

Abstract
BACKGROUND AND OBJECTIVE:
To date, there are no published studies from Saudi Arabia on the incidence or etiology of craniofacial anomalies. This study aimed to report the patterns of craniofacial anomalies in Saudi Arabia.
DESIGN AND SETTING:
Hospital-based, descriptive study conducted during 2002 to 2009 in the Cleft Lip/Palate and Craniofacial Anomalies Registry at King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia.
PATIENTS AND METHODS:
Data was collected on craniofacial patients in the registry.
RESULTS:
Of the 447 craniofacial patients (male, 242; female, 205), 109 (24.4%) had only cranial anomalies, 261 (58.4%) had only facial anomalies and 77 (17.2%) had both of these conditions. Craniosynostosis was seen in 33.3% of the total patients (81 males and 68 females). Of the 65 craniosynostosis syndromic patients, 25 (38.5%) had Apert syndrome and 18 (27.7%) had Crouzon syndrome. Among facial anomalies, 47 (19.4%) had dysmorphic features, followed by 35 (14.5%) with micrognathia. Among facial syndromes, 72 (59.0%) were observed to have Pierre-Robin sequence, 17 (13.9%) had Goldenhar syndrome and another 17 (13.9%) had Van der Woude syndrome. Cleft palate was more common in 171 (56.8%) patients as an associated deformity, followed by cleft lip with cleft palate in 99 (32.9%) and cleft lip in 23 (7.6%) patients. Of the 224 patients having other congenital anomalies, the cardiovascular system was most commonly affected, with 46 (20.5%) children diagnosed with congenital heart disease. A family history of anomalies was observed more in children born to parents of a consanguineous marriage than in those whose parents were unrelated (P=.01).
CONCLUSIONS:
Additional efforts should be made towards creating awareness among the general population about these deformities in relation to consanguinity.
Craniofacial anomalies (CFAs) are a highly diverse group of complex congenital anomalies. Over the years, efforts have been made to record the frequency of birth defects.1 Data on the frequency of CFAs are still lacking in many parts of the world, particularly in Africa, Asia and Eastern Europe.2 Reliable data on the prevalence of facial clefts in the Middle East is not available at present. However, a few published articles give a rough idea on the incidence of facial cleft in the region. Fida et al,3 found 1.9 orofacial malformations per 1000 live births in western Saudi Arabia. Another hospital-based series from Riyadh (Women's Specialised Hospital in King Fahad Medical City), however, reported a high prevalence (7.98 per 1000 pregnancies) of cranial anomalies.4 In the neighboring emirate, the national congenital anomalies registry of the United Arab Emirates showed that the prevalence of orofacial cleft was 0.3 per 1000 births.5 The prevalence of oral clefts in Oman is 1.5 per 1000 live births;6 and in Jordan, 2.4 facial clefts per 1000 live births.7 Craniosynostosis, premature cranial suture fusion, occurs approximately in 1 in 2500 live births.8 In addition to information on environmental exposures, genetic predisposition is important and requires research on genetic polymorphisms and gene-environment interaction. Using a combination of gene-targeting technology and traditional developmental techniques both in mice and baby chickens, significant progress has been made in the identification of numerous genes and gene pathways critical for craniofacial development. These include transcription factors, growth factors, cell-signaling molecules, folate pathway genes and detoxification enzymes, some of which are carried in the human population samples also. Studies on syndromic genes and their molecular pathways will provide a useful and informative route to gain a better understanding of human craniofacial pathology.9,10 Having an understanding of multifactorial etiology helps in implementing measures for prevention of craniofacial anomalies.
The exact number of people with craniofacial anomalies in Saudi Arabia is unknown due the lack of a system for registering birth defects and the absence of national surveys on the topic. The main purpose of this article is to report the distribution of CFA and its correlates based on data from a hospital-based registry at King Faisal Specialist Hospital and Research Center (KFSHRC), a tertiary care hospital in Riyadh. To the best of the our knowledge, there are no previous studies exclusively on craniofacial anomalies, either on their incidence or on their etiology, from Saudi Arabia. With a large sample, the results of this study may provide a groundwork for additional etiologic studies, including genetic studies.
