumour-antigen presenting cells and T lymphocyte transfer. Blood. 2007;110:2838–2845. doi: 10.1182/blood-2007-05-091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matera L. The choice of the antigen in the dendritic cell-based vaccine therapy for prostate cancer. Cancer Treat Rev. 2010;36:131–141. doi: 10.1016/j.ctrv.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to co-express tumour-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossig C, Brenner MK. Genetic modification of T lymphocytes for adoptive immunotherapy. Mol Ther. 2004;10:5–18. doi: 10.1016/j.ymthe.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumour. Cancer Res. 2010;70:9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukaemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vera JF, Brenner MK, Dotti G. Immunotherapy of human cancers using gene modified T lymphocytes. Curr Gene Ther. 2009;9:396–408. doi: 10.2174/156652309789753338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 11.Raff AB, Gray A, Kast WM. Prostate stem cell antigen: a prospective therapeutic and diagnostic target. Cancer Lett. 2009;277:126–132. doi: 10.1016/j.canlet.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbisan F, Mazzucchelli R, Santinelli A, Scarpelli M, Lopez-Beltran A, Cheng L, et al. Expression of prostate stem cell antigen in high-grade prostatic intraepithelial neoplasia and prostate cancer. Histopathology. 2010;57:572–579. doi: 10.1111/j.1365-2559.2010.03666.x. [DOI] [PubMed] [Google Scholar]
- 13.Vera JF, Brenner LJ, Gerdemann U, Ngo MC, Sili U, Liu H, et al. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex) J Immunother. 2010;33:305–315. doi: 10.1097/CJI.0b013e3181c0c3cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leyton JV, Olafsen T, Lepin EJ, Hahm S, Bauer KB, Reiter RE, et al. Humanized radioiodinated minibody for imaging of prostate stem cell antigen-expressing tumours. Clin Cancer Res. 2008;14:7488–7496. doi: 10.1158/1078-0432.CCR-07-5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leyton JV, Olafsen T, Sherman MA, Bauer KB, Aghajanian P, Reiter RE, et al. Engineered humanized diabodies for microPET imaging of prostate stem cell antigen-expressing tumours. Protein Eng Des Sel. 2009;22:209–216. doi: 10.1093/protein/gzn055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vera JF, Hoyos V, Savoldo B, Quintarelli C, Giordano Attianese GM, Leen AM, et al. Genetic manipulation of tumour-specific cytotoxic T lymphocytes to restore responsiveness to IL-7. Mol Ther. 2009;17:880–888. doi: 10.1038/mt.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumour-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–2802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leen AM, Sili U, Vanin EF, Jewell AM, Xie W, Vignali D, et al. Conserved CTL epitopes on the adenovirus hexon protein expand subgroup cross-reactive and subgroup-specific CD8+ T cells. Blood. 2004;104:2432–2440. doi: 10.1182/blood-2004-02-0646. [DOI] [PubMed] [Google Scholar]
- 19.Wente MN, Jain A, Kono E, Berberat PO, Giese T, Reber HA, et al. Prostate stem cell antigen is a putative target for immunotherapy in pancreatic cancer. Pancreas. 2005;31:119–125. doi: 10.1097/01.mpa.0000173459.81193.4d. [DOI] [PubMed] [Google Scholar]
- 20.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu Z, Yamashiro J, Kono E, Reiter RE. Anti-prostate stem cell antigen monoclonal antibody 1G8 induces cell death in vitro and inhibits tumour growth in vivo via an Fc-independent mechanism. Cancer Res. 2005;65:9495–9500. doi: 10.1158/0008-5472.CAN-05-2086. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad S, Casey G, Sweeney P, Tangney M, O'Sullivan GC. Prostate stem cell antigen DNA vaccination breaks tolerance to self-antigen and inhibits prostate cancer growth. Mol Ther. 2009;17:1101–1108. doi: 10.1038/mt.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:20–22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 25.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heslop HE. Safer CARS. Mol Ther. 2010;18:661–662. doi: 10.1038/mt.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsueda S, Yao A, Ishihara Y, Ogata R, Noguchi M, Itoh K, et al. A prostate stem cell antigen-derived peptide immunogenic in HLA-A24− prostate cancer patients. Prostate. 2004;60:205–213. doi: 10.1002/pros.20038. [DOI] [PubMed] [Google Scholar]
- 28.Matsueda S, Kobayashi K, Nonaka Y, Noguchi M, Itoh K, Harada M. Identification of new prostate stem cell antigen-derived peptides immunogenic in HLA-A2(+) patients with hormone-refractory prostate cancer. Cancer Immunol Immunother. 2004;53:479–489. doi: 10.1007/s00262-003-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–373. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 31.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]