

Abstract
The quiver/sleepless (qvr/sss) gene encodes a small, glycosylphosphatidylinositol-anchored protein that plays a critical role in the regulation of sleep in Drosophila. Loss-of-function mutations in qvr/sss severely suppress sleep and effect multiple changes in in situ Shaker K+ currents, including decreased magnitude, slower time-to-peak, and cumulative inactivation. Recently, we demonstrated that SLEEPLESS (SSS) protein modulates Shaker channel activity, possibly through a direct interaction at the plasma membrane. We show here that SSS accelerates the activation of heterologously expressed Shaker channels with no effect on deactivation or fast N-type inactivation. Furthermore, this SSS-induced acceleration is sensitive to the pharmacological disruption of lipid rafts and sufficiently accounts for the slower time-to-peak of in situ Shaker currents seen in qvr/sss mutants. We also find that SSS decreases the rate of C-type inactivation of heterologously expressed Shaker channels, providing a potential mechanism for the cumulative inactivation phenotype induced by qvr/sss loss-of-function mutations. Kinetic modeling based on the in vitro results suggests that the SSS-dependent regulation of channel kinetics accounts for nearly 40% of the decrease in Shaker current magnitude in flies lacking SSS. Sleep duration in qvr/sss-null mutants is restored to normal by a qvr/sss transgene that fully rescues the Shaker kinetic phenotypes but only partially rescues the decrease in current magnitude. Together, these results suggest that the role of SSS in the regulation of sleep in Drosophila correlates more strongly with the effects of SSS on Shaker kinetics than current magnitude.
Introduction
Humans experience an increasing pressure to sleep when they remain awake for longer than 16 h (Van Dongen et al., 2003). This phenomenon, called sleep homeostasis, is a conserved hallmark of sleep (Borbély and Achermann, 1999), but its molecular mechanism is unknown. Loss-of-function mutations in the Drosophila gene quiver/sleepless (qvr/sss) profoundly decrease sleep and, unlike sleep-reducing alleles of other genes (e.g., Shaker, dopamine transporter), severely impair homeostatic recovery sleep following sleep deprivation (Cirelli et al., 2005; Koh et al., 2008; Wu et al., 2008). Therefore, uncovering the actions of the protein product of qvr/sss, SLEEPLESS (QVR/SSS or SSS), may lend the first insight into the cellular machinery underlying sleep homeostasis.
SSS is a small (∼15 kDa), glycosylphosphatidylinositol (GPI)-anchored protein predicted to contain a three-fingered Ly6 domain (Koh et al., 2008; Wu et al., 2010). Similar to the GPI-anchored, Ly6-containing protein Lynx1 that modulates acetylcholine-gated ion channels (Miwa et al., 1999), SSS appears to interact with the canonical voltage-gated K+ channel Shaker, forming a stable complex when coexpressed with Shaker in vitro (Wu et al., 2010). Mutations in qvr/sss have multiple effects on Drosophila Shaker K+ currents, including decreased magnitude, delayed time-to-peak, and a cumulative inactivation upon repetitive depolarization (Wang et al., 2000; Wang and Wu, 2010; Wu et al., 2010). We recently demonstrated that SSS cell autonomously modulates Shaker currents in Drosophila and that SSS coexpression alters the kinetics of heterologously expressed Shaker channels in a manner consistent with the in situ findings (Wu et al., 2010).
Several questions remain about the SSS–Shaker interaction. For example, the kinetic processes (i.e., activation, deactivation, inactivation) affected by qvr/sss are unknown. Additionally, because qvr/sss mutants express less Shaker protein (Wu et al., 2010), the contributions of the SSS-dependent kinetic effects to the net loss of Shaker current magnitude and the sleep phenotype are unclear. Finally, while lipid rafts facilitate the effects of some GPI-anchored proteins on ion channel signaling [e.g., the α2δ subunit of voltage-gated Ca2+ channels (A. Davies et al., 2010)], the role of rafts in the interactions between SSS and Shaker has not been explored.
We show here that SSS accelerates activation of heterologously expressed Shaker channels with little effect on deactivation or N-type inactivation. The SSS-induced activation effect may account for the slower time-to-peak of Shaker currents in qvr/sss mutants and is sensitive to the depletion of membrane cholesterol. Coexpression with SSS also slows entry of heterologously expressed Shaker channels into the C-type inactivated state, providing a potential mechanism for the cumulative inactivation phenotype in loss-of-function qvr/sss mutants. Furthermore, kinetic modeling based on the in vitro results suggest that the effects of SSS on Shaker activation and C-type inactivation account for nearly 40% of the total loss of Shaker current magnitude in qvr/sss mutants. We propose that the regulation of sleep in Drosophila by SSS is more dependent on the modulation of Shaker current kinetics than on Shaker current magnitude.
Materials and Methods
Fly stocks and maintenance.
The transgenic fly line bearing a P-element insertion in the open reading frame (exon 5) of the gene designated CG33472 (qvr/sss) by the Drosophila Genome Project has been previously described as sssP1 (Koh et al., 2008). The sssP1 strain was outcrossed five times to an isogenic white background stock (iso31) obtained from the Bloomington Stock Center. Individual fly lines containing the 24B-GAL4 driver and UAS-sss were generated as previously described (Wu et al., 2010) and crossed into an sssP1 background. Fly stocks were maintained at 25°C on standard molasses–yeast–cornmeal food and entrained to a 12:12 h light:dark schedule.
Cells, cell transfection, and DNA constructs.
HEK-tsA cells were cultured in standard media (DMEM, 10% FBS, 1% penicillin/streptomycin) at 37°C and 5% CO2. Cells were transfected by FuGENE6 (Roche) with a plasmid DNA coding for GFP (to identify transfected cells) and equal quantities (1.78 μg) of plasmid DNAs coding for SSS and K+ channels in pGWI or pcDNA3 expression vectors. We used the following wild-type K+ channels: Drosophila Shaker B (ShB; AAF48785), Drosophila Eag (dEag; AAA28495), and human large-conductance Ca2+-activated K+ channels (hSlo1; AAB65837). The truncated and mutant channel variants used were ShBΔN (deletion of residues 6–46) (Hoshi et al., 1990), ShBΔN T449A, ShBΔN W434F, and hKv1.3 H399Y (AAH35059; a gift from Dr. C. Deutsch, University of Pennsylvania, Philadelphia, PA). Substitutions in the pore region T449A and H399Y accelerate and slow the rate of C-type inactivation, respectively (López-Barneo et al., 1993), while substitution W434F results in a nonconducting ShB channel (Perozo et al., 1993).
In situ electrophysiology.
For in situ recordings, muscle ionic currents were recorded from third-instar Drosophila larvae during the middle 6 h of the light cycle, in a manner previously described (Wu et al., 2010). Larvae were dissected at room temperature in a Ca2+-free, hemolymph-like saline [HL3.1; consisting of the following (in mm): 70 NaCl, 5 KCl, 4 MgCl2, 10 NaHCO3, 5 trehalose, 115 sucrose, 5 HEPES, pH 7.1 with N-methyl-d-glucamine (NMG)](Feng et al., 2004) to gain access to muscle 6 of hemisegments A3–A5. The dissection solution was replaced with a Ca2+-free HL3.1 recording solution containing 5 mm EGTA and either 5 mm (physiological) or 30 mm (supraphysiological) KCl. The two ends of the target muscle were impaled with a voltage-sensing electrode and a current-injecting electrode, both coated with Sylgard (Dow Corning). When filled with 2 m KCl (with 10 mm EGTA, pH 7.2 with KOH), the electrodes had initial resistances of 8–20 and 4–10 MΩ, respectively. Signals were amplified (OC-725C; Warner), low-pass filtered at 1 kHz (8-pole Bessel, model 902; Frequency Devices), digitized using an AD/DA converter (ITC16; HEKA), and recorded/stored via custom data acquisition software. Igor Pro 6.1 (WaveMetrics) was used for post hoc analysis of the data.