PATIENTS AND METHODS
The Cleft Lip/Palate and Craniofacial Anomalies Registry at KFSHRC, Riyadh, began collecting information in June 1999. Though registration of all cleft lip/palate patients seen in this hospital (irrespective of date of reporting or date of admission) commenced at that time, the registration for craniofacial anomalies was confined to patients reporting on or after January 2002. This registry was approved by the Research Advisory Council, the institutional review board at KFSHRC, Riyadh, which includes the approval of the Clinical Research Committee, Bio-Ethics Committee and the Basic Research Committee. The information collected by the registry includes, but is not limited to, cleft type, syndromic status, name, age, sex, national identity number, parental consanguinity, the presence of other anomalies, and a family history of cleft, if any. The diagnoses of craniofacial anomalies were coded according to the International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9 CM). A centralized, web-based registration system was used for data entry, updates and validation. The diagnosis validation rules, data entry validation checks and warning messages that were integrated into the software restricted users from making any data-entry mistakes and confirmed accuracy of the data. Further details about the registry can be found elsewhere.11
This study includes all the patients registered during the period of 1 January 2002 to 31 December 2009. In this report, the term “craniofacial anomalies” literally encompasses all deformities of the cranium and the face. More specifically, the term has come to imply those congenital anomalies of the head that interfere with physical and mental well-being. A pediatric geneticist systematically diagnosed and evaluated all the patients to identify their syndromic condition. Frequency tables and descriptive statistics were used to analyze the condition of anomalies with respect to demographic variables and some of the possible risk factors. The statistical significance between the variables was obtained using the chi-square test, and P<.05 was considered statistically significant.
RESULTS
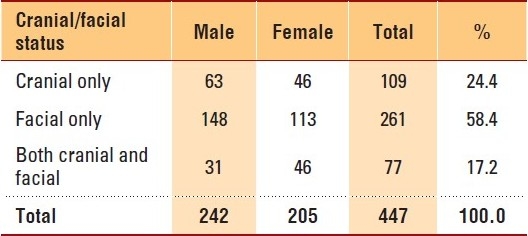
During the 8-year period (2002-2009), this registry registered 447 patients diagnosed with craniofacial anomalies, with a male-to-female ratio of 1.18:1. More facial conditions than cranial and more males than females among facial patients were observed (Table 1). There were 426 Saudis (males, 234; females, 192) and 21 non-Saudi (males, 8; females, 13) patients in our study group. Most of the patients (440; 98.4%) were from Saudi Arabia, and only 7 patients came from outside the country. Furthermore, about 30% of the patients were from Riyadh, the capital of Saudi Arabia, where the study took place. Another 16.6% and 12.0% of the patients were from the Eastern Province and the Asir province, respectively, both areas adjacent to Riyadh province.
Table 1.
Distribution of craniofacial patients by cranial/ facial status and gender: 2002-2009, KFSHRC, Riyadh.
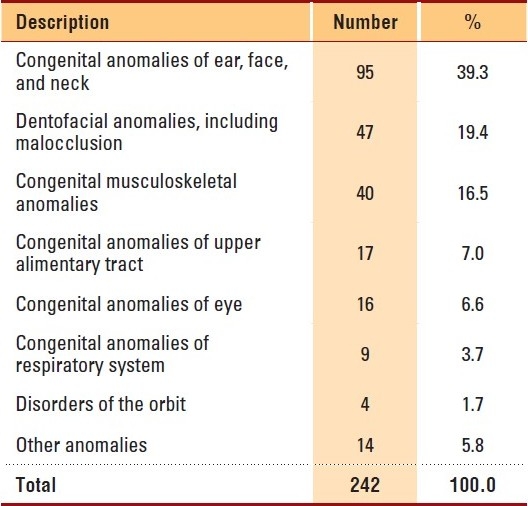
Craniosynostosis was seen in 81 males and 68 females, accounting for 33.3% of the total patients. There were more non-syndromic craniosynostosis patients than syndromic patients (Table 2), and there was no significant difference in syndromic status between genders (P=.8). Among craniosynostosis patients, Apert syndrome was seen predominantly (25, 38.5%), followed by Crouzon syndrome (18, 27.7%) and Saethre-Chotzen syndrome (8, 12.3%). Table 3 shows the broad categories of facial conditions; showing congenital anomalies of the ear, face and neck as the major facial conditions, followed by dentofacial anomalies, including malocclusion and congenital musculoskeletal anomalies. Specifically, dysmorphic features were often observed (47, 19.4%) among facial anomalies, followed by micrognathia (35, 14.5%), hypertelorism (29, 12.0%), protruded premaxilla (21, 8.7%) and low set ears (20, 8.3%). The common facial syndromes observed in our study were Pierre-Robin sequence (72, 59.0%), Goldenhar syndrome (17, 13.9%), Van der Woude syndrome (17, 13.9%) and Treacher-Collins syndrome (10, 8.2%). Almost equal numbers of patients were observed in both genders in all the syndromes except in Goldenhar syndrome (males, 13; females, 4); and orofaciodigital syndrome, which occurred in 5 female patients only.
