

Abstract
Brain-derived neurotrophic factor (BDNF) and its cognate receptor, TrkB, regulate a wide range of cellular processes, including dendritic spine formation and functional synapse plasticity. However, the signaling mechanisms that link BDNF-activated TrkB to F-actin remodeling enzymes and dendritic spine morphological plasticity remain poorly understood. We report here that BDNF/TrkB signaling in neurons activates the Vav family of Rac/RhoA guanine nucleotide exchange factors through a novel TrkB-dependent mechanism. We find that Vav is required for BDNF-stimulated Rac-GTP production in cortical and hippocampal neurons. Vav is partially enriched at excitatory synapses in the postnatal hippocampus but does not appear to be required for normal dendritic spine density. Rather, we observe significant reductions in both BDNF-induced, rapid, dendritic spine head growth and in CA3-CA1 theta burst-stimulated long-term potentiation in Vav-deficient mouse hippocampal slices, suggesting that Vav-dependent regulation of dendritic spine morphological plasticity facilitates normal functional synapse plasticity.
Introduction
A vast majority of excitatory synapses in the mature brain are formed onto dendritic spines (Harris and Kater, 1994). The structure of a dendritic spine is highly dynamic, exhibiting morphological plasticity under both basal and activity-induced conditions (Dillon and Goda, 2005). Increasing evidence suggests that dendritic spine structural dynamics are coupled with functional synapse plasticity, particularly for durable plasticity events such as long-term potentiation (LTP), which is correlated with dendritic spine head enlargement, and long-term depression, a phenomenon accompanied by a reduction in spine head size (Yuste and Bonhoeffer, 2001; Dillon and Goda, 2005; Cingolani and Goda, 2008). Brain-derived neurotrophic factor (BDNF) and its high-affinity receptor, TrkB, are well established positive modulators of LTP. BDNF- or TrkB-deficient mice exhibit impaired hippocampal LTP, and the addition of exogenous BDNF enhances tetanus and theta burst stimulation (TBS)-induced hippocampal LTP in a Trk-dependent manner (Shen and Cowan, 2010). Recent work in hippocampal slices suggests that BDNF/TrkB facilitates TBS-LTP, at least in part, through regulation of F-actin remodeling and dendritic spine structural dynamics (Rex et al., 2007). Furthermore, pairing local glutamate uncaging with postsynaptic spikes induces long-lasting dendritic spine enlargement through a mechanism that requires BDNF/TrkB kinase-dependent signaling (Tanaka et al., 2008). While several studies describe a requirement for new protein synthesis in BDNF-induced, long-lasting, functional and structural synapse plasticity (Bramham, 2008), an additional likely mechanism by which BDNF/TrkB regulates plasticity is through activation of Rho-family GTPases. These are critical regulators of the actin cytoskeleton, and have documented roles in dendritic spine formation, motility, and morphology (Luo, 2002; Tashiro and Yuste, 2004). Activation of Rac and Cdc42 GTPases promotes spine formation and enlargement, while activation of RhoA promotes dendritic spine instability (Nakayama et al., 2000; Tashiro et al., 2000). Guanine-nucleotide exchange factors (GEFs) render GTPases active by accelerating the exchange of GDP for GTP (Cerione and Zheng, 1996). However, the molecular links between BDNF/TrkB receptors and activation of Rho-family GTPases, and a potential role for this pathway in synapse plasticity, are poorly understood.
The Vav-family GEFs comprise 3 distinct genes (Vav1–3) in vertebrates (Bustelo, 2001). While Vav1 expression occurs nearly exclusively in hematopoietic cells, Vav2 and Vav3 exhibit a more ubiquitous pattern of expression, and are highly expressed in the brain during embryonic and early postnatal development (Turner and Billadeau, 2002; Cowan et al., 2005). Vav GEFs are activated upon phosphorylation of conserved acidic domain tyrosines (Y142, Y159, and Y172) that disrupt an autoinhibitory interaction between the acidic domain and the GEF catalytic DH (Dibble homology) domain (Aghazadeh et al., 2000).
We report here a novel functional interaction between BDNF/TrkB signaling and Vav GEFs. Vav is partially enriched in developing hippocampal synapses and is regulated by BDNF and TrkB kinase activity. We show that Vav is required for BDNF-induced Rac-GTP induction and rapid dendritic spine growth of hippocampal CA1 pyramidal neurons. Finally, we show that hippocampal CA1 LTP is impaired in Vav-deficient neurons, suggesting a functional link among actin-remodeling enzymes, dendritic spine growth, and activity-dependent functional synapse plasticity in the hippocampus.
Materials and Methods
DNA constructs.
Full-length and point mutant forms of Vav2 were generated as described previously (Cowan et al., 2005). Deletion constructs were generated using PCR-based cloning strategies. All Vav2 deletion plasmids contain a T7 epitope tag at the C terminus. Detailed plasmid maps are available upon request. Full-length and mutant rat TrkB receptors are subcloned into pcDNA3 vectors and contain an N-terminal Flag epitope tag that begins after the signal sequence cleavage site. The Flag-TrkB (rat) Y490F, Y670F, Y674/675F, Y670/674/675F, Y785F, and K571N (amino acid positions refer to homologous positions on TrkA) were generated using modified QuikChange mutagenesis (Stratagene).
Protein reagents.
The anti-Vav2 total antibody (1:1000 dilution) was described previously (Cowan et al., 2005). Anti-P-Y172 Vav2 (1:1000; 1 μg/ml) was generated in rabbits using KLH coupling using the following synthesized phosphorylated peptide (C-AEGDEIYEDLMRL) and was affinity purified by standard procedures using a Pierce Sulfo-link resin and 100 mm glycine, pH 2.3, elution. The following are commercial antibodies: anti-phosphotyrosine (clone 4G10, pan anti-phosphotyrosine antibody), 1:1000 (Millipore); anti-Rac1 (clone 23A8), 1:1000 (Millipore); anti-TrkB, 1:1000 (BD Biosciences PharMingen); anti-P-TrkA (Y490), 1:1000 (Cell Signaling Technology); anti-Flag (clone M2), 1:2000 (Millipore); anti-T7, 1:10,000 (Novagen); anti-MAPK, 1:1000 (Cell Signaling Technology); anti-P-MAPK (T202/Y204), 1:1000 (Cell Signaling Technology); anti-P-Src (Y418), 1:1000 (Invitrogen); and anti-Src, 1:1000 (Cell Signaling Technology). BDNF (Peprotech) was dissolved in 0.1% (w/v) BSA in 10 mm Tris-HCl, pH 7.4.
Dissociated cortical cultures.
Embryonic cortical neurons were cultured from embryonic day 16.5 (E16.5) mice or E18 rats. The cortices were dissected and treated with 100 U of papain (Worthington) for 3 min. The digestion was terminated by the addition of trypsin inhibitor (Sigma). The tissue was washed for a total of 3× with trypsin inhibitor, followed by 3 washes with plating medium consisting of DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 1% l-glutamine (Sigma), and 1% penicillin-streptomycin (Sigma). Neurons were mechanically dissociated with a pipette and plated on polyornithine (Sigma)-coated 10 cm dishes at a density of 8 million cells per dish. The culture medium was replaced with Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen), l-glutamine (Sigma), and 1% penicillin-streptomycin (Sigma) 24 h after plating.