Muscles were voltage-clamped at −80 mV and depolarized (e.g., to 10 mV) either directly from holding potential to elicit IA and IK K+ currents or after a 200 ms prepulse to −20 mV to elicit IK alone. Subtraction of the two series of sweeps yielded IA alone. Unless specified, pulses were applied every ∼2 s.
Ionic currents were normalized by cell capacitance, calculated from currents recorded during a voltage ramp by the following relationship: Iramp − C(dV/dt) = Istep, where Iramp is recorded at a particular voltage during a ramp of dV/dt, and Istep is recorded at steady state at the same voltage (Wu et al., 2010).
Macroscopic capacitive and leak currents were subtracted using a P/4 protocol. All data are presented as mean ± SEM. The statistical significance of differences in electrophysiology metrics was determined by two-tailed Student's t tests. Comparisons involving multiple groups were analyzed by two-way ANOVA, followed by Student's t tests corrected with the Bonferroni method. Statistical significance was assumed at p ≤ 0.05.
In vitro electrophysiology.
Inside-out and whole-cell patch-clamp recordings were conducted at room temperature (21–23°C) 2 d after transfection. The extracellular recording solution contained the following (in mm): 130 NaCl, 10 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 15 glucose (pH 7.4 with NMG). The intracellular solution contained the following (in mm): 140 KCl, 2 MgCl2, 11 EGTA, and 10 HEPES (pH 7.2 with NMG). Output signals from the patch-clamp amplifier (Axopatch 200A; Molecular Devices) were filtered at 10 kHz and digitized at 100 kHz. Data were recorded, stored, and analyzed as described above. Ionic and gating currents were elicited by depolarizing pulses from a holding voltage of −100 mV. Unless specified, the typical interpulse interval was ∼3 s. Macroscopic capacitive and leak currents were subtracted using a P/8 protocol. Data were analyzed as above for in situ electrophysiology.
Modeling of Shaker currents.
Zagotta et al. (1994a) proposed a model of Shaker gating based on experimental observations of ShB expressed in Xenopus oocytes. An abbreviated form of the 4 × 2-step model is outlined in Figure 3A and further described in Results, below. The original rate constant values were as follows (in s−1): α = 1120, β = 373, γ = 2800, and δ = 21.2. The rate constants for transitions after channel opening (not depicted in the abbreviated form) were O→Cf = 600 and Cf→O = 3800. Adjusting these parameters to fit our experimental observations only required adjusting α to 616. Further manipulations of the forward rate constants α and γ are described in the Results. The voltage dependence of the rate constants was not altered.
Figure 3.
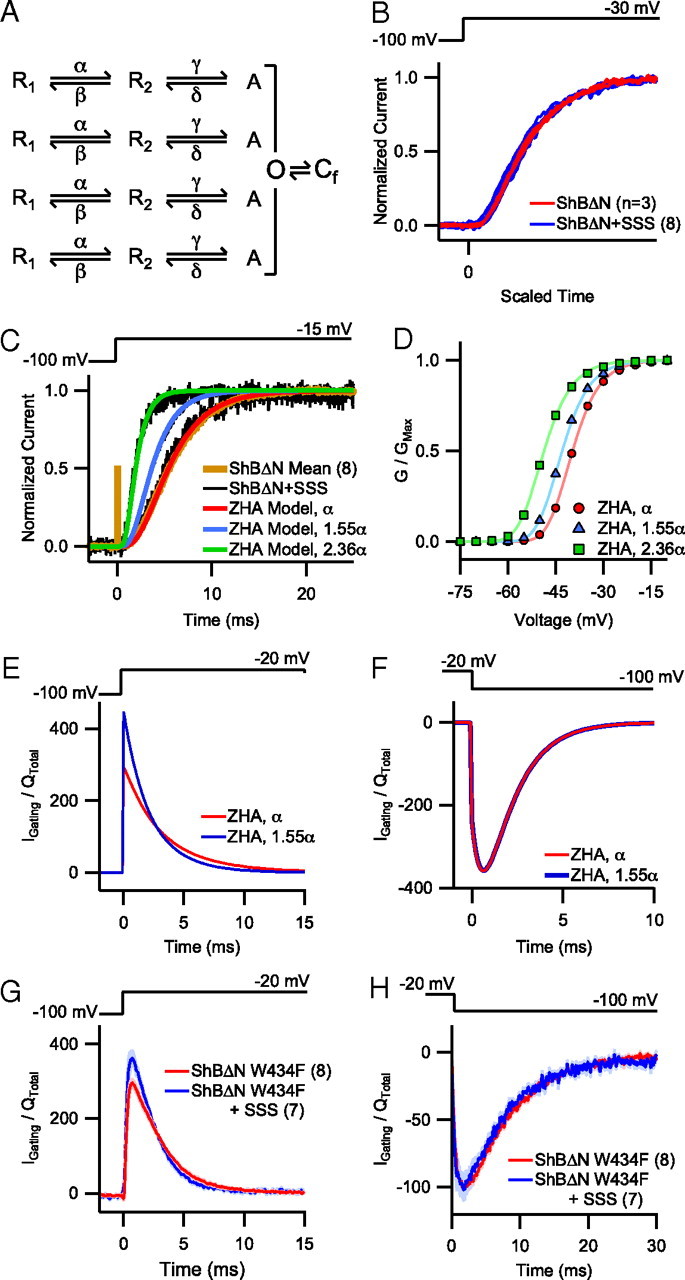
Increasing the activation rate in kinetic models of Shaker accurately predicts GV and gating current effects. A, Abbreviated form of the 4 × 2-step model (see Class D in Zagotta et al., 1994a) depicting two voltage-dependent conformational changes per subunit before a final, concerted transition. α and γ are forward rate constants (i.e., rate constants of activation) and δ and β are backward rate constants (i.e., rate constants of deactivation). B, Sigmoidicity comparison of individual traces of ShBΔN with and without SSS coexpression. As described by Zagotta et al. (1994b), 11 individual current traces were scaled by normalizing the vertical and horizontal axes so that the maximum current and slope at half-maximum current were the same for each. C, Individual traces depicting patch-clamp recordings of ShBΔN current with and without SSS (black and brown, respectively) overlayed with simulated traces generated from the ZHA two-transition kinetic model of Shaker (red, blue, green). A baseline ZHA trace (red) was established by fitting the model to the pooled ShBΔN trace (brown). No change in forward rate constant α was required to fit the baseline ZHA trace to the slow ShBΔN + SSS trace (slowest black trace). Multiplying α by 1.55 and 2.36 (blue and green, respectively) was needed to match the moderate and fast ShBΔN + SSS traces, respectively. D, GV relationship for ZHA modeled channels based on a previously established rate constant for α compared with values of 1.55α and 2.36α. E, F, ON (E) and OFF (F) gating current traces normalized by total charge movement generated by the ZHA model at baseline and 1.55α. G, Normalized gating current from pooled whole-cell recordings (mean ± SEM) of ShBΔN W434F with and without SSS coexpression upon depolarization to −20 mV. H, Normalized current from pooled whole-cell recordings (mean ± SEM) of ShBΔN W434F with and without SSS coexpression upon repolarization to −100 mV.
Drosophila sleep assay.