Table 2.
Syndromic status of craniosynostosis patients by gender: 2002-2009, KFSHRC, Riyadh.
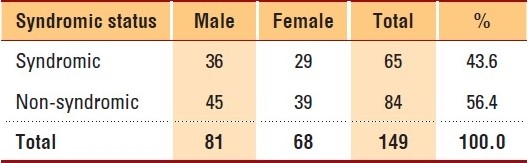
Table 3.
Facial description of the patients: 2002-2009, KFSHRC, Riyadh.
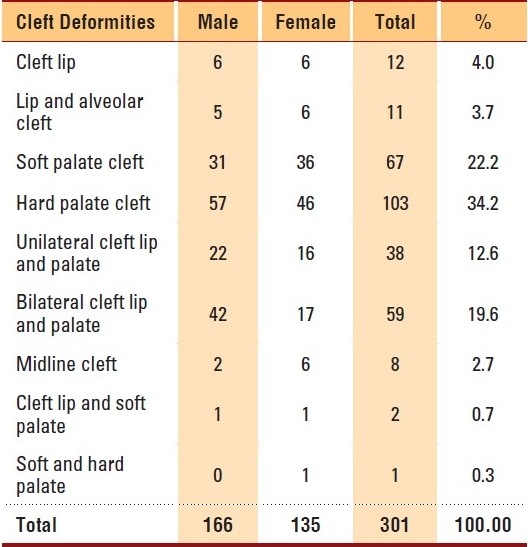
Almost two-thirds of craniofacial patients had associated deformities of cleft lip with or without cleft palate (CLP). There were 166 male and 135 female patients of CLP, with a male-to-female ratio of 1.23:1 (Table 4). Overall, cleft palate (171, 56.8%) was more common, followed by cleft lip with cleft palate (99, 32.9%), and cleft lip (23, 7.6%). Only 8 patients had midline cleft. In our study, it was noted that 50% (224) of the patients had other congenital anomalies. The most common organ system affected was the cardiovascular system, and congenital heart disease was seen in 46 (20.5%) children with associated anomalies. Syndactyly of the foot or hand was seen in 35 (15.6%) patients. Other conditions such as clubfeet and failure to thrive were present in a few patients.
Table 4.
Distribution of associated cleft deformities among craniofacial anomalies patients by gender (2002-2009, KFSHRC, Riyadh).
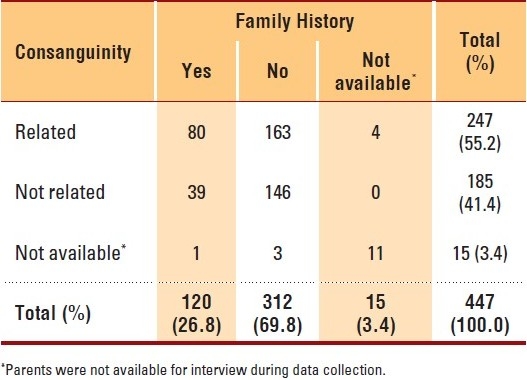
In this study, about 55% of the patients were born of parents that had consanguineous marriages, and more than one quarter of the patients had a family history of cleft deformities (Table 5). Siblings of the patients were mostly affected (74, 61.7%) compared to other relations. A family history of anomalies was observed to a significantly greater extent (P=.01) in children born to consanguineous parents in comparison with children born to non-consanguineous parents.
Table 5.
Number of craniofacial patients by their parental consanguinity and family history of craniofacial anomalies (2002-2009, KFSHRC).
DISCUSSION
From birth to maturity, children with orofacial clefts undergo multiple surgical and nonsurgical treatments, with considerable disruption to their lives, and often with adverse psychological consequences for themselves and for their families. Accurate data on the incidence are important not only for documenting the burden in relation to the planning of public health services, but also because they form the basis for research into the causes. Lack of reliable data on the magnitude of CFAs in Saudi Arabia and the scarcity of epidemiological data led to the establishment of Cleft Lip/Palate and Craniofacial Anomalies Registry at KFSHRC, Riyadh. The registry includes all patients diagnosed with CLP or craniofacial anomalies in other hospitals and referred to KFSHRC, as well as patients born at KFSHRC with CLP or craniofacial anomalies. The fact that orofacial clefts are readily diagnosed in newborns makes their registry relatively more reliable as compared to registries of some other congenital birth defects.12 In the present study, more facial than cranial anomalies were observed. There were more males than females, with both cranial or facial anomalies; however, when both conditions presented concomitantly, more females were seen.
