

Abstract
Although the importance of the pituitary gland for growth was recognized in late 19th century, Growth hormone (GH) therapy was made available for severely GH-deficient children and adolescents only in late 1950s. Use of GH for other conditions was limited because of the limited supply of human pituitary-derived hormone. With unlimited availability of recombinant human GH (rhGH), the scenario of GH treatment has been changed enormously. Currently there is ever increasing list of indications of GH treatment in children, adolescents, and adults.
Keywords: Growth hormone, history, pituitary
INTRODUCTION
The struggle for discovery of growth hormone (GH) was protracted and hard. The fascinating circuitous road to discovery involved a host of clinicians, chemists, physiologists, and pathologists. The tale is crammed with amazing insights as well as striking errors, serendipity and unsuccessful labors, and triumphs and defeats.
HISTORY OF GROWTH HORMONE
The efforts to acquire pituitary GH for the treatment of GH-deficient children started in the mid-1940s. Initial work was on subprimates for nearly a decade. These efforts led to the purification of bovine GH by Li and Evans at the University of California, Berkeley, and Fishman at Yale. And porcine GH was purified by Raben and Westermeyer, at Tufts. But none of these preparations showed a significant biochemical or metabolic activity because the effects of GH is species specific.[1]
Growth hormone (GH) first was isolated from the human pituitary gland in 1956, by both Li and Papkoff, in California, and Raben, in Massachusetts, but its biochemical structure was not elucidated until 1972. In 1958, Raben reported the results of the first trial to show the effects of human GH on growth. By 1960 it was clear that GH-deficient children would benefit from pituitary GH. In 1960, National Pituitary Agency (NPA) was formed to further the goals of coordinating pituitary collection and extraction to support both basic and clinical research.[1,2] Between 1963 and 1985 the NPA supervised almost all of the GH treatment in the United States. And during this period about 7700 children in the United States and 27,000 children worldwide were given GH extracted from human pituitary glands to treat severe Growth hormone deficiency (GHD).[3,4]
By 1985, pituitary GH was in use for nearly 30 years in the United States and Canada, either for research purpose or therapeutically. In 1985, US Food and Drug Administration (US FDA) received reports of four young adults in the United States with the fatal, slow viral (prion-mediated) Creuzfeldt Jacob Disease (CJD), who had been treated with GH from the NPA in the 1960s. The connection was recognized on reviewing data within a few months by FDA and NIH. On April 19, 1985, distribution of pituitary GH was suspended and use of human pituitary GH rapidly ceased. And an exciting and important era in pediatric endocrinology came to an abrupt finish.[5]
Identification of the biochemical structure of GH in 1972 became the catalyst for the development of recombinant DNA-derived human GH, the gene for which was cloned for the first time in 1979. Genentech (San Francisco, California), developed in 1981 the first recombinant human GH (rhGH) by a biosynthetic process. Later, an improved process to develop rhGH was developed called protein secretion technology. Wherein, the vector plasmid is isolated from a strain of E. coli and the DNA strand to be cloned is derived from the appropriate source. Both the plasmid and the required DNA strand are cleaved by restriction enzymes, joined together and then reformed into a circular structure. The recombinant plasmid is inserted into E. coli, which is then transformed to synthesize the desired protein This is currently the most common method used to synthesize rhGH, known generically as somatotropin. Discontinuation of human cadaveric GH led to rapid US Food and Drug Administration (FDA) approval of Genentech's synthetic methionyl GH, which was introduced in the United States in 1985 for the therapy of severe childhood GHD.[6,7]
Earlier to the discovery of rhGH, the GH treatment was reserved for only the most severe cases of GHD and, because of scarce supplies. With the development of rhGH, an unlimited commercial source became available, allowing for an ever-growing list of FDA-approved indications for GH use in non–GH-deficient children and for additional indications in adults.[8,9]
HISTORY OF INDICATIONS FOR GROWTH HORMONE THERAPY IN CHILDREN
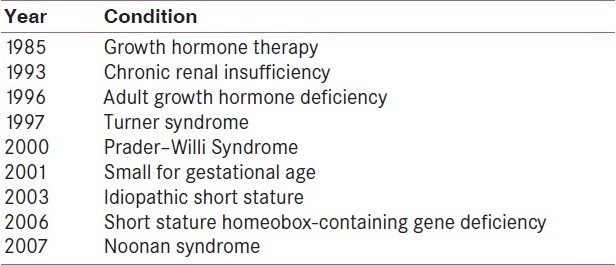
Growth hormone deficiency (1985)
GHD may result from a disruption of the GH axis in the hypothalamus or pituitary gland. This etiology of the dysfunction may be congenital or acquired in etiology. The classical presentation of severe GHD is characterized by short stature, slow growth, and delayed skeletal maturation, with reduced secretion of GH in response to provocative stimulation. In spite of this expansive list of etiologies, the cause of GHD in most children is idiopathic. GH treatment of GHD in children is, to a certain extent, standardized worldwide. rhGH is injected once daily by the subcutaneous route, usually in the evening to simulate normal physiology.[3,9]