Adult female Drosophila (3- to 5-d-old) were individually loaded into glass locomotor tubes compatible with the Drosophila Activity Monitoring System (Trikinetics); one end of each tube was filled with yarn while the other contained a 5%:2% sucrose:agar solution with or without additional KCl supplementation. After 1 d of acclimation, locomotor activity was recorded over 4 consecutive days, processed via DAMFileScan (Trikinetics), and analyzed via pySolo sleep analysis software (Gilestro and Cirelli, 2009). As has been established, sleep was defined as periods of inactivity lasting at least 5 min (Pfeiffenberger et al., 2010).
Sodium bicarbonate extraction of lipid rafts.
HEK-tsA cells in 100 mm culture dishes were transfected as described above. Culture medium was replaced with the extracellular recording solution described above and allowed to stand at room temperature. After 30 min, cells were washed twice with ice-cold PBS and homogenized over ice in 2.1 ml of 500 mm NaHCO3 (pH 11 with NaOH). The homogenate was then sonicated by six 5 s pulses with a wand sonicator (Model 100; Fisher). Two milliliters of the homogenate were mixed with an equal volume of 90% sucrose (w/v) in MES-buffered saline (MBS; Boston Bioproducts) and placed in an UltraClear ultracentrifuge tube (Beckman Coulter). The tube was then layered with 4 ml of 30% sucrose in MBS, 250 mm NaHCO3, ∼5 ml of 5% sucrose in MBS, and 250 mm NaHCO3. The tube was centrifuged at >175,000 g (sw40-Ti rotor, L8-M UC; Beckman Coulter) for 20–23 h at 4°C. Serial fractions of 0.5–1 ml were harvested from the top of the sucrose gradient and frozen at −80°C for later analysis by Western blotting.
Western blot analysis.
Sucrose gradient aliquots were thawed, supplemented with SDS sample buffer, and boiled for 5 min before being loaded onto a 4–15% Tris-HCl SDS-PAGE gel (Bio-Rad). Identical volumes were loaded in each lane. The proteins were transferred to nitrocellulose membranes (Bio-Rad), which were detected by primary antibodies to Sh (1:1000; UPGP57), SSS (1:1000; UPGP69), caveolin (1:2000; polyconal #610060, BD Transduction Laboratories), and β-adaptin (1:2000; #610382, BD Transduction Laboratories) and secondary antibodies at 1:2000–3000. Membranes were stripped with Western blot stripping buffer (Thermo Scientific) before exposure to a second primary antibody. Immunoreactivity was visualized by chemiluminescence reagents (ECLPlus; GE Life Sciences) and exposure to x-ray film. X-ray film was scanned into Photoshop where only rudimentary manipulations (i.e., brightness and contrast) were applied to the entire blot.
Results
SSS specifically affects the activation kinetics of Shaker K+ channels
Shaker channels are the sole source of the fast-activating, fast-inactivating K+ current (A-type current, IA) in Drosophila larval muscles (Wu and Haugland, 1985). As previously reported (Wu et al., 2010), larvae bearing mutations in qvr/sss exhibit substantially decreased IA magnitude (control: 23.84 ± 3.15 nA/nC; sssP1: 3.87 ± 0.44 nA/nC; p < 0.001) (Fig. 1A) and a slower IA time-to-peak (control: 2.60 ± 0.10 ms; sssP1: 4.08 ± 0.17 ms; p < 0.001), while in vitro SSS coexpression significantly decreases wild-type ShB time-to-peak (ShB: 6.71 ± 0.57 ms; ShB + SSS: 4.58 ± 0.38 ms; p < 0.001) (Fig. 1B). Concomitant with time-to-peak, SSS also significantly accelerated the rate of ShB current decay (Fig. 1B,F) at certain voltages, but did not affect the rate of recovery from decay (Fig. 1G). The lack of an effect of SSS coexpression on the activation time courses of wild-type dEAG, wild-type hSlo1, or hKv1.3 H499Y currents (Fig. 1C–E) demonstrated the preferred action of SSS on Shaker channels.
Figure 1.
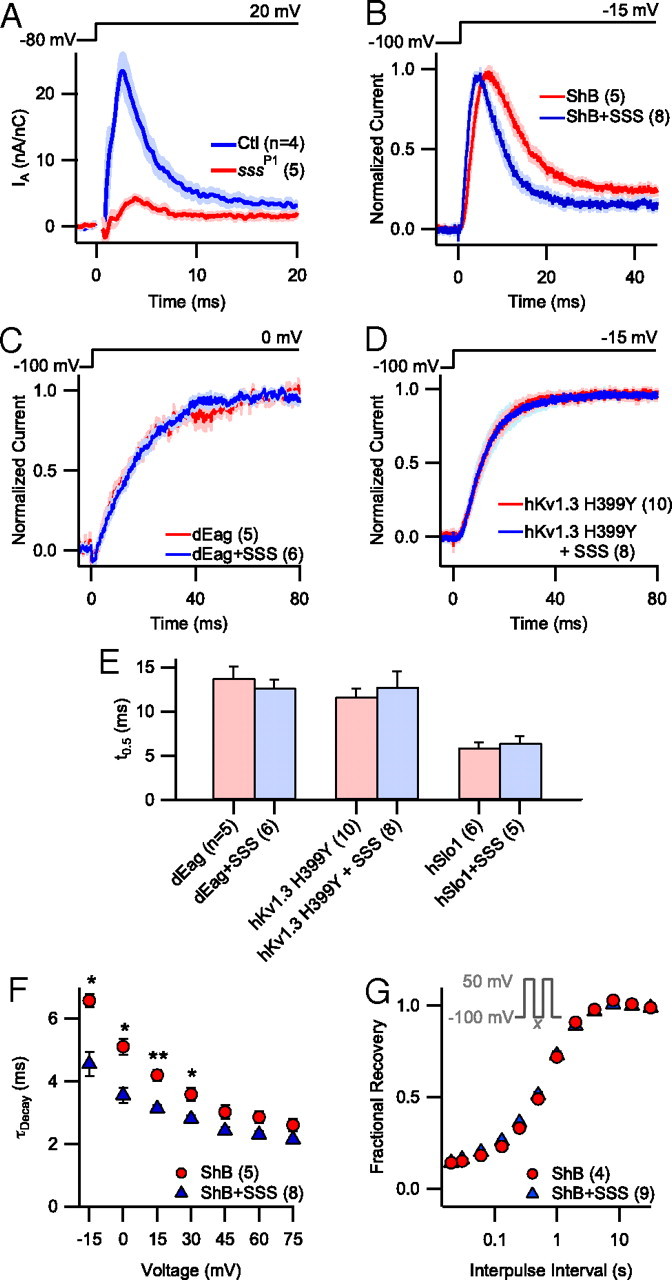
SSS produces similar kinetic effects in vitro and in vivo. A, Pooled IA traces (mean ± SEM) collected by two-electrode voltage-clamp recordings from sssP1 mutant and control (Ctl) Drosophila larval muscles at 20 mV. The capacitative transients were removed for clarity. B–D, Pooled current traces (mean ± SEM) collected by patch-clamp recordings from HEK cells heterologously expressing K+ channels with and without SSS. B, ShB currents were recorded from inside-out patches at −15 mV. C, Whole-cell dEag currents were recorded at 0 mV. D, hKv1.3 H399Y currents were recorded from inside-out patches at −15 mV. E, Time-to-half-maximum (t0.5) of dEag, hKv1.3, and hSlo1 currents at 0, −15, and 100 mV, respectively, with and without SSS coexpression. F, Rate of decay of ShB current (τDecay) with and without SSS coexpression at voltages between −15 and 75 mV. τDecay was measured by fitting a single exponential curve to the falling phase of ShB current traces at the indicated voltage. Comparisons of τDecay were only significantly different at voltages ≤30 mV. G, Rate of recovery of ShB from inactivation. Fractional recovery of ShB current observed during the second test pulse as a function of interpulse interval duration (x in the Inset). Inset, Standard two-pulse protocol in which patches were delivered two 50 ms test pulses to 50 mV separated by a variable interpulse interval (ranging from 0.16 to 32 s) at −100 mV. *p < 0.05, **p < 0.01.