Out of the 13 administrative regions in Saudi Arabia, the three regions of Riyadh, Eastern Province and Asir provinces comprised 58.1% of the craniofacial patients; particularly Riyadh, with 29.5%. The higher proportion of patients being from the Asir and Eastern Provinces compared to the rest of the country may be due to the fact that these areas are adjacent to Riyadh. In the absence of reliable information on the number of birth defects by region, it is difficult to attribute these differences to the prevailing birthrates across regions. These discrepancies may also be due to variations in prevailing risk factors, such as consanguinity in marriages or factors such as the unavailability of health facilities to treat these patients in those regions or differences in referral pattern across regions. However, future studies are necessary to identify and confirm this.
The evaluation of patients with multiple congenital anomalies is of critical importance, since the description of component anomalies in patients with multiple congenital anomalies may help in identifying recognizable entities and delineating new syndromes. This knowledge can be used to better understand the needs of the population (i.e., diagnosis, prognosis and counseling) and to develop suitable policies for health care.13 There is a great paucity of literature from the Middle East on any association of CFAs with other congenital malformations; and no literature at all on such associations from Saudi Arabia. Though there is a close association of CFA with CLP,14 it has not yet been established whether clefts are definitely related to specific types of anomalies. In our study, among the three cleft types (cleft lip, cleft palate, and cleft lip with cleft palate), the highest rate of associated deformities was seen in those with cleft palate only; followed by cleft lip with cleft palate; and cleft lip only. Apart from CLP, congenital heart disease was by far the most common associated malformation, present in 20.5% of the children, followed by syndactyly of the foot or hand. Ascertaining associated anomalies in a tertiary care center, particularly for those defects that are more difficult to detect at birth, is likely to be more complete and comprehensive.
Consanguineous marriages are commonly practiced in Saudi Arabia, and the prevalence remains high. In a survey of a representative sample of Saudi families identified by a multistage random sampling procedure representing both urban and rural settlements, the prevalence of consanguinity was 56%.15 Furthermore, the information on the relationship between the husband and wife showed first-cousin marriages to be more common (33.6%) than all other relationships (22.4%). The overall prevalence was significantly more common in rural (59.5%) than in urban settlements (54.7%). There are regions with a high prevalence (67.2%), such as Madina; and regions with a significantly lower prevalence (42.1%), such as Al-Baha. These results place Saudi Arabia among the countries of the world with a high rate of consanguinity. In our study, it was observed that 55.2% of the patients were born of consanguineous marriages, which is closer to what was observed by El-Mouzan et al.15 The role of consanguinity in congenital malformations has been studied by several authors from countries where the consanguinity rate is high.16–18 There are several underlying factors which may encourage consanguineous marriages, one of which is education level. Illiterate and elementary-educated individuals had significantly higher frequencies of consanguinity than better-educated couples.19 Consanguineous marriages are an important factor in the development of cleft anomalies, as well as a host of other genetic abnormalities, and should be discouraged.20 Al-Bustan et al21 evaluated epidemiological factors such as gender and consanguinity that may be associated with cleft lip or cleft palate in Kuwait and found no definite association between consanguinity and the occurrence of facial clefts, but our study showed a statistically significant association between consanguinity and occurrence of craniofacial anomalies.