Organotypic slice cultures.
Hippocampal organotypic slices were cultured from P6 mice of either sex following the protocol of (Stoppini et al., 1991). The mice were rapidly decapitated, and the brains were extracted and placed in chilled dissection medium (1 mm CaCl2, 5 mm MgCl2, 10 mm glucose, 4 mm KCl, 26 mm NaHCO3, and 40 mm sucrose) oxygenated with 95% O2/5% CO2. Hippocampi were dissected and sectioned coronally into 400-μm-thick sections using a McIlwain tissue chopper. Slices were transferred onto porous Millicell membranes in a six well plate with 750 μl of medium (MEM, Invitrogen) with 25% horse serum, 13 mm glucose, 5 mm NaHCO3, 30 mm HEPES, 1 mm CaCl2, 2 mm MgSO4, 1 mm l-glutamine, 0.00125% ascorbic acid, 1 μg/ml insulin, pH 7.28. Slices were maintained at this liquid–air interface at 37°C with 5% CO2.
Western blotting.
Samples were run on SDS-PAGE gels and transferred to PVDF membrane (GE Healthcare). The membranes were blocked in 10% milk for 1 h and probed with 1° antibody for 2 h at room temperature or overnight at 4°C. The membranes were then incubated with 2° antibody (1:10,000 G ∝ R or G ∝ M, Jackson ImmunoResearch Laboratories) for 1 h at room temperature and developed with a homemade enzymatic chemiluminescence (ECL) solution or ECL Plus Western Blotting Detection System (GE Healthcare).
GST-PBD assay.
Dissociated cortical neurons (plated at a density of 8 million cells per dish in a 10 cm dish) or hippocampal organotypic slices, prepared as described above, were treated with BDNF (0.1 μg/ml and 0.25 μg/ml, respectively; Peprotech) over a time course at 6 and 9 d in culture, respectively. The neurons were lysed in a buffer consisting of 50 mm Tris, pH 7.2, 1% Triton X-100, 250 mm NaCl, and 10 mm MgCl2. Each time point was incubated with 10 μg of GST-PBD (PAK1 p21-binding domain) and 20 μl of glutathione beads (50% slurry; GE Healthcare) for 1 h at 4°C. The beads were washed 3× with a wash buffer consisting of 50 mm Tris, pH 7.2, 1% Triton X-100, 150 mm NaCl, and 5 mm MgCl2. Samples were loaded onto SDS-PAGE gels for Western blotting.
RNA isolation and reverse transcription.
Whole hippocampi were dissected from mice of either sex at various ages and processed immediately or snap frozen on ethanol/dry ice. The samples were homogenized in TRIzol (Invitrogen) using a tissue homogenizer, and the RNA was precipitated with chloroform (Sigma). The remaining steps were performed using the RNeasy Micro kit (Qiagen). The RNA concentration of each sample was determined using a NanoDrop spectrophotometer and was reverse transcribed using the Superscript III First-Strand Synthesis System for RT-PCR (Invitrogen).
Quantitative real-time PCR.
All primers were designed to amplify a 100–150 bp product. The primers used to amplify Vav2 RNA were 5′-GCTCTGAAGTCCACCTCTGG-3′ (forward) and 5′-TCCTTGGTGCTCTGAATGTG-3′ (reverse). The primers used to amplify Vav3 RNA were 5′-AAAAGAGGCTCATGCTCAGG-3′ (forward) and 5′-CGAAATCACGAAAGCTGTGA-3′ (reverse). The primers used to amplify cyclophilin were 5′-CATCTATGGTGAGCGCTTCCC-3′ (forward) and 5′-GCCTGTGGAATGTGAGGGGTG-3′ (reverse). The reactions were performed using the SYBR Green PCR Master Mix (Ambion) and the ABI 7500 real-time PCR thermal cycler. Vav2 and Vav3 expression in the hippocampus were determined by running reactions of 50 ng of cDNA. Fold changes relative to cyclophilin were determined using the ΔΔCt method, in which the mean fold change (2−ΔΔCtAVE) and SEM (abs(((2−ΔΔCtAVE × 2−ΔΔCtSEM) − (2−ΔΔCtAVE/2−ΔΔCtSEM))/2)) were determined.
Synaptosome preparation.
Whole hippocampi were dissected from P15 mice of either sex and homogenized in buffer consisting of 7 mm Tris-HCl, pH 7.5, 0.36 m sucrose, 0.5 mm EGTA, and 0.25 mm DTT using a Wheaton glass mortar and Teflon pestle homogenizer. The samples were spun at 3500 rpm for 2 min at 4°C to pellet nuclei. The supernatant was collected and spun at 23,000 × g for 6 min at 4°C, resulting in the formation of a membrane fraction-containing pellet. The pellet was resuspended in homogenization buffer and layered onto a 5%/13% discontinuous Ficoll gradient. The sample was spun at 45,000 × g for 45 min at 4°C. Synaptosomes were collected from the 5 and 13% Ficoll interface, washed in ice-cold PBS, spun at 23,000 × g for 20 min at 4°C, and resuspended in ice-cold PBS. The postsynaptic density (PSD) was collected by homogenizing an aliquot of the synaptosome fraction in homogenization buffer with 0.5% Triton X-100. The sample was vortexed repeatedly during a 15 min incubation on ice, then spun at 33,000 × g for 20 min at 4°C. The pellet that formed following the centrifugation was PSD.
Slice culture transfection and live-cell imaging.
After 6 DIV, slices were transfected with GFP using the biolistic gene transfer method (Bio-Rad). All experiments, acquisition of images, and analyses were performed blind to genotype. GFP-positive pyramidal neurons were selected for time-lapse imaging after 72 h of gene expression. Time-lapse images were collected from the secondary and tertiary basal dendrites of CA1 hippocampal neurons on a Zeiss LSM 510 inverted microscope using an Achroplan 40 × 0.8 NA water-immersion objective. Hippocampal slices were maintained in the liquid–air interface environment for the duration of the imaging session with 5% CO2 at 34°C. GFP was visualized at 900 nm with a Chameleon Ti:Sapphire 2 photon laser (Coherent). Images were collected in stacks at an interval of 0.5 μm with a resolution of 0.09 pixels/μm. Image stacks were collapsed into maximum projections and analyzed. Dendritic spine head fluorescence intensity was normalized to an adjacent area of the dendrite. Spine head size was defined as the area of thresholded pixels and measured with ImageJ software. BDNF (250 ng/ml) was directly applied as a 1 μl drop to the top of the slice. Each sample represents an average of five randomly chosen, isolated dendritic spines from one dendritic length. One dendritic length per slice was imaged and averaged to give an average spine behavior per dendrite.
Intracellular recordings.