The faster time-to-peak and decay of ShB currents in the presence of SSS could be caused by changes in the activation, deactivation, and/or inactivation kinetics of the ShB channel. Although SSS coexpression accelerated the rate of ShB current decay at voltages ≤30 mV, the effect of SSS diminished at increasingly depolarized voltages, failing to reach statistical significance at voltages >30 mV (Fig. 1F). This observed convergence in current decay rates at depolarized voltages is consistent with the possibility that SSS primarily affects the rate of ShB activation. Activation and N-type inactivation of the Shaker channel are tightly coupled, and the rate constant of entry into the inactivated state is largely independent of voltage (Zagotta et al., 1989; Hoshi et al., 1990). Thus, at relatively hyperpolarized voltages at which the kinetics of activation and inactivation are comparable, activation kinetics often influence the time course of current decay. However, at depolarized voltages, where activation occurs much faster than inactivation, the rate of current decay is less affected.
To assess the effect of SSS on ShB channel kinetics in the absence of inactivation, we used the ShBΔN channel, a well characterized deletion mutant in which N-type inactivation is disrupted (Hoshi et al., 1990). SSS coexpression significantly decreased the time-to-half-maximum of ShBΔN currents (ShBΔN: 8.88 ± 0.67 ms; ShBΔN + SSS: 5.38 ± 0.38 ms; p < 0.001) (Fig. 2A) and the time constant of activation was significantly smaller throughout the entire voltage range from −30 to 75 mV (Fig. 2B). For example, at 75 mV, where the opening rate constants dominate and the closing rate constant values are negligible, SSS decreased the activation time constant by ∼22%. The voltage dependence of the activation time constant was not altered by SSS. Furthermore, despite the significant effect of SSS on the ShBΔN current activation, the rate of deactivation remained unaffected (Fig. 2C). In summary, these data are consistent with a SSS-induced acceleration of Sh activation kinetics, with little to no effect on inactivation or deactivation.
Figure 2.
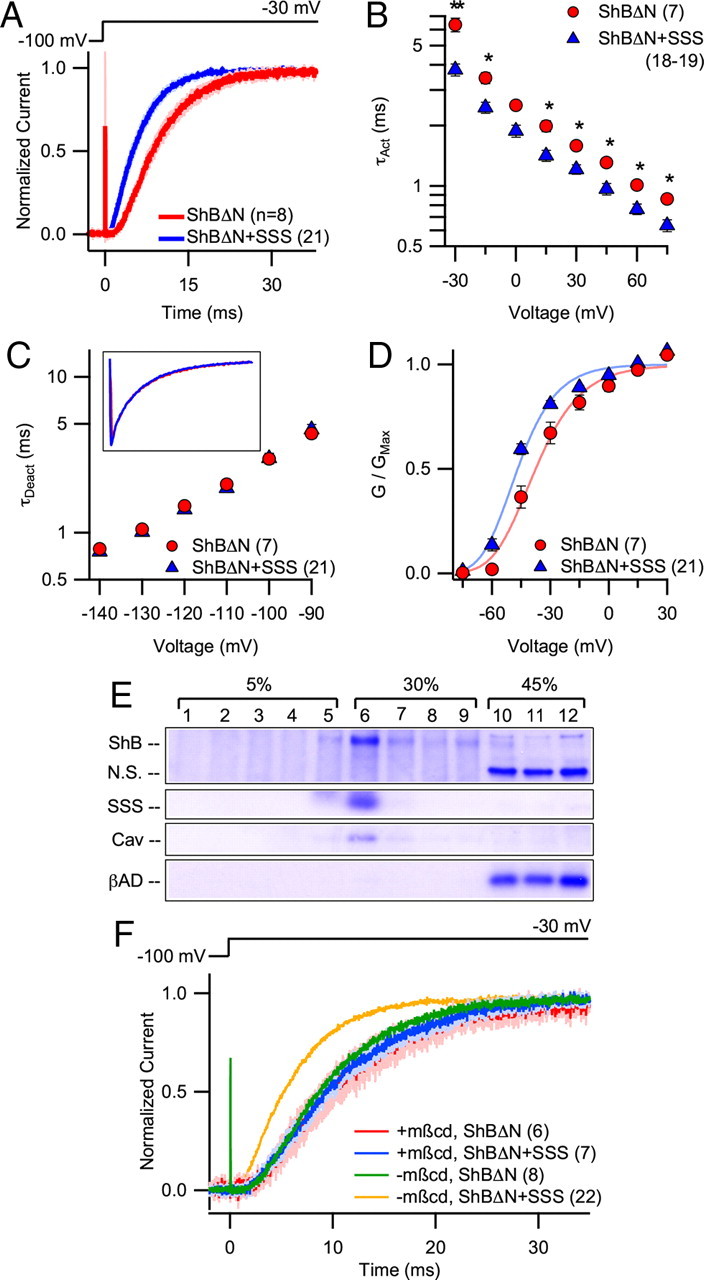
SSS coexpression accelerates activation of ShBΔN in a cholesterol-dependent manner. A, Pooled current traces (mean ± SEM) at −30 mV collected by inside-out patch-clamp recordings from HEK cells heterologously expressing ShBΔN with and without SSS. B, ShBΔN activation rate (τAct) with and without SSS coexpression measured at voltages between −30 and 75 mV. τAct was measured by fitting a single exponential curve to the second half of the rising phase (i.e., from half-maximum to maximum amplitude) of ShBΔN current traces. C, ShBΔN deactivation rate (τDeact) with and without SSS coexpression measured at voltages between −140 and −90 mV. τDeact was measured by fitting a single exponential curve to the decay of ShBΔN current trace from a test pulse of 45 mV to the indicated voltage. Inset, Pooled ShBΔN deactivation current trace (mean) at −130 mV (nShBΔN = 8; nShBΔN + SSS = 21). D, GV curves for ShBΔN with and without SSS coexpression. V0.5 was calculated after fitting a Boltzmann function raised to the fourth power to GV curves generated from tail currents for each patch. E, Western blot of proteins from HEK cells transfected with ShB and SSS. Cells were homogenized in a 500 mm NaHCO3 (pH 11) solution and segregated via sucrose gradient centrifugation. A single nitrocellulose membrane was probed for each of the following proteins: ShB, SSS, caveolin (Cav), and β-adaptin (βAD). Nonspecific bands (N.S.) also appear upon labeling with Shaker antibody. F, Pooled current traces (mean ± SEM) at −30 mV collected by inside-out patch-clamp recordings of ShBΔN with and without SSS from HEK cells preincubated for 30 min in 5 mm mβcd (blue ± light blue and red ± pink, respectively). Mean traces without mβcd preincubation (as in Fig. 2A) are provided for comparison (green and orange). *p < 0.05, **p < 0.01.
Analysis of the conductance-voltage (GV) relationships from ShBΔN ± SSS traces showed that SSS shifts the half-activation voltage (V0.5) of GV toward the negative direction (i.e., a left-shift) by ∼10 mV (ShBΔN: −37.0 ± 2.8 mV; ShBΔN + SSS: −46.7 ± 1.1 mV, p < 0.001) (Fig. 2D). Single-channel current–voltage relationship (iV) was not affected (data not shown).