When a child is born with an oral cleft, the parents are usually concerned about its cause. The parents may experience a range of negative emotional feelings (e.g., rejection, guilt, anguish) before accepting the situation and dealing with the child's problems. Oral cleft occurrence may be due to either genetic or environmental factors. If the cleft is genetic in origin and associated with a syndrome (e.g., Van der Woude syndrome, Apert syndrome), the etiology can easily be determined by offering molecular testing for these disorders, which is becoming increasingly available. In an isolated cleft, the etiology may be more difficult to determine unless there was a specific teratogen involved in the pregnancy or there existed a medical problem during the pregnancy. There is a general consensus that heredity is the most significant etiology of clefts. This study, similar to others,22 found an excess of male patients compared to female patients with craniosynostosis. Of those diagnosed with craniosynostosis, approximately 15% were syndromic;23 and of the more than 100 syndromes described within craniosynostosis, the Crouzon, Apert, Saethre-Chotzen, and Pfeiffer syndromes were the most common syndromes.23–25 In this study, two-thirds of the syndromic patients were affected either by Apert or Crouzon syndrome. We observed more syndromic patients than Jadico et al23 (43.6% vs. 15%). Also, the prevalence of Crouzon syndrome and the Apert syndrome, of all craniosynostosis observed in our series, was much higher compared to the 4.8% of Crouzon syndrome observed by Cohen et al.,24 and the 4.5% of Apert syndrome observed by Cohen and Kreiborg.25
In Saudi Arabia, primary and secondary care is mostly provided by the Ministry of Health (MOH) or other ministries (such as Ministry of Interior, Defense and National Guard) with minimal participation by the private sector, particularly in urban areas. All services provided by all the hospitals that are supported by government are free for nationals and for those working in the government sector, irrespective of their socioeconomic status or educational level. Though the clinical and epidemiologic studies of defined geographic populations can serve as a means of establishing data important for genetic counseling and as a first step in identifying strategies best suited for identification of causes,26 hospital-based studies substitute to a great extent in the absence of such population-based studies. The pattern of CFA observed in this study was confined to one tertiary care hospital, thus failing to take into consideration those clefts registered outside this hospital system. But being a tertiary care center, KFSHRC receives patients from all over Saudi Arabia, like any other MOH hospitals, and there was no difference in case definition and inclusion/exclusion criteria over the study period. KFSHRC provides interdisciplinary (cleft lip, palate and craniofacial) team services to patients with craniofacial anomalies. These results provide groundwork for additional etiologic studies, including genetic studies. In addition, future studies focusing on specific environmental and genetic factors are necessary to facilitate health-related policies that focus on CFA prevention and care. Increasing the availability and use of relevant information for programs and policies is essential if health care for newborn babies and their mothers is to be improved. The present study points to the need of establishing a national Cleft Lip/Palate and Craniofacial Anomalies Registry to ensure reliable recording of data of patients with these congenital anomalies. Additional efforts should be made to create awareness and educate the public and patients about these deformities in relation to consanguinity.
REFERENCES
- 1.WHO 2001. Global strategies to reduce the health-care burden of craniofacial anomalies: report of WHO Meetings on International Collaborative Research on Craniofacial Anomalies, Geneva, Switzerland, 5-8 November 2000; Park City, Utah, U. S. A., 24-26 May 2001. [DOI] [PubMed]
- 2.Global registry and database on craniofacial anomalies: Report of a WHO Registry Meeting on Craniofacial Anomalies WHO Registry Meeting on Craniofacial Anomalies. Bauru, Brazil: 2001. WHO 2001. [Google Scholar]
- 3.Fida NM, Al-Aama J, Nichols W, Nichols W, Alqahtani M. A prospective study of congenital malformations among live born neonates at a University Hospital in Western Saudi Arabia. Saudi Med J. 2007;28:1367–73. [PubMed] [Google Scholar]
- 4.Sallout BI, Al-Hoshan MS, Attyyaa RA, Al Suleimat AA. Antenatal diagnosis, prevalence and outcome of major congenital anomalies in Saudi Arabia: A hospital-based study. Ann Saudi Med. 2008;28:272–6. doi: 10.5144/0256-4947.2008.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Hosani H, Salah M, Abu-Zeid H, Farag HM, Saade D. The National Congenital Anomalies Register in the United Arab Emirates. East Mediterr Health J. 2005;11:690–9. [PubMed] [Google Scholar]