Hippocampal organotypic cultures were prepared from wild-type (WT) and Vav2−/−3−/− mice of either sex at P6, as described previously. At 9 DIV, slices were transferred to a recording chamber and perfused with artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 2.6 NaH2PO4, 1.3 MgCl2, and 2.5 CaCl2, saturated with 95%O2/5%CO2. Picrotoxin (100 μm, Sigma-Aldrich) and tetrodotoxin (0.2 μm, Ascent Scientific) were added to block GABAA receptors and Na channels, respectively. Neurons were voltage-clamped at −60 mV through whole-cell recording pipettes (4–6 MΩ) filled with internal solution containing the following (in mm): 130 K-gluconate, 10 KCl, 10 HEPES, 3 MgCl2, 2 Mg-ATP, and 1 Na-GTP, pH 7.30, 280 ± 4 mOsm. CA1 pyramidal neurons were visualized using a Zeiss Axoskop 2A equipped with infrared differential interference contrast optics and a contrast gradient light source. Whole-cell voltage-clamp recordings were obtained using a Multiclamp 700A amplifier (Molecular Devices). For all recordings, the first 10 min were discarded to allow the internal solution time to dialyze the neuron and reach equilibrium. The following 15 min were considered, and cells were discarded if <100 events were recorded. mEPSCs were detected and analyzed using an automatic detection program (MiniAnalysis; Synaptosoft Inc). Seventy-five events were randomly selected per recording, and the average amplitude and interevent interval (IEI) were calculated. Following mEPSC collection, Schaffer collaterals were stimulated using custom-made stimulation electrodes, with constant current discovered by AMPI stimulus isolators. The stimulus intensity was adjusted to evoke a monosynaptic EPSC, and the stimulus was delivered twice at 100 ms (10 Hz) intervals. This was repeated at least seven times every 30 s. Off-line analysis was performed using scientific analysis software (Igor Pro, Wavemetrics). Significant differences between wild-type and Vav2−/−3−/− slices were determined using a t test.
Slice preparation and TBS-LTP electrophysiology.
All studies used postnatal day 15 (P15) mice of either sex. For slice preparation, animals were rapidly decapitated, and the brains were removed and placed in ice-cold, oxygenated aCSF containing the following: 25 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 1 mm MgCl2 · 6H2O, 2 mm CaCl2 · 2H2O, 25 mm NaHCO3, and 25 mm dextrose. A Vibratome 1500 sectioning system was used to prepare transverse hippocampal slices of 300 μm thickness, and the slices were immediately transferred to a holding chamber containing room-temperature aCSF. Slices were allowed to recover for at least 1.5 h before recording. Slices were transferred to a recording chamber perfused with aCSF maintained at 30°C. fEPSPs were recorded from CA1 in response to the stimulation of Schaffer collaterals. Pulses were delivered through the stimulation electrode at 0.05 Hz. After establishment of a 10 min stable baseline, two theta bursts were delivered, with each burst consisting of four pulses at 100 Hz and separated by 200 ms.
HEK293 cell transfections and immunoprecipitations.
HEK293T cells were transfected using a calcium phosphate method. After 24 h, cells were lysed in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.5% DOC, and 0.1% SDS). Flag-tagged TrkB protein was precipitated using 20 μl of anti-FLAG agarose (50% slurry; Sigma).
Results
BDNF stimulates the transient activation of Vav in neurons
As Vav GEFs are activated by several receptor tyrosine kinases (Pandey et al., 2000; Cowan et al., 2005; Hunter et al., 2006; Garrett et al., 2007), we speculated that BDNF/TrkB signaling might regulate Vav. To test this possibility, we stimulated cultured primary neurons (E18 + 6 DIV, rat cortical) with recombinant BDNF (100 ng/ml) over a 1 h time course. Endogenous Vav2 was immunoprecipitated with specific antibodies (Fig. 1A), and Western blotting was performed to detect Vav2 tyrosine phosphorylation. We found that BDNF induced a rapid and transient tyrosine phosphorylation of Vav2, with phosphorylation peaking at ∼2 min and remaining elevated above prestimulation levels through at least 30 min (Fig. 1B). Consistent with these observations, TrkB receptors also activate Vav GEFs in HEK293T cells (Fig. 1C). Under these conditions, transient expression of wild-type TrkB displays high basal activity without the addition of exogenous BDNF, as revealed by Western blotting with the P-Y490 Trk receptor-specific antibody. Compared withVav2 or Vav3 expressed alone, coexpression with wild-type TrkB dramatically increased tyrosine phosphorylation of Vav2 and Vav3 using either a general phosphotyrosine antibody (4G10) or using a site-specific antibody that is specific for Vav2 P-Y172, which is a key GEF activity-regulating residue in the Vav acidic domain (Crespo et al., 1997; Han et al., 1997; Schuebel et al., 1998). In addition, the coexpression of kinase-active TrkB receptor with either Vav2 or Vav3 resulted in altered cell morphology of transfected HEK293T cells compared with cells expressing TrkB or Vav2/3 alone (data not shown), suggesting a functional interaction between TrkB and Vav in living cells.
Figure 1.

BDNF stimulates the transient activation of Vav2 in neurons. A, Vav2 antibodies are highly specific. Western blot of whole hippocampi from P15 WT or Vav2−/−3−/− (DKO) mice with anti-Vav2 antibody (Cowan et al., 2005). B, E18 rat cortical neurons were cultured for 6 d and then stimulated with BDNF (100 ng/ml) over a time course. Vav2 was immunoprecipitated and blotted with antiphosphotyrosine antibody (4G10). Total cell lysates (TCLs) were blotted with anti-phospho-TrkB (Y490) and anti-TrkB antibodies. The results are a representative finding of five independent experiments. C, Coexpression of Vav2 or Vav3 and TrkB increases phospho-Vav in HEK293T cells. HEK293T cells were transfected with T7-tagged Vav2 or HA-tagged Vav3 and Flag-tagged TrkB. To examine Vav2 Y172 phosphorylation, whole-cell lysates were blotted with anti-phospho-Vav2 (Y172) antibody. To examine Vav3 phosphorylation, total cell lysates were immunoprecipitated with anti-HA antibody, then immunoblotted with antiphosphotyrosine (4G10) and anti-HA (Vav) antibodies. Total cell lysates were blotted with anti-pTrkB (Y490) antibody. The results are a representative finding of three independent experiments. D, BDNF stimulates activation of Vav2 independently of Src kinase activity. E18 cortical neurons were cultured for 6 d and then incubated with DMSO, PP2 (5 μm), or PP3 (5 μm) for 30 min before stimulation with BDNF (100 ng/ml) over a time course. Vav2 was immunoprecipitated and blotted with antiphosphotyrosine antibody (4G10). Total cell lysates were blotted with anti-phospho-TrkB (Y490), anti-TrkB, anti-phospho-ERK1/2 (T202/Y204), anti-ERK1/2, anti-phospho-Src (Y418), and anti-Src antibodies. The results are a representative finding of two independent experiments.