The kinetic effect of SSS on ShBΔN is cholesterol-dependent
SSS is a GPI-anchored protein, and many GPI-anchored proteins are thought to be associated with cholesterol-rich membrane domains in Drosophila (Rietveld et al., 1999). These domains, called lipid rafts, are implicated in the modulation of voltage-gated K+ channel activity (Martens et al., 2001), leading us to hypothesize a role for lipid rafts in the interaction between SSS and Shaker channels. To determine whether SSS and/or ShB can traffic to lipid rafts, we performed a detergent-free isolation of lipid rafts from HEK cells heterologously expressing ShB and SSS. Like caveolin, a known component of lipid rafts, both ShB and SSS localized preferentially to low-density, lipid raft-containing membrane fractions, while β-adaptin, a protein typically excluded from lipid rafts, did not (Fig. 2E).
We also investigated the importance of membrane cholesterol for the effect of SSS on Shaker by using methyl-β-cyclodextrin (mβcd), a cholesterol-chelating agent (Yancey et al., 1996). Incubation with mβcd (5 mm) for 30 min had minimal effects on ShBΔN activation kinetics at −30 mV; however, it was sufficient to completely ablate the SSS-induced acceleration of ShBΔN activation (Fig. 2F).
Qualitative modeling of ShBΔN activation predicts SSS-induced changes in the GV relationship and gating charge movement
Measurements of single-channel currents and macroscopic ionic and gating currents of ShB channels led Zagotta and colleagues (Hoshi et al., 1994; Zagotta et al., 1994a,b) to propose a model of Shaker activation in which opening of the ion conduction gate is preceded by two coupled transitions in each of the four independent subunits. An abbreviated form of this model (the ZHA model) is depicted in Figure 3A. While a recent study suggests that each subunit may undergo four transitions before the main gate opens (Tao et al., 2010), the ZHA model adequately describes most aspects of ShB channel gating in an experimentally constrained manner. Similarly, with only minor modifications of the rate constant values originally derived from data collected from Xenopus oocytes, the ZHA model described ShBΔN ionic currents measured from patches from HEK-tsA cells (see Materials and Methods, above). Thus, we sought to identify the minimal change in the ZHA model required to account for the effects of SSS on ShBΔN kinetics.
To proceed, we first determined whether SSS affected the sigmoidicity of ShBΔN current activation. Introduced by Zagotta et al. (1994b), sigmoidicity of a current trace quantifies the relative amount and distribution of delay in ion channel opening. Thus, sigmoidicity comparisons may detect alterations in the number of conformational changes that a channel undergoes before it opens or indicate unequal changes in the kinetics of individual subunits. Recordings from patches of ShBΔN with and without SSS coexpression exhibited very similar sigmoidicities (Fig. 3B), consistent with the idea that ShB, with or without SSS, undergoes the same number of transitions and that all four subunits are equally affected by SSS. The sigmoidicity comparison and aforementioned observation that coexpression with SSS affects activation kinetics without affecting the voltage dependence of activation time constant (Fig. 2B) prompted us to increase the value of the forward rate constant α in the ZHA model (Fig. 3A) without altering its partial charge movement. Deactivation was not affected by SSS (Fig. 2C) and consequently the backwards rate constants were unaltered. Because we noticed considerable patch-to-patch variability in the speed of Shaker current activation upon SSS coexpression, we selected three sample traces (Fig. 3C, black) that covered the continuous range of speeds that we observed: slow (from the 0–25th percentile), moderate (from the 40–60th percentile), and fast (from the 75–100th percentile). While no increase in α was necessary to fit our ZHA baseline trace with the slow ShBΔN + SSS trace, increasing α by 55% (Fig. 3C, blue) and 136% (Fig. 3C, green) was necessary to match the moderate and fast ShBΔN + SSS traces. Increasing α in the ZHA model by 55% and 136% also left-shifted the GV V0.5 values from −39.7 mV to −42.9 mV and −48.6 mV, respectively (Fig. 3D). These predicted voltage shifts are similar to the experimentally observed left shift in the V0.5 of the ShB + SSS group (Fig. 2D).
The ZHA model also predicted that increasing α would affect the time course of gating charge movement upon depolarization. The 55% increase in α required to simulate moderate ShBΔN + SSS activation kinetics was predicted to accelerate the ON gating current (Fig. 3E) with minimal effect on the OFF gating current (Fig. 3F). Gating currents measured from whole cells expressing ShBΔN W434F showed that SSS coexpression accelerated ON gating current kinetics in a manner similar to the ZHA predictions without appreciably affecting the OFF gating current (Fig. 3G,H). The ZHA model also predicted an approximate 4 mV left-shift in the Q/V curve; however, we were unable to resolve such a difference and suspect that the small change was below our experimental detection limit (data not shown).
As described above, the effects of SSS on ShB activation kinetics are well accounted for by increasing α, one of the two forward rate constants of the ZHA model (Fig. 3A). In contrast, simulations in which we instead increased the other forward rate constant γ did not readily produce currents consistent with the experimental observations. Specifically, the increases in γ necessary to match the SSS-induced acceleration of activation at −30 mV do not continue to produce appreciably faster currents at very depolarized voltages (e.g., 60 mV) (data not shown), which is inconsistent with experimental observations (Fig. 2B). Furthermore, increasing γ produces a noticeable rising phase in ON gating current, which was absent from our gating current recordings (Fig. 3G). While we cannot exclude the possibility that SSS affects both α and γ, it is clear that α manipulations alone are sufficient to reproduce the SSS effects that we have observed.
SSS confers resistance to C-type inactivation in vitro and in situ
Shaker channels show two distinct inactivation processes, N-type and C-type inactivation (Hoshi et al., 1991). We evaluated whether SSS affects the rate of C-type inactivation in the ShB channel. C-type inactivation in the wild-type ShB channel is slow, occurring on the order of many seconds and masked by faster N-type inactivation (Hoshi et al., 1991), making it difficult to study experimentally. In the mutant ShBΔN T449A, C-type inactivation occurs in a more experimentally manageable range (López-Barneo et al., 1993). Coexpression with SSS significantly slowed the time course of C-type inactivation in the ShBΔN T449A channel (τinact,C = 0.393 ± 0.027 vs 0.245 ± 0.008 s, p > 0.001) (Fig. 4A), but did not affect the time course of recovery from C-type inactivation (Fig. 4B).
Figure 4.