Chronic renal insufficiency (1993)
Children suffering from chronic kidney disease (CKD) are prone to develop severe growth failure. The etiology of uremic growth failure is multifactorial. GH therapy is an accepted measure to increase final height. GH treatment in CKD has improved final height in various studies. In prepubertal CKD patients, growth response is positively associated with duration of GH therapy and initial degree of growth failure. It was negatively associated with the duration of dialysis. Doses recommended in children who have CKD are higher than in patients who have GHD. Among children treated with GH, treatment usually is discontinued after transplantation, but sometimes it is reinstituted if the growth rate remains low. Better outcomes are seen in children who begin treatment earlier, who are younger, and who have milder deterioration in renal function.[10,11]
Turner syndrome (1997)
Turner syndrome (TS) affects about one in 1500–2500 live-born females. One of the most prevalent and salient features of the syndrome is extremely short stature. Untreated women are approximately 20–21 cm shorter than normal women within their respective populations. rhGH has been used to increase growth and final height in girls who have TS. Although organic GH deficiency is not a feature of TS, the efficacy of rhGH in increasing linear growth and final height in TS patients is well documented.[12,13]
Prader–Willi syndrome (2000)
Prader–Willi syndrome (PWS) is the first human disorder attributed to genomic imprinting in which genes are expressed differentially based on the parent of origin. Children with PWS demonstrate profoundly abnormal body composition, with a similar phenotype as is seen in classic GHD. Administration of GH to GHD children not only restores linear growth but also promotes growth of lean body mass, decreases fat mass by increasing fat oxidation and total body energy expenditure, increases bone mineral density and improves cardiovascular risk factors. While GH therapy is currently approved for growth failure secondary to PWS, these other benefits alone may justify a trial of GH therapy.[14,15]
Small for gestational age (2001)
Small for gestational age (SGA) is defined as birth weight or length at least 2 SD score (SDS) below the mean gestational age. Independently of whether these children are born prematurely or at term, most SGA infants experience postnatal growth sufficient to normalize their height by 2 years of age. This is referred to as catch-up growth. Several studies have shown that most of SGA children benefit from GH therapy and attaining a normal adult height.[16,17]
Idiopathic short stature (2003)
Idiopathic short stature (ISS) is defined as a condition in which the height of an individual is more than 2 SDS below the corresponding mean height for a given age, sex, and population group without evidence of systemic, endocrine, nutritional, or chromosomal abnormalities. Specifically, children with ISS have normal birth weight and are GH sufficient. GH therapy has shown to increase the mean adult height attributable to GH therapy (average duration of 4–7 years) in children with ISS by 3.5–7.5 cm compared with historical controls, with patients’ own pretreatment predicted adult heights, or with nontreatment control or placebo control groups. There have been no unusual GH safety issues in patients who have ISS.[18,19]
Short stature homeobox-containing gene deficiency (2006)
In 2006, the US FDA approved the use of rhGH in patients with short stature homeobox-containing gene (SHOX) deficiency. Haploinsufficiency of the SHOX gene is associated with short stature, Madelung deformity, and high-arched palate and is thought to be the principal cause of short stature in girls with TS. The SHOX gene are a fairly frequent cause of short stature and that GH treatment is effective in improving the linear growth of patients with various forms of SHOX-D.[19,20]
Noonan syndrome (2007)
Noonan syndrome is a clinically heterogeneous disorder characterized by proportionate postnatal short stature, dysmorphic facial features, chest deformities, and congenital heart disease. Apart from facial features and cardiac disease, one of the cardinal signs of NS is short stature. The cause of growth impairment is unclear, and trials with rhGH administration have been performed. Several studies indicate that short-term hGH therapy (1–4 years) is able to increase growth velocity and improve height SD score (SDS).[19,21]
Adult growth hormone deficiency
GH treatment of GH-deficient adults was approved by the FDA in 1996. Subsequently, much clinical experience has been evaluated regarding the treatment of adults with GHD. Although treatment appears to be safe overall, certain areas require long-term surveillance, such as risks of glucose intolerance, pituitary/hypothalamic tumor recurrence, and cancer. Benefits of GH treatment of GH-deficient adults have been found in body composition, bone health, cardiovascular risk factors, and quality of life indicators. However, reductions in cardiovascular events and mortality have yet to be demonstrated, and treatment costs remain high [Table 1].[22]
Table 1.