In the immune system, Src family kinases phosphorylate Vav1 at acidic domain tyrosines in response to T-cell receptor activation (Bustelo, 2000). However, incubating cortical neurons with the Src inhibitor PP2 did not block BDNF-induced endogenous Vav2 tyrosine phosphorylation (Fig. 1D), indicating a Src kinase-independent mechanism. Together, these experiments indicate that BDNF/TrkB activation increases tyrosine phosphorylation and activation of Vav2 and Vav3 GEFs, which suggests a potential role for this novel BDNF/TrkB signaling pathway in neurons.
BDNF stimulates Rac-GTP formation through a Vav-dependent mechanism
Since Vav GEFs are activated by BDNF/TrkB signaling (Fig. 1) and are strong activators of Rac (Abe et al., 2000; Heo et al., 2005), we speculated that Vav GEFs might contribute to the BDNF-induced activation of Rac-GTP in neurons. To test this idea, we treated WT or Vav2−/−3−/− hippocampal slice cultures (P6 + 9 DIV), dissociated cortical neurons (E16.5 + 6 DIV), or acute hippocampal slices (adult) with recombinant BDNF for various periods of time and measured the production of Rac-GTP (Fig. 2A,C,D). In wild-type hippocampal slices (Fig. 2A) or dissociated neuron cultures (Fig. 2C), we observed that exogenous BDNF addition induced a rapid and transient increase in Rac-GTP levels, with peak levels occurring ∼2–5 min after stimulation (Fig. 2). The kinetics of Rac-GTP activation were very similar to the kinetics of BDNF-induced Vav2 tyrosine phosphorylation (compare Figs. 1B, 2C). In contrast, BDNF stimulation of Vav-deficient neurons produced a dramatically reduced induction of Rac-GTP (Fig. 2A,C,D), indicating that Vav GEFs are required for normal activation of Rac in these neuronal populations. We observed normal induction of P-ERK1/2 in the Vav-deficient neurons (Fig. 2B), indicating that Vav GEFs are not required for all BDNF/TrkB forward-signaling events. While we observed a significant reduction in BDNF-induced Rac activation in Vav-deficient cortical cultures or hippocampal slice cultures, the BDNF-induction of Rac-GTP is not completely abolished. Two recent studies indicated that the Rac GEF Tiam1 can be activated by BDNF/TrkB signaling and contribute to Rac-GTP levels in Cos-7 cells or in cerebellar granule neurons (Miyamoto et al., 2006; Zhou et al., 2007); therefore, the residual BDNF-induced Rac activation is likely accounted for by Tiam1 or a similar Rac GEF.
Figure 2.
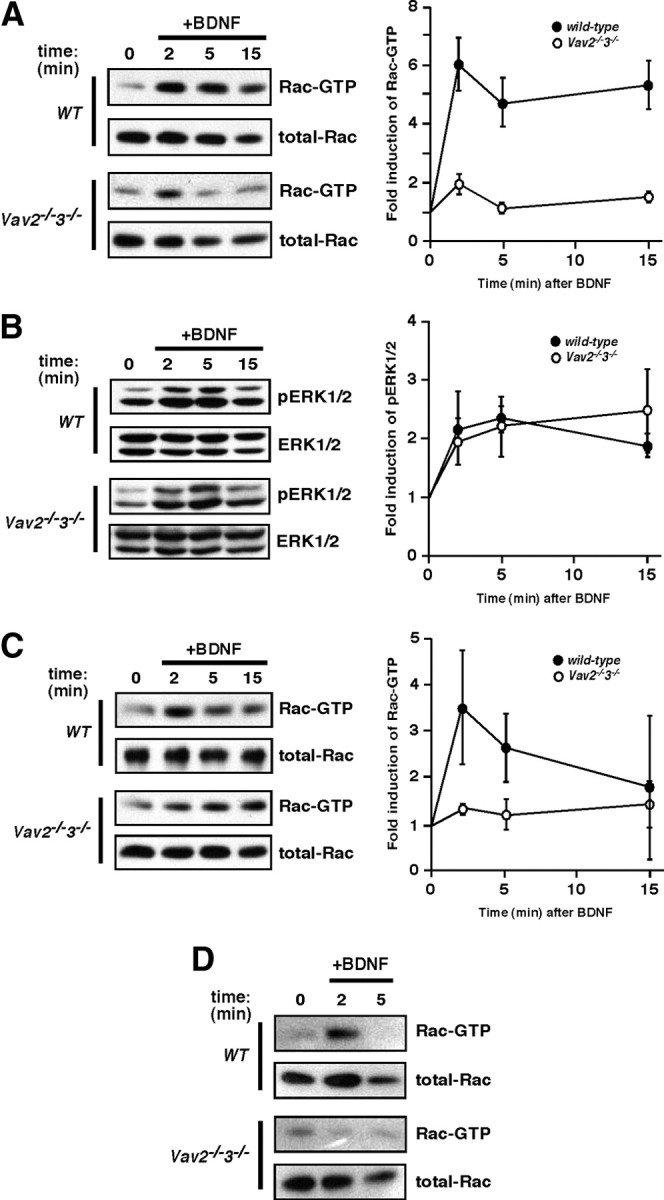
BDNF stimulates Rac-GTP formation through a Vav-dependent mechanism. A, Hippocampal organotypic slices (400 μm) were cultured from wild-type or Vav2−/−3−/− mice at P6. At 9 DIV, slices were stimulated with BDNF (250 ng/ml) over a time course. Rac1-GTP was precipitated using GST-PBD fusion protein and glutathione Sepharose. Total cell lysates were blotted with anti-Rac1 antibody. Rac1-GTP was normalized to total Rac1 levels and expressed as a fold change compared with the 0 min time point (p < 0.001, genotype by two-way ANOVA). Wild-type and Vav2−/−3−/− data are from four and five independent experiments, respectively. B, Erk1/2 pathway is activated normally in Vav2−/−3−/− hippocampi (as in A) in response to BDNF stimulation. Total cell lysates were blotted with anti-phospho-ERK1/2 (T202/Y204) and anti-ERK1/2 antibodies. Wild-type and Vav2−/−3−/− data are from three and four independent experiments, respectively. C, Rac1-GTP stimulation by BDNF was performed as in A, except that cultured primary cortical neurons (E16.5 + 6DIV) from either wild-type or Vav2−/−3−/− mice were used and treatment was administered with 100 ng/ml BDNF for the indicated times. Wild-type and Vav2−/−3−/− data are from four independent experiments. D, Acute hippocampal slices (300 μm) from adult (3 month) wild-type or Vav2−/−3−/− mice were stimulated with BDNF (100 ng/ml) over a time course. Rac1-GTP was precipitated using GST-PBD fusion protein and glutathione Sepharose. Wild-type and Vav2−/−3−/− results are representative findings from three independent experiments.