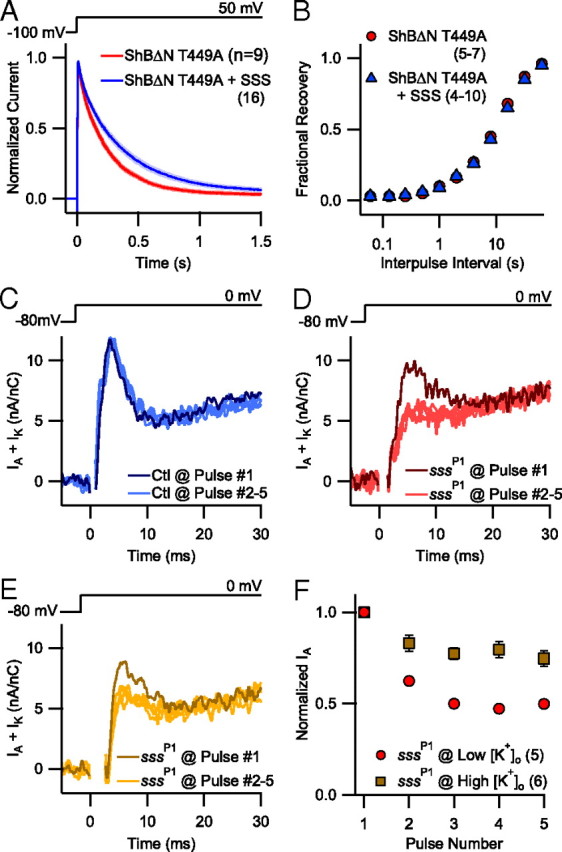
An SSS effect on Shaker C-type inactivation can explain the emergence of cumulative inactivation of IA in sss mutants. A, Pooled current traces (mean ± SEM) collected by inside-out patch-clamp recordings from HEK cells heterologously expressing ShBΔN T449A with and without SSS. B, Fractional recovery of ShBΔN T449A channels as a function of time between two 50 ms test pulses at 50 mV. Channels were allowed to recover at −100 mV with interpulse intervals ranging between 0.06 and 64 s. C–E, Representative serial K+ current traces collected by two-electrode voltage-clamp recordings from larval Drosophila muscle at physiological (C, D) and supraphysiological (E) [K+]o (5 vs 30 mm). K+ currents were elicited from control (Ctl) (C) and sssP1 mutant animals (D, E) by repeated depolarization to 0 mV every 4 s from a resting potential of −80 mV. The Shaker-dependent component of K+ current (IA) is the fast-activating, fast-inactivating component in the depicted traces. Traces from the first five consecutive pulses (Pulse #1 in darker colors than Pulses #2–5) are superimposed to visualize changes in current magnitude. Capacitative transients were deleted to improve clarity. F, Normalized peak IA magnitude upon repeated depolarization in physiological (5 mm) and supraphysiological (30 mm) [K+]o. Data were collected as in C–E. *p < 0.05, **p < 0.01.
Earlier reports noted that qvr/sss mutant flies exhibit cumulative inactivation of IA upon repetitive depolarization that is not observed in wild-type animals (Wang et al., 2000; Wang and Wu, 2010). The cumulatively inactivated component was named IAS, but the mechanism of loss is unknown. Because we did not observe any changes in the rate of recovery from N-type inactivation in ShB expressed in HEK cells (Fig. 1G), we hypothesize that Shaker channels in the absence of SSS, as in Drosophila sssP1 mutants, more readily proceed to a C-type inactivated state.
Measuring the rate of C-type inactivation in Drosophila larval muscles is technically challenging, as the current size following N-type inactivation is miniscule at best. We looked instead for a key characteristic of C-type inactivation: sensitivity to extracellular K+. Previous studies have demonstrated that elevating extracellular [K+] ([K+]o) slows the time course of C-type inactivation in Shaker channels (Choi et al., 1991; López-Barneo et al., 1993) and we thus measured the amplitude of IA upon repetitive depolarization at different concentrations of [K+]o. At a physiological level of [K+]o (5 mm), the IA component in wild-type flies recovered from inactivation completely between the depolarizing pulses so that the peak current magnitude was stable with repeated stimulation (Fig. 4C). Similar results were found at supraphysiological (30 mm) [K+]o (data not shown). In contrast, the IA component in sssP1 flies dramatically decreased after the first depolarization pulse (Fig. 4D,F), in a manner previously described for a qvr/sss hypomorph (Wang and Wu, 2010). As expected for the antagonistic effect of high extracellular K+ on C-type inactivation, IA in sssP1 flies was more resistant to cumulative inactivation when [K+]o was increased to 30 mm (51.1 ± 1.3% vs 25.2 ± 4.3%, p < 0.001) (Fig. 4E,F). Because extracellular K+ ions could decrease fast inactivation by removing the inactivation particle from its binding pocket (Demo and Yellen, 1991), we also measured the rate of IA decay at 5 and 30 mm [K+]o, but found no significant difference (τDecay = 2.6 ± 0.1 vs 2.6 ± 0.2 ms, respectively). Separately, expression of SSS in muscles of sssP1 mutants significantly decreased the proportion of cumulatively inactivated current upon repetitive stimulation at 5 mm [K+]o (sssP1/sssP1, UAS-sss; 24B-GAL4/+ = 5.0 ± 3.2% vs sssP1; 24B-GAL4/+ = 39.3 ± 3.9%, p < 0.001) while not affecting the initial IA magnitude (7.8 ± 0.6 vs 7.4 ± 0.8 nA/nC, p = 0.72).
The finding that high [K+]o partially antagonizes cumulative inactivation of IA in sssP1 flies suggested that the shortened sleep phenotype of sssP1 flies may also be sensitive to [K+]o. As small molecules can be introduced effectively into live Drosophila to manipulate behavioral phenotypes (e.g., octopamine) (Monastirioti et al., 1996), we supplemented the fly food with up to 100 mm KCl but found no rescue of sleep using the accepted Drosophila sleep-recording techniques (Pfeiffenberger et al., 2010).
Discussion
In our in vitro system, SSS increased the rate of Shaker channel activation without affecting the rate of deactivation. While SSS also increased the rate of decay of heterologously expressed wild-type Shaker currents at voltages <30 mV, this may be an indirect effect of enhancing the rate of channel opening. As illustrated by kinetic modeling in Figure 5A, enhancing the rate of activation of inactivating channels not only hastens the time-to-peak, but also increases the rate of current decay. Thus, an increase in activation alone is sufficient to explain the in situ and in vitro effects of SSS on wild-type Shaker channels.
Figure 5.
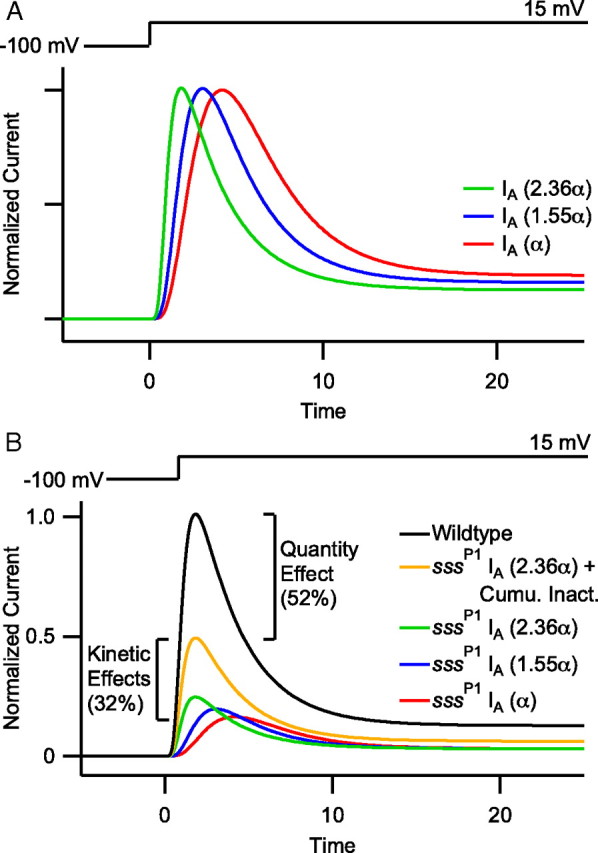
The SSS-dependent effects on Shaker kinetics comprise a significant proportion of in situ IA magnitude. A, Simulated ZHA traces in which an inactivation process has been restored, thus describing wild-type ShB currents. Enhancing the forward rate constant α decreases the time to peak and elicits a secondary increase in the rate of current decline (apparent τDecay = 3.4, 3.2, and 2.8 ms at α, 1.55α, and 2.36α, respectively). These traces were normalized to peak current to illustrate the kinetic effect. B, Simulated ZHA traces to depict the differential contributions of changes in Shaker kinetics and quantity to the overall sssP1 IA phenotype in situ. After repetitive depolarization, sssP1 mutants exhibit an approximate 84% decrease in IA current in situ. We have simulated this loss using the ZHA model (black vs red). Relative to the sssP1 IA (red), the ZHA model predicts that the SSS-induced enhancement in activation kinetics may yield up to a 40% increase in Shaker current magnitude and a faster time-to-peak (blue and green). Furthermore, recovery of current lost to cumulative inactivation upon repeated stimulation nearly doubles the total IA (orange vs green). Thus, the combined kinetic effects of sssP1 make up an approximate 32% decrease in IA relative to wild-type. Presumably the remaining 52% of IA absent in sssP1 flies is due to a decrease in Shaker quantity, consistent with the approximate 60% decrease in Shaker quantity observed by Western blot (Wu et al., 2010).