History of growth hormone therapy
CONCLUSIONS
GH is an important growth-promoting factor. Until 1985, GH was obtained through extraction from cadaveric pituitary glands. With the availability of unrestricted quantities of rhGH, there is ever growing list of indications of non–GH-deficient children and adults. Apart from the conditions mentioned above, rhGH is also approved for HIV-associated wasting and in short bowel syndrome. There are now reports on the use of GH in cystic fibrosis, inflammatory bowel disease, juvenile rheumatoid arthritis, osteoporosis, and in patients who require chronic glucocorticoid administration. Treatment with rhGH does increase adult height in several conditions and seems to be a safe therapy. However, there was a recent alert by the US FDA[23] to the public on a French observational (SAGhE) study's results describing a small increased risk of death in adults who were treated with rhGH during childhood for certain types of short stature. At this time, FDA believes the benefits of rhGH continue to outweigh its potential risks. As more is learned about GH, it is unavoidable that its uses will persist to broaden.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Frasier SD. The not-so-good old days: Working with pituitary growth hormone in North America, 1956 to 1985. J Pediatr. 1997;131:S1–4. doi: 10.1016/s0022-3476(97)70001-5. [DOI] [PubMed] [Google Scholar]
- 2.Sumner JY. Human growth hormone: Current status of availability and usefulness. Pediatrics. 1969;44:766–7. [Google Scholar]
- 3.Franklin SL, Geffner ME. Growth hormone: The expansion of available products and indications. Endocrinol Metab Clin North Am. 2009;38:587–611. doi: 10.1016/j.ecl.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Escamilla RF, Hutchings JJ, Deamer WC, Li CH, Forsham PH. Long-term effects of human growth hormone (Li) in a pituitary dwarf. J Clin Endocrinol Metab. 1961;21:721–6. doi: 10.1210/jcem-21-6-721. [DOI] [PubMed] [Google Scholar]
- 5.Report of the Committee on Growth Hormone Use of the Lawson Wilkins Pediatric Endocrine Society, May 1985. Degenerative neurologic disease in patients formerly treated with human growth hormone. J Pediatr. 1985;107:10–2. [PubMed] [Google Scholar]
- 6.Cronin MJ. Pioneering recombinant growth hormone manufacturing: Pounds produced per mile of height. J Pediatr. 1997;131:S5–7. doi: 10.1016/s0022-3476(97)70002-7. [DOI] [PubMed] [Google Scholar]
- 7.Flodh H. Human growth hormone produced with recombinant DNA technology: Development and production. Acta Paediatr Scand Suppl. 1986;325:1–9. doi: 10.1111/j.1651-2227.1986.tb10356.x. [DOI] [PubMed] [Google Scholar]
- 8.Takeda A, Cooper K, Bird A, Baxter L, Frampton GK, Gospodarevskaya E, et al. Recombinant human growth hormone for the treatment of growth disorders in children: A systematic review and economic evaluation. Health Technol Assess. 2010;14:1. doi: 10.3310/hta14420. [DOI] [PubMed] [Google Scholar]
- 9.Hindmarsh PC, editor. Endocrine Development. 2nd ed. Vol. 18. Basel: Karger Publishers; 2010. Current Indications for Growth Hormone Therapy; pp. 92–108. [DOI] [PubMed] [Google Scholar]
- 10.Nissel R, Lindberg A, Mehls O, Haffner D. Pfizer International Growth Database (KIGS) International Board. Factors predicting the nearfinal height in growth hormone-treated children and adolescents with chronic kidney disease. J Clin Endocrinol Metab. 2008;93:1359–65. doi: 10.1210/jc.2007-2302. [DOI] [PubMed] [Google Scholar]