Vav GEFs in the hippocampus colocalize with excitatory synaptic proteins
Since BDNF/TrkB signaling activates Rac-GTP in large part through a Vav-dependent mechanism, we speculated that Vav GEFs might contribute to morphological or functional synaptic changes induced by BDNF/TrkB signaling in hippocampal neurons. Toward this end, we first analyzed expression levels of Vav2 and Vav3 during the postnatal time frame in which robust synaptogenesis, dendritic spinogenesis and synaptic remodeling are occurring in vivo (P4–P21) and in adult hippocampus. Using reverse transcription and qRT-PCR, we found that Vav2 mRNA levels are high during the early postnatal phase (P4–P7) and then slightly decline during the next 2 weeks (P14–P21) before stabilizing at their adult hippocampal levels, which are ∼50% of peak levels (Fig. 3A). In comparison, hippocampal Vav3 mRNA levels do not change from P4 to adult (Fig. 3A). By relative comparison, Vav2 is expressed at a level that is approximately twofold to fourfold higher than Vav3 mRNA in the hippocampus, depending on the postnatal age (Fig. 3B). Together, these data reveal that Vav2 and Vav3 are both highly expressed in the developing hippocampus.
Figure 3.
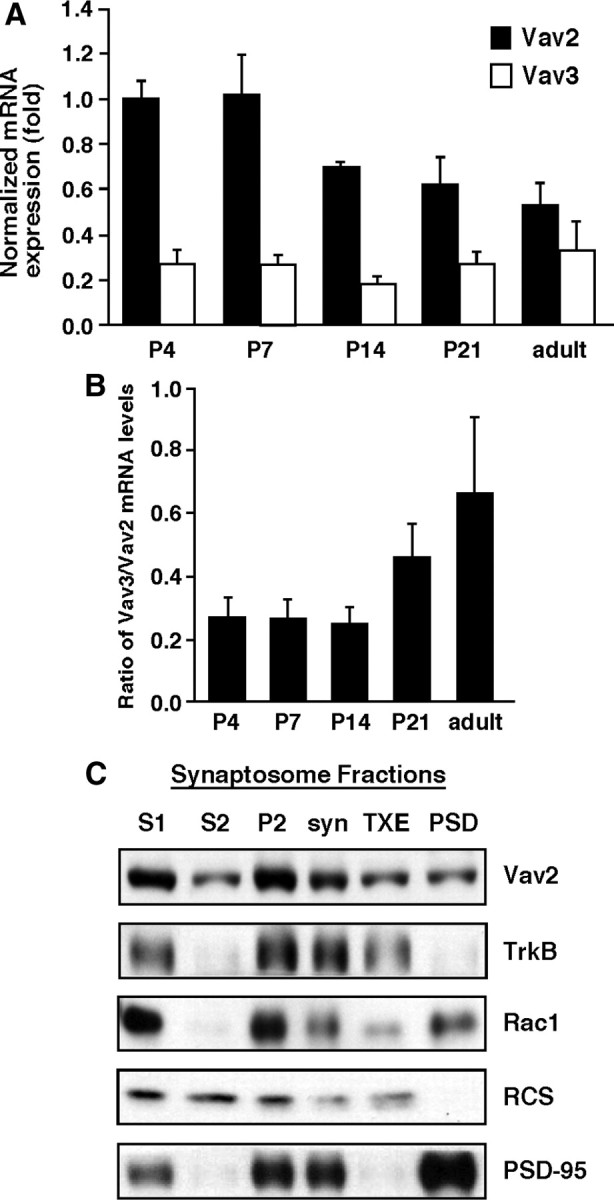
Vav GEFs colocalize with excitatory synaptic proteins in the hippocampus. A, Relative hippocampal expression of Vav2 and Vav3 mRNA at indicated time points was determined using real-time qPCR. Results were normalized to cyclophilin mRNA levels. For A and B, data are from three independent experiments, each of which was conducted in triplicate. B, Ratio of Vav3/Vav2 mRNA expression at indicated time points. C, Synaptosomes were prepared from whole hippocampi of P15 mice. Vav2 localized to the membrane (P2) and synaptosome fractions. S1, Total hippocampal homogenate with nuclei removed; S2, cytosol; P2, crude membrane fraction; syn, synaptosomes collected following Ficoll gradient fractionation; TXE, 1% Triton X-100-soluble fraction of syn; PSD, pellet after 0.5% Triton X-100 extraction. Synaptosome fractions were blotted with anti-Vav2, anti-TrkB, anti-Rac, anti-RCS (regulator of calmodulin signaling) (Pulipparacharuvil et al., 2008), and anti-PSD-95 antibodies. The results are a representative finding of two independent experiments.
BDNF/TrkB signaling is an important modulator of synapse formation, and morphological (dendritic spine) and functional synapse plasticity in the hippocampus (Korte et al., 1995; Figurov et al., 1996; Patterson et al., 1996; Minichiello et al., 1999; Pozzo-Miller et al., 1999; Kossel et al., 2001; Luikart et al., 2005, 2008; Rex et al., 2007; Tanaka et al., 2008). To test whether Vav GEFs are found near excitatory synapses, and therefore in a position to mediate BDNF-dependent synaptic effects, we used a biochemical fractionation procedure to isolate membranous fractions enriched for glutamatergic synaptic proteins (i.e., synaptosomes) (Rao and Steward, 1991) from whole mouse hippocampi (P15) and then analyzed Vav2 protein cofractionation in the various cellular fractions by Western blotting with a highly specific Vav2 antibody (Fig. 1A). Interestingly, Vav2 was strongly enriched in proteins associated with the non-nuclear cell membrane pellet (P2) and synaptosome, similar to the fractionation profiles of TrkB, Rac1, and PSD-95 (Fig. 3C), despite the fact that Vav2 lacks known membrane-spanning domains or membrane-anchoring post-translational modifications. To determine whether Vav2 is a component of the PSD in synaptosome preparations, we performed mild detergent (0.5% Triton X-100) extraction of the synaptosomal fraction. Interestingly, Vav2 partially extracted from the synaptosomal fraction, indicating that a large amount of synaptosomal Vav2 is associated with the PSD (Fig. 3C). In comparison, TrkB was almost completely detergent extracted from the synaptosome fraction, whereas PSD-95 was not extracted from synaptosomes under these conditions (Fig. 3C), as expected. Together, these data suggest that hippocampal Vav2 is largely membrane associated in synapse-containing fractions, is a partial constituent of the PSD, and is thus in a position to mediate BDNF/TrkB-mediated morphological and/or functional plasticity at the excitatory synapse.