Consistent with previous reports (Wang et al., 2000; Wang and Wu, 2010), we found that repetitive depolarization of qvr/sss mutant larval muscles leads to a significant loss of Shaker current. The cumulatively inactivated component, recently named IAS (Wang and Wu, 2010), is eliminated by muscle-directed restoration of SSS in sssP1 mutants, suggesting that SSS normally prevents the cumulative inactivation of Shaker currents in wild-type flies. In our in vitro studies, SSS did not affect recovery from either N- or C-type inactivation, but it decreased the rate of entry into the C-type inactivated state. SSS may also slow C-type inactivation of Shaker in Drosophila, as the cumulative inactivation phenotype observed in qvr/sss mutants could be caused by an absence of SSS-dependent resistance to C-type inactivation. This hypothesis is supported by the reduction of cumulative inactivation at supraphysiological concentrations of extracellular K+. However, direct quantitative comparisons between the in situ Shaker currents and the in vitro currents of ShBΔN T449A cannot be made, as the precise Shaker isoforms underlying the former are unknown, and the latter has a fundamentally different rate of C-type inactivation than wild-type channels. Similarly, testing the specific contribution of C-type inactivation to the sssP1 behavioral phenotypes may also be difficult. We were unable to rescue the sssP1 phenotype with a high K+ diet but suspect that systemic K+ concentration in Drosophila is tightly regulated, as it is in mammals.
Mutations in sss profoundly decrease Drosophila IA magnitude (Wang et al., 2000; Wu et al., 2010); however, it is unclear whether the loss of Shaker current in qvr/sss mutants is caused by decreases in Shaker expression or changes in Shaker kinetics. Kinetic modeling based on in vitro experiments suggest that kinetic effects of SSS account for nearly 40% of the total loss in IA magnitude exhibited by sssP1 mutants (Fig. 5B); the rest is likely due to a decrease in channel quantity. Interestingly, the cumulative inactivation of IA comprises a large portion of the in situ current loss. Because the rescue of the cumulative inactivation phenotype nearly doubles the Shaker magnitude after repetitive depolarization, this kinetic rescue could account for the entirety of the previously reported partial rescue of total Shaker current magnitude (Wu et al., 2010). Therefore, in sssP1 mutants, targeted expression of SSS completely rescues altered Shaker gating kinetics, cumulative inactivation, and slower tPeak, as previously reported (Wu et al., 2010), while only minimally rescuing total Shaker current magnitude. Nevertheless, SSS expression fully rescues the sssP1 mutant sleep phenotype when driven broadly throughout the Drosophila brain as well as selectively in cholinergic neurons (Wu et al., 2010). Thus, the severity of the sleep phenotype in sssP1 mutants may better correlate with an alteration in Shaker current kinetics than magnitude. For example, SSS overexpression in wild-type flies reduces Shaker current in Drosophila muscles but does not induce a sleep phenotype, even when expressed using drivers that rescued the sleep phenotype in sssP1 flies (Wu et al., 2010). Additionally, Drosophila bearing Shaker-null alleles exhibit a less severe sleep phenotype than sssP1 mutants despite having no Shaker current (Cirelli et al., 2005; Koh et al., 2008). Because organisms can compensate for constitutive changes in ion channel quantity to preserve a neuron's overall excitability (MacLean et al., 2003; Lee et al., 2008), we speculate that static decreases in Shaker quantity are easier to compensate for than the dynamic, use-dependent loss of Shaker K+ current seen in qvr/sss mutants. Future studies of in vivo central neurons in adult Drosophila (Crocker et al., 2010) may better elucidate the neurophysiological significance of the kinetic effects of SSS on Shaker.
The Shaker β-subunit Hyperkinetic (Hk) accelerates rate of Shaker activation (Chouinard et al., 1995) to a degree similar to that induced by SSS; however, Hk is an intracellular protein whereas SSS is tethered to the extracellular side of the membrane by a GPI-anchor. Although GPI-anchored auxiliary subunits have not yet been proposed for voltage-gated K+ channels, a recent study found that the voltage-gated Ca2+ channel auxiliary subunit α2δ is indeed GPI-anchored (A. Davies et al., 2010). Just as adequate Ca2+ channel expression depends on α2δ, a deficiency of SSS leads to decreases in Shaker protein levels in vivo (Wu et al., 2010). Additionally, α2δ-containing Ca2+ channels concentrate in lipid rafts, much like SSS and Shaker. Furthermore, the SSS-induced increase in activation of heterologously expressed Shaker channels was sensitive to cholesterol depletion, similar to K+ currents in ex vivo neurons from the Drosophila mushroom bodies (Gasque et al., 2005), raising the possibility that the mechanism by which SSS interacts with Shaker involves lipid rafts. Thus, GPI-anchored proteins like SSS and α2δ may be critical regulators of ion channel function and quantity at the cell surface.
SSS enhances gating charge movement, but the mechanism and stoichiometry by which SSS affects the charge-bearing S4 transmembrane segment remain unknown. Because SSS and Shaker coimmunoprecipitate when coexpressed in a heterologous system (Wu et al., 2010), one potential mechanism is a direct protein–protein interaction between SSS and the extracellular loops of Shaker. The S1–S2 linker is a promising candidate, as a threonine to isoleucine substitution in this loop (Shmns) also induces a sleep phenotype; however, the functional consequences of that allele have not yet been thoroughly characterized (Cirelli et al., 2005).
Increasing the forward rate constant α in the two-step model of Shaker activation (Zagotta et al., 1994a) was sufficient to reproduce the effects of SSS on the activation of ionic currents and the time course of gating charge movement. A recent structure–function study of Shaker gating by Tao et al. (2010) suggests that activation requires at least four transitions between five discrete states of the S4 voltage sensor. The forward rate constant α in the ZHA model may correspond to the initial transitions of the voltage sensor, and thus SSS may act upon the early intermediate states of S4, including destabilizing the resting state when the first charge of S4 is located below the S2 phenylalanine cap.
There are also potential mechanisms for the SSS-dependent modulation of activation and C-type inactivation aside from a direct SSS–Sh interaction. For instance, SSS may indirectly act upon Shaker through another protein, as even in an in vitro system like HEK cells, native proteins found in lipid rafts (e.g., caveolin) can modulate K+ channels (L. M. Davies et al., 2010). Another potential effect of SSS may be to increase local extracellular K+ accumulation upon channel opening and K+ efflux, therefore indirectly slowing C-type inactivation. SSS may even affect channel kinetics by altering interactions between surrounding lipids and Shaker channels, an important consideration given that membrane phosopholipids play critical roles in ion channel function (Xu et al., 2008). A lipid-channel interaction mediated by SSS is consistent with our finding that the sigmoidicity of macroscopic current activation was unaltered by SSS, regardless of the degree of acceleration that SSS induced. Had the interaction between SSS and Shaker followed a strict stoichiometry in which individual subunits within a tetramer were selectively enhanced, then it is unlikely that activation sigmoidicity could have been preserved over the range of activation speeds observed. Because sigmoidicity is preserved over the entire range of activation speeds, we suspect that every subunit of each channel in a patch is equally affected by the variable expression levels of SSS. The effect may be unique to Shaker, as human Slo1 activation kinetics are not affected by SSS expression despite also trafficking to lipid rafts (Weaver et al., 2007), suggesting that either Shaker is particularly sensitive to lipid membrane composition or that cholesterol plays a permissive role in the effects of SSS on Shaker kinetics. Nevertheless, we cannot entirely exclude effects of SSS on channels other than Shaker.