- 6.Rajab A, Thomas C. Oral clefts in the Sultanate of Oman. European J Plastic Surg. 2001;24:230–3. [Google Scholar]
- 7.Aqrabawi HE. Facial cleft and associated anomalies: Incidence among infants at a Jordanian medical centre. East Mediterr Health J. 2008;14:356–9. [PubMed] [Google Scholar]
- 8.Wilkie AO. Craniosynostosis: Genes and mechanisms. Hum Mol Genet. 1997;6:1647–56. doi: 10.1093/hmg/6.10.1647. [DOI] [PubMed] [Google Scholar]
- 9.Mossey P. Epidemiology underpinning research in the aetiology of orofacial clefts. Orthod Craniofac Res. 2007;10:114–20. doi: 10.1111/j.1601-6343.2007.00398.x. [DOI] [PubMed] [Google Scholar]
- 10.Stanier P, Moore GE. Genetics of cleft lip and palate: Syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13:R73–81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- 11.Al Johar A, Ravichandran K, Subhani S. Pattern of cleft lip and palate in hospital-based population in Saudi Arabia: Retrospective study. Cleft Palate Craniofac J. 2008;45:592–6. doi: 10.1597/06-246.1. [DOI] [PubMed] [Google Scholar]
- 12.Tolarová MM, Cervenka J. Classification and birth prevalence of orofacial clefts. Am J Med Genet. 1998;75:126–37. [PubMed] [Google Scholar]
- 13.Sárközi A, Wyszynski DF, Czeizel AE. Oral clefts with associated anomalies: Findings in the Hungarian Congenital Abnormality Registry. BMC Oral Health. 2005;5:4. doi: 10.1186/1472-6831-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beriaghi S, Myers SL, Jensen SA, Kaimal S, Chan CM, Schaefer GB. Cleft lip and palate: Association with other congenital malformations. J Clin Pediatr Dent. 2009;33:207–10. doi: 10.17796/jcpd.33.3.c244761467507721. [DOI] [PubMed] [Google Scholar]
- 15.El-Mouzan MI, Al-Salloum AA, Al-Herbish AS, Qurachi MM, Al-Omar AA. Regional variations in the prevalence of consanguinity in Saudi Arabia. Saudi Med J. 2007;28:1881–4. [PubMed] [Google Scholar]
- 16.Abdulrazzaq YM, Bener A, Al-Gazali LI, Al-Khayat AI, Micallef R, Gaber T. A study of possible deleterious effects of consanguinity. Clin Genet. 1997;51:167–73. doi: 10.1111/j.1399-0004.1997.tb02447.x. [DOI] [PubMed] [Google Scholar]
- 17.Jaber L, Halpern GJ, Shohat M. The impact of consanguinity worldwide. Community Genet. 1998;1:12–7. doi: 10.1159/000016130. [DOI] [PubMed] [Google Scholar]
- 18.Sawardekar KP. Profile of major congenital malformations at Nizwa Hospital, Oman: 10-year review. J Paediatr Child Health. 2005;41:323–30. doi: 10.1111/j.1440-1754.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 19.Al Husain M, Al Bunyan M. Consanguineous marriages in a Saudi population and the effect of inbreeding on prenatal and postnatal mortality. Ann Trop Paediatr. 1997;17:155–60. doi: 10.1080/02724936.1997.11747879. [DOI] [PubMed] [Google Scholar]
- 20.Elahi MM, Jackson IT, Elahi O, Khan AH, Mubarak F, Tariq GB, et al. Epidemiology of cleft lip and cleft palate in Pakistan. Plast Reconstr Surg. 2004;113:1548–55. doi: 10.1097/01.prs.0000117184.77459.2b. [DOI] [PubMed] [Google Scholar]
- 21.Al-Bustan SA, El-Zawahri MM, Al-Adsani AM, Bang RL, Ghunaim I, Maher BS, et al. Epidemiological and genetic study of 121 cases of oral clefts in Kuwait. Orthod Craniofac Res. 2002;5:154–60. doi: 10.1034/j.1600-0544.2002.02203.x. [DOI] [PubMed] [Google Scholar]
- 22.Singer S, Bower C, Southall P, Goldblatt J. Craniosynostosis in Western Australia, 1980-1994: A population-based study. Am J Med Genet. 1999;83:382–7. doi: 10.1002/(sici)1096-8628(19990423)83:5<382::aid-ajmg8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 23.Jadico SK, Young DA, Huebner A, Edmond JC, Pollock AN, McDonald-McGinn DM, et al. Ocular abnormalities in Apert syndrome: Genotype/phenotype correlations with fibroblast growth factor receptor type 2 mutations. J AAPOS. 2006;10:521–7. doi: 10.1016/j.jaapos.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 24.Cohen MM, Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erickson JD, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992;42:655–9. doi: 10.1002/ajmg.1320420505. [DOI] [PubMed] [Google Scholar]
- 25.Cohen MM, Jr, Kreiborg S. Birth prevalence studies of the Crouzon syndrome: Comparison of direct and indirect methods. Clin Genet. 1992;41:12–5. doi: 10.1111/j.1399-0004.1992.tb03620.x. [DOI] [PubMed] [Google Scholar]
- 26.Murray JC, Daack-Hirsch S, Buetow KH, Munger R, Espina L, Paglinawan N, et al. Clinical and epidemiologic studies of cleft lip and palate in the Philippines. Cleft Palate Craniofac J. 1997;34:7–10. doi: 10.1597/1545-1569_1997_034_0007_caesoc_2.3.co_2. [DOI] [PubMed] [Google Scholar]