Vav GEFs are required for BDNF-induced dendritic spine head growth in hippocampal CA1 neurons
Several recent studies reveal an important role for BDNF/TrkB signaling in hippocampal dendritic spine growth, F-actin remodeling and functional synapse plasticity (Korte et al., 1995; Figurov et al., 1996; Luikart et al., 2005, 2008; Rex et al., 2007; Tanaka et al., 2008). Since (1) Vav GEFs are activated by BDNF/TrkB signaling, (2) Vav GEFs are required for BDNF-induced Rac-GTP production, and (3) Vav2 is colocalized with TrkB in synapse-enriched fractions, we speculated that Vav GEFs might be involved in BDNF/TrkB-induced morphological synapse plasticity. To test this idea, we treated WT and Vav2−/−3−/− mouse organotypic hippocampal slice cultures (P6 + 9 DIV) with recombinant BDNF and monitored dendritic spines using 2 photon confocal live cell imaging (Fig. 4A). Based on the rapid kinetics of BDNF-induced Vav2 phosphorylation and Rac-GTP activation, we monitored randomly chosen dendritic regions on secondary branches for 10 min before and 15 min after the addition of exogenous BDNF (Fig. 4A). Analysis of CA1 dendritic spines revealed that the existing spines demonstrated highly dynamic behavior in the basal state, with individual spine heads enlarging and shrinking rapidly within minutes (data not shown). However, the mean dendritic spine head size was fairly stable over the 10 min of pre-BDNF treatment (Fig. 4A, bottom). Upon the addition of BDNF to the wild-type hippocampal slice, we observed a significant increase in spine head area (Fig. 4A, bottom) (p < 0.05, two-way ANOVA) that peaked by 8 min and maintained an enlarged spine head area throughout the duration of the experiment (Fig. 4A). In contrast, analysis of hippocampal slices from the Vav-deficient mice showed no significant change in spine head area after addition of BDNF (Fig. 4A,B), indicating that Vav GEFs are required for BDNF-induced dendritic spine head growth.
Figure 4.
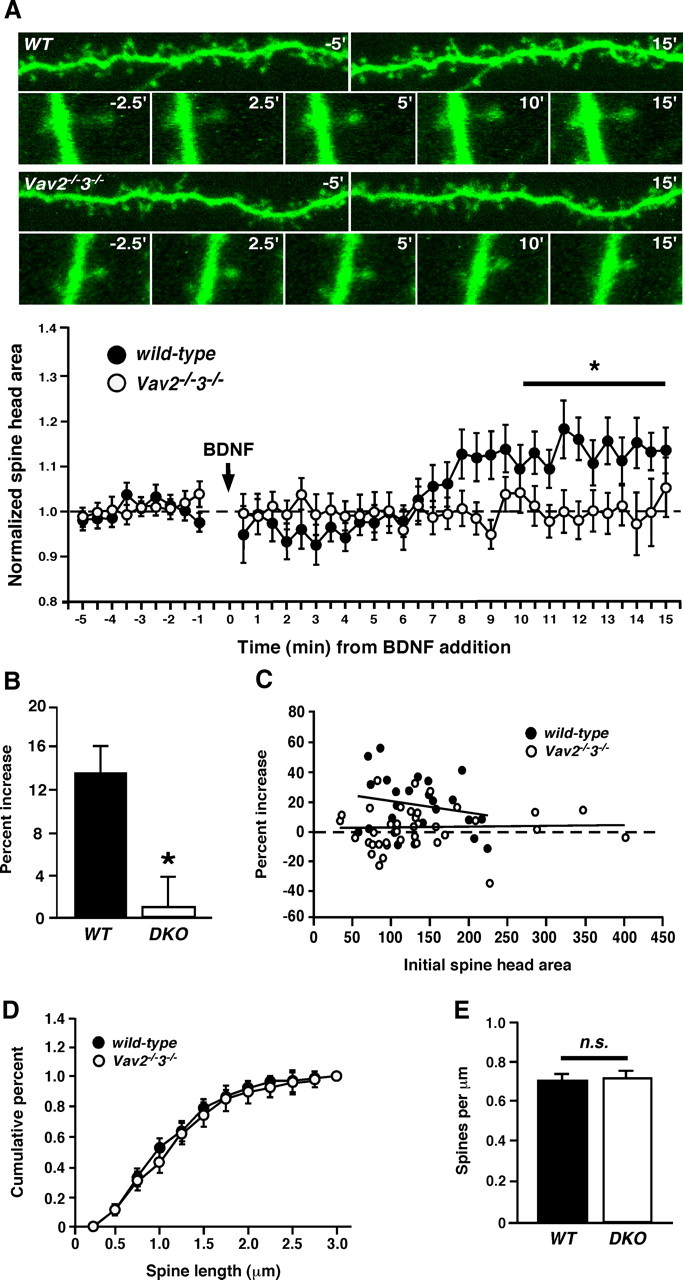
Vav GEFs are required for BDNF-induced dendritic spine head growth in hippocampal CA1 neurons. A, Top, Hippocampal organotypic slices (400 μm) were cultured from wild-type or Vav2−/−3−/− mice at P6. At 9 DIV, slices were stimulated with BDNF (250 ng/ml) over a time course and imaged using 2 photon microscopy. Representative photomicrographs showing dendritic segments and dendritic spine heads from wild-type or Vav2−/−3−/− mice at indicated times from BDNF stimulation. Data plot: normalized spine head areas from wild-type and Vav2−/−3−/− mice at times before and after BDNF addition to slice. *p < 0.05, two-way ANOVA; genotype and time, n = 6 (wild-type), n = 8 (Vav2−/−3−/−). For all experiments (A–E), n = 6 (wild-type) and n = 8 (Vav2−/−3−/−). Each n represents an average of five randomly chosen dendritic spines from one dendritic length. One dendritic length per slice was imaged and averaged to give an average spine behavior per dendrite. B, Percentage increase of mean spine head size at 10–15 min post-BDNF compared with the first 5 min after BDNF application (*p < 0.05, Student's t test). C, Plot of the percentage increase in spine head size versus initial spine head area, demonstrating that a similar range of spine heads was analyzed and that the failure of Vav KO spine head growth with BDNF treatment is not due to occlusion. D, Plot of cumulative dendritic spine length of wild-type and Vav2−/−3−/− neurons from pre-BDNF treated spines in A. E, Dendritic spine densities of wild-type and Vav2−/−3−/− neurons, cultured and imaged in A.
Because initial spine head area may affect a dendritic spine's relative capacity to change (occlusion), we analyzed pre-BDNF dendritic spine area in WT and Vav2/3 KO slices, but observed no significant differences between the genotypes (Fig. 4C). Similarly, there were no differences in cumulative dendritic spine length between wild-type and Vav-deficient neurons (Fig. 4D). We also did not observe evidence of new dendritic spines forming during the 15 min BDNF treatment (data not shown), indicating that BDNF-induced synaptogenesis requires a longer time scale (e.g., 24–48 h), as reported by others (Tyler and Pozzo-Miller, 2001, 2003).