In summary, SSS regulates the kinetics of Shaker channels on timescales ranging from milliseconds to hundreds of milliseconds. These kinetic effects, in addition to the SSS-dependent effects on Shaker expression and subcellular localization (Wu et al., 2010), suggest that SSS is an important regulator of Shaker and that the modulation of Shaker K+ conductance is a critical component of the sleep behavior in Drosophila.
Footnotes
This work was supported by grants from the National Institutes of Health (T32HL007953 and 1F30HL099407-01 to T.D., R01GM057654 and R01GM078579 to T.H., and P01AG017628 to A.S.). A.S. is an investigator of the Howard Hughes Medical Institute. We thank Carol Ma of the Levitan laboratory at Jefferson University for demonstrating how to record from Drosophila larval muscles.
The authors declare no competing financial interests.
References
- Borbély AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14:557–568. doi: 10.1177/074873099129000894. [DOI] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci U S A. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouinard SW, Wilson GF, Schlimgen AK, Ganetzky B. A potassium channel beta subunit related to the aldo-keto reductase superfamily is encoded by the Drosophila hyperkinetic locus. Proc Natl Acad Sci U S A. 1995;92:6763–6767. doi: 10.1073/pnas.92.15.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G. Reduced sleep in Drosophila Shaker mutants. Nature. 2005;434:1087–1092. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- Crocker A, Shahidullah M, Levitan IB, Sehgal A. Identification of a neural circuit that underlies the effects of octopamine on sleep:wake behavior. Neuron. 2010;65:670–681. doi: 10.1016/j.neuron.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, Bauer CS, Pratt WS, Dolphin AC. The α2δ subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A. 2010;107:1654–1659. doi: 10.1073/pnas.0908735107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LM, Purves GI, Barrett-Jolley R, Dart C. Interaction with caveolin-1 modulates vascular ATP-sensitive potassium (KATP) channel activity. J Physiol. 2010;588:3255–3266. doi: 10.1113/jphysiol.2010.194779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Feng Y, Ueda A, Wu CF. A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. J Neurogenet. 2004;18:377–402. doi: 10.1080/01677060490894522. [DOI] [PubMed] [Google Scholar]
- Gasque G, Labarca P, Darszon A. Cholesterol-depleting compounds modulate K+-currents in Drosophila Kenyon cells. FEBS Lett. 2005;579:5129–5134. doi: 10.1016/j.febslet.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Gilestro GF, Cirelli C. pySolo: a complete suite for sleep analysis in Drosophila. Bioinformatics. 2009;25:1466–1467. doi: 10.1093/bioinformatics/btp237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Shaker potassium channel gating. I. Transitions near the open state. J Gen Physiol. 1994;103:249–278. doi: 10.1085/jgp.103.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Joiner WJ, Wu MN, Yue Z, Smith CJ, Sehgal A. Identification of SLEEPLESS, a sleep-promoting factor. Science. 2008;321:372–376. doi: 10.1126/science.1155942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ueda A, Wu CF. Pre- and post-synaptic mechanisms of synaptic strength homeostasis revealed by slowpoke and shaker K+ channel mutations in Drosophila. Neuroscience. 2008;154:1283–1296. doi: 10.1016/j.neuroscience.2008.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Activity-independent homeostasis in rhythmically active neurons. Neuron. 2003;37:109–120. doi: 10.1016/s0896-6273(02)01104-2. [DOI] [PubMed] [Google Scholar]
- Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations: targeting of Kv1.5 to caveolae. J Biol Chem. 2001;276:8409–8414. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- Miwa JM, Ibanez-Tallon I, Crabtree GW, Sánchez R, Sali A, Role LW, Heintz N. Lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron. 1999;23:105–114. doi: 10.1016/s0896-6273(00)80757-6. [DOI] [PubMed] [Google Scholar]
- Monastirioti M, Linn CE, Jr, White K. Characterization of Drosophila tyramine beta-hydroxylase gene and isolation of mutant flies lacking octopamine. J Neurosci. 1996;16:3900–3911. doi: 10.1523/JNEUROSCI.16-12-03900.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E, MacKinnon R, Bezanilla F, Stefani E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358. doi: 10.1016/0896-6273(93)90190-3. [DOI] [PubMed] [Google Scholar]
- Pfeiffenberger C, Lear BC, Keegan KP, Allada R. Processing sleep data created with the Drosophila activity monitoring (DAM) system. Cold Spring Harb Protoc. 2010;2010:pdb.prot5520. doi: 10.1101/pdb.prot5520. [DOI] [PubMed] [Google Scholar]
- Rietveld A, Neutz S, Simons K, Eaton S. Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J Biol Chem. 1999;274:12049–12054. doi: 10.1074/jbc.274.17.12049. [DOI] [PubMed] [Google Scholar]
- Tao X, Lee A, Limapichat W, Dougherty DA, MacKinnon R. A gating charge transfer center in voltage sensors. Science. 2010;328:67–73. doi: 10.1126/science.1185954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26:117–126. doi: 10.1093/sleep/26.2.117. [DOI] [PubMed] [Google Scholar]
- Wang JW, Wu CF. Modulation of the frequency response of Shaker potassium channels by the quiver peptide suggesting a novel extracellular interaction mechanism. J Neurogenet. 2010;24:67–74. doi: 10.3109/01677061003746341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Humphreys JM, Phillips JP, Hilliker AJ, Wu CF. A novel leg-shaking Drosophila mutant defective in a voltage-gated K+ current and hypersensitive to reactive oxygen species. J Neurosci. 2000;20:5958–5964. doi: 10.1523/JNEUROSCI.20-16-05958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver AK, Olsen ML, McFerrin MB, Sontheimer H. BK channels are linked to inositol 1,4,5-triphosphate receptors via lipid rafts: a novel mechanism for coupling [Ca2+]i to ion channel activation. J Biol Chem. 2007;282:31558–31568. doi: 10.1074/jbc.M702866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CF, Haugland FN. Voltage clamp analysis of membrane currents in larval muscle fibers of Drosophila: alteration of potassium currents in Shaker mutants. J Neurosci. 1985;5:2626–2640. doi: 10.1523/JNEUROSCI.05-10-02626.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Koh K, Yue Z, Joiner WJ, Sehgal A. A genetic screen for sleep and circadian mutants reveals mechanisms underlying regulation of sleep in Drosophila. Sleep. 2008;31:465–472. doi: 10.1093/sleep/31.4.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Joiner WJ, Dean T, Yue Z, Smith CJ, Chen D, Hoshi T, Sehgal A, Koh K. SLEEPLESS, a Ly-6/neurotoxin family member, regulates the levels, localization and activity of Shaker. Nat Neurosci. 2010;13:69–75. doi: 10.1038/nn.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ramu Y, Lu Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 2008;451:826–829. doi: 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey PG, Rodrigueza WV, Kilsdonk EP, Stoudt GW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins: demonstration of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proc Natl Acad Sci U S A. 1989;86:7243–7247. doi: 10.1073/pnas.86.18.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating. III. Evaluation of kinetic models for activation. J Gen Physiol. 1994a;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Dittman J, Aldrich RW. Shaker potassium channel gating. II. Transitions in the activation pathway. J Gen Physiol. 1994b;103:279–319. doi: 10.1085/jgp.103.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]