- 11.Franke D, Zivicnjak M, Ehrich JH. Growth hormone treatment of renal growth failure during infancy and early childhood. Pediatr Nephrol. 2009;24:1093–6. doi: 10.1007/s00467-009-1190-1. [DOI] [PubMed] [Google Scholar]
- 12.Baxter L, Bryant J, Cave CB, Milne R. Recombinant growth hormone for children and adolescents with turner syndrome. Cochrane Database Syst Rev. 2007:CD003887. doi: 10.1002/14651858.CD003887.pub2. [DOI] [PubMed] [Google Scholar]
- 13.Bolar K, Hoffman AR, Maneatis T, Lippe B. Long-term safety of recombinant human growth hormone in turner syndrome. J Clin Endocrinol Metab. 2008;93:344–51. doi: 10.1210/jc.2007-1723. [DOI] [PubMed] [Google Scholar]
- 14.Carrel AL, Myers SE, Whitman BY, Allen DB. Benefits of long-term GH therapy in Prader-Willi syndrome: A 4-year study. J Clin Endocrinol Metab. 2002;87:1581–5. doi: 10.1210/jcem.87.4.8414. [DOI] [PubMed] [Google Scholar]
- 15.Whitman BY, Myers S, Carrel A, Allen D. The behavioral impact of growth hormone treatment for children and adolescents with Prader-Willi syndrome: A 2-year, controlled study. Pediatrics. 2002;109:E35. doi: 10.1542/peds.109.2.e35. [DOI] [PubMed] [Google Scholar]
- 16.Maiorana A, Cianfarani S. Impact of growth hormone therapy on adult height of children born small for gestational age. Pediatrics. 2009;124:e519–31. doi: 10.1542/peds.2009-0293. [DOI] [PubMed] [Google Scholar]
- 17.Lee PA, Chernausek SD, Hokken-Koelega AC, Czernichow P. International Small for Gestational Age Advisory Board Consensus Development Conference Statement: Management of short children born small for gestational age, April 24-October 1, 2001. Pediatrics. 2003;111:1253–61. doi: 10.1542/peds.111.6.1253. [DOI] [PubMed] [Google Scholar]
- 18.Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. 2007 ISS Consensus Workshop participants.Consensus statement on the diagnosis and treatment of children with idiopathic short stature: A summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008;93:4210–7. doi: 10.1210/jc.2008-0509. [DOI] [PubMed] [Google Scholar]
- 19.Collett-Solberg PF. Update in growth hormone therapy of children. J Clin Endocrinol Metab. 2011;96:573–9. doi: 10.1210/jc.2010-1131. [DOI] [PubMed] [Google Scholar]
- 20.Blum WF, Crowe BJ, Quigley CA, Jung H, Cao D, Ross JL, et al. SHOX Study Group.Growth hormone is effective in treatment of short stature associated with short stature homeobox- containing gene deficiency: Two-year results of a randomized, controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92:219–28. doi: 10.1210/jc.2006-1409. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira LV, Souza SA, Arnhold IJ, Mendonca BB, Jorge AA. PTPN11 (Protein Tyrosine Phosphatase, Nonreceptor Type 11) mutations and response to growth hormone therapy in children with Noonan syndrome. J Clin Endocrinol Metab. 2005;90:5156–60. doi: 10.1210/jc.2004-2559. [DOI] [PubMed] [Google Scholar]
- 22.Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Shalet SM, Vance ML. Endocrine Society's Clinical Guidelines Subcommittee, Stephens PA. Evaluation and treatment of adult growth hormone deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2006;91:1621–34. doi: 10.1210/jc.2005-2227. [DOI] [PubMed] [Google Scholar]
- 23.Drug Safety Communication 12/22/2010. [Last accessed on 2011 May 1]. Available from: http://www.fda.gov .