BDNF and TrkB signaling are required for dendritic filopodial motility and synapse formation in early postnatal periods of hippocampal development in CA1 neurons (Luikart et al., 2005, 2008). To test whether Vav GEFs are required for these TrkB-influenced processes, we investigated morphological and functional synapses in wild-type and Vav-deficient neurons in hippocampal slice cultures. However, we observed no significant differences in dendritic spine density between wild-type and Vav-deficient CA1 neurons from analysis of dendritic regions used for the live-cell imaging study above (Fig. 4E). Using whole-cell patch-clamp recordings from WT and Vav-deficient CA1 pyramidal neurons in hippocampal slice cultures, we observed no significant differences in mEPSC frequency (Fig. 5A) or amplitude (Fig. 5B), and no differences in presynaptic vesicle release probability as determined from the paired-pulse ratio of evoked postsynaptic responses with a 100 ms stimulus interval (Fig. 5C). Therefore, while Vav GEFs are important for BDNF-induced rapid induction of Rac-GTP and morphological dendritic spine plasticity, they appear to be dispensable for early hippocampal synaptogenesis or basal synaptic function. As such, we sought to test whether Vav might function in existing dendritic spines to modulate or facilitate functional synapse plasticity.
Figure 5.

Vav GEFs mediate synaptic plasticity in the hippocampus. A, Average IEI per neuron from wild-type or Vav2−/−3−/− CA1 pyramidal neurons (n.s., Student's t test). B, Average mEPSC amplitude per neuron from same recordings as in B (n.s., Student's t test). C, Paired-pulse stimulation (10 Hz). Average paired-pulse ratio measured as the percentage of the second pulse relative to the first (n.s., Student's t test). D, Left, fEPSPs were recorded from CA1 of P15 wild-type or Vav2−/−3−/− acute hippocampal slices (300 μm) in response to two theta burst stimulations of Schaffer collaterals. Right, The fEPSP slopes were normalized for each genotype. Normalized fEPSP enhancement at 40–50 min is shown (*p < 0.05, Student's t test).
Vav GEFs mediate synaptic plasticity in the hippocampus
F-actin remodeling and BDNF/TrkB signaling are required for induction of TBS-LTP at CA1 glutamatergic synapses (Chen et al., 1999; Kim and Lisman, 1999; Krucker et al., 2000). To test whether Vav GEFs are required for TBS-LTP, we stimulated Schaffer collateral axons with a weak theta-burst stimulus (2× TBS) and recorded fEPSPs from the CA1 region of wild-type or Vav-deficient acute hippocampal slices at P15. Weak TBS stimulation of wild-type slices resulted in significant LTP as determined from evoked field responses during the last 10 min of a 50 min post-tetanus recording period. TBS-LTP was also significantly generated in the Vav-deficient slices, but the LTP level was significantly reduced compared with wild-type slices (Fig. 5D) (wild-type, 43.5%±8.7; Vav2−/−3−/−, 15.9%±4.4; p < 0.05, Student's t test). Together, these findings reveal a novel role for Vav GEFs in the process of functional synaptic plasticity, and suggest that Vav GEFs facilitate TBS-LTP by regulating structural dynamics at functional synapses.
Vav2 activation requires TrkB kinase activity, but not autophosphorylation of the Shc/Frs2- and PLCγ-interacting TrkB phosphotyrosines
To better define how BDNF/TrkB signaling regulates Vav GEF activity, we transiently transfected HEK293T cells with plasmids expressing Vav2 alone or together with wild-type or mutant TrkB receptors. As mentioned previously, coexpression of wild-type TrkB with Vav2 resulted in elevated tyrosine phosphorylation levels using either a general phosphotyrosine-specific antibody (Fig. 1B) or a specific anti-P-Y172-specific antibody (Figs. 1C, 6A). However, the TrkB-dependent increase in P-Y172 Vav2 required TrkB activity (Fig. 6A) [WT vs K571N (kinase dead, KD)]. Interestingly, the TrkB kinase-dependent increase in Vav2 P-Y172 levels did not require tyrosine autophosphorylation at TrkB Y490 (Shc/Frs2 binding site) or Y785 (PLCγ binding site) (Fig. 6A). Similarly, mutating one TrkB kinase domain autophosphorylation site (Y670F) did not alter P-Y172 levels (Fig. 6A). However, mutating multiple TrkB kinase domain autophosphorylation sites, Y674/675F, did reduce Vav2 P-Y172 levels, but these TrkB mutations caused similar magnitude reductions of TrkB P-Y490 autophosphorylation (data not shown), suggesting that the decreased Vav2 phosphorylation was due to indirect effects of these mutations on intrinsic TrkB kinase activity. Finally, the TrkB kinase-dependent increase in P-Y172 Vav2 was largely independent of the Vav2 SH2 domain (Fig. 6A, right), which was previously shown to be important for activation by Eph receptors (Cowan et al., 2005). Together, these data indicate that Vav GEFs are activated by a TrkB-dependent mechanism that is independent of the two best-studied TrkB autophosphorylation sites (Y490 and Y785) and independent of the Vav SH2 domain.
Figure 6.

Kinase-active TrkB activates Vav2 independently of the ERK/PI3K- and PLCγ-interacting sites. A, Coexpression of TrkB and Vav2 increases phospho-Vav2 (Y172) in HEK293T cells. HEK293T cells were transfected with T7-tagged wild-type or ΔSH2 and Flag-tagged wild-type or mutant TrkB. Total cell lysates were blotted with anti-phospho-Vav2 (Y172), anti-T7 (Vav2), anti-phospho-TrkB (Y490), and anti-TrkB antibodies. A–E, Results are representative findings from three independent experiments. B, TrkB binds Vav2 and Vav3 in HEK293T cells. HEK293T cells were transfected with HA-tagged Vav2 or Vav3 and Flag-tagged TrkB. Total cell lysates were immunoprecipitated with anti-Flag antibody, then immunoblotted with anti-HA (Vav) and anti-Flag (TrkB) antibodies. Whole-cell lysates were blotted with anti-HA (Vav) antibody. C, TrkB binds Vav2 independently of TrkB kinase activity. T7-tagged Vav2 was coexpressed with Flag-tagged TrkB (wild-type), TrkB-KD (K571N), TrkB-Y490F, or TrkB-Y785F. Total cell lysates (TCLs) were immunoprecipitated with anti-Flag antibody, then immunoblotted with anti-Vav2 and anti-Flag (TrkB) antibodies. Whole-cell lysates were blotted with anti-Vav2 antibody. D, Deletion analysis of Vav2 identifies the DH/PH/ZF region as sufficient to mediate coassociation with TrkB. Domain composition of full-length and Vav2 deletion mutants that were generated. C, Calponin homology domain; A, acidic domain; DH, Dbl homology domain; P, pleckstrin homology domain; Z, zinc finger domain; P, proline-rich domain; 3, Src homology type 3 domain (SH3); 2, Src homology type 2 domain (SH2). HEK293T cells were transfected with Flag-tagged TrkB (wild-type) and T7-tagged Vav2 (wild-type), Vav2-ΛCH/AD, Vav2-Λadaptor, or Vav2-DH/PH/ZF. Total cell lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with anti-T7 (Vav2) and anti-Flag (TrkB) antibodies. Whole-cell lysate was blotted with anti-T7 (Vav2) antibody. E, TrkB's kinase domain is required for its interaction with Vav2. T7-tagged Vav2 was coexpressed with wild-type or Δ kinase domain (ΔKD)-TrkB. Total cell lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with anti-Vav2 and anti-Flag (TrkB) antibodies. Whole-cell lysate was blotted with anti-Vav2 antibody.
To determine whether Vav GEFs physically interact with TrkB receptors, we expressed wild-type Vav2 or Vav3 alone or together with TrkB in HEK293T cells and performed coimmunoprecipitation assays. We found that TrkB specifically coimmunoprecipitated with either Vav2 or Vav3 (Fig. 6B) under stringent detergent lysis/IP buffer conditions (i.e., RIPA buffer; see Materials and Methods). Unlike the regulation of Vav2 P-Y172 levels, we found that this interaction occurred independently of TrkB activity since Vav2 and kinase-inactive TrkB (K571N) coimmunoprecipitated together to a similar extent (Fig. 6C). Similarly, TrkB autophosphorylation site mutants, Y490F and Y785F, also coimmunoprecipitated with Vav2 at similar levels as wild-type TrkB (Fig. 6C).
To further define the nature of this novel TrkB–Vav2 interaction, we generated a series of deletion mutants of Vav2 lacking the N-terminal calponin homology and acidic domain regions, or lacking the C-terminal adaptor domain, which comprises two SH3 domains flanking the SH2 domain. We also analyzed Vav2 lacking both of these regions, which leaves only the core DH/pleckstrin homology (PH)/zinc finger (ZF) regions intact (Fig. 6D). All of these Vav2 mutants interacted strongly with TrkB, indicating that the TrkB kinase-independent interaction between Vav2 and TrkB is mediated by residues within the DH/PH/ZF region. Deletion of the TrkB kinase domain disrupted the interaction (Fig. 6E), suggesting that the Vav DH/PH/ZF region mediates the interaction with the TrkB domain. These findings reveal a strong kinase-independent binding mechanism between Vav and TrkB in cells, and suggest that activation of TrkB kinase activity stimulates Vav2 tyrosine phosphorylation and GEF activity.
Discussion
In this study, we report an essential role for Vav GEFs as important mediators of BDNF/TrkB receptor-induced activation of Rac GTPases and morphological plasticity of hippocampal dendritic spines, and a novel role in functional synapse plasticity (Fig. 7). Specifically, we report that BDNF/TrkB signaling increases Vav tyrosine phosphorylation, and that Vav GEFs are required for BDNF-induced production of Rac-GTP and spine head growth in hippocampal neurons. Vav2 copurifies with biochemically enriched excitatory postsynaptic proteins, and Vav2 and Vav3 are necessary for normal TBS-LTP, supporting a role for F-actin remodeling enzymes in functional synaptic plasticity. Together, these data suggest that Vav GEF-dependent regulation of Rac activity and F-actin assembly in existing dendritic spines is an important process for the establishment and maintenance of LTP.
Figure 7.

Model for the role of Vav GEFs in BDNF/TrkB-induced dendritic spine growth and functional synapse plasticity.
While our findings link Vav GEFs to regulated F-actin dynamics in dendritic spines and functional synapses, and suggest a possible mechanism by which BDNF/TrkB signaling may contribute to the establishment of stable LTP, there remain a number of important questions about the role of Vav GEFs in BDNF-dependent structural and functional synapse plasticity for future investigation. Our findings demonstrate an important role for Vav in the BDNF induction of Rac-GTP, but there is clearly a significant residual activation of Rac that remains in the Vav-deficient mice. Two recent studies reported that BDNF/TrkB signaling activates Tiam1 in neurons and non-neuronal cells (Miyamoto et al., 2006; Zhou et al., 2007). Like Vav GEFs, Tiam1 is a member of the Dibble homology family of Rho GEFs and a potent regulator of Rac activity. In Cos-7 cells, BDNF/TrkB signaling stimulates tyrosine phosphorylation of Tiam1 and activation of Rac-GTP, and in dissociated cortical neurons, dominant-negative Tiam1 expression reduced BDNF-induced neurite outgrowth (Miyamoto et al., 2006). Similarly, using an RNA interference-based approach, BDNF-regulated migration of cerebellar granule cell precursors was shown to require Tiam1-dependent activation of Rac (Zhou et al., 2007). As such, we speculate that Tiam1, which is expressed in the developing hippocampus (Ehler et al., 1997), likely contributes to the residual Rac-GTP activation in Vav2/3-null mice. Depending on the developmental stage and neuronal cell type, Vav and Tiam GEFs may serve in a functionally redundant pathway for Rac activation, but either alone is not sufficient for full Rac activation. In the future, it will be important to study the relationship between these GEFs in BDNF-dependent F-actin dynamics and related biological processes.
Recently, Zhou et al. (2007) reported that PI3-K activity was not required for BDNF-induced activation of Rac in cerebellar GCPs, which is consistent with the idea that Rac activation is not downstream of PI3-K activity, but is rather an essential parallel signaling process important for cell morphological changes. A recent study reported an important role for TrkB P-Y490 and PI3-K activity in the process of filopodial motility and hippocampal synaptogenesis (Luikart et al., 2008). In general, our findings are consistent with these observations in that Vav GEFs, while important for BDNF-induced Rac activation, are not required for early hippocampal dendritic spinogenesis through postnatal day 15 (Fig. 4E). Similarly, P-Y490 TrkB is not required for Vav2 activation in non-neuronal cells (Fig. 6A), nor is it required for LTP induction (Minichiello et al., 2002; Gruart et al., 2007), suggesting that TrkB-mediated enhancement of filopodial motility and synaptogenesis is a distinct process from BDNF/Vav-dependent Rac activation, spine head growth, and maintenance of LTP. Interestingly, unlike TrkB receptor knock-out mice, CA1 hippocampal neurons of conditional brain-specific BDNF knock-out mice do not have a reduced dendritic spine density, but instead have fewer mushroom-type spines and more thin-type spines (Rauskolb et al., 2010), suggesting the possibility that TrkB receptors may regulate CA1 hippocampal neuron dendritic spine density in a BDNF-independent manner in vivo.
Many previous studies have documented the importance of F-actin assembly/remodeling for LTP maintenance (Kim and Lisman, 1999; Krucker et al., 2000; Fukazawa et al., 2003; Rex et al., 2007), and a strong correlation has been established between spine head volume and functional synapse plasticity (Matsuzaki et al., 2004; Okamoto et al., 2004; Honkura et al., 2008; Tanaka et al., 2008). In addition, BDNF enhances TBS-induced F-actin content in dendritic spines and the inhibition of endogenous BDNF reduces TBS-LTP (Rex et al., 2007), supporting the hypothesis that BDNF/TrkB signaling enhances TBS-LTP by facilitating F-actin assembly in stimulated glutamatergic spine synapses. Rac activation in particular seems to be important for dendritic spine enlargement and stabilization (Nakayama et al., 2000; Tashiro et al., 2000), and as a major effector of Vav GEFs (Crespo et al., 1997; Abe et al., 2000; Zeng et al., 2000), the dramatic reduction in BDNF-induced Rac-GTP levels in Vav-deficient neurons likely contributes to the defect in BDNF-induced spine head enlargement and TBS-LTP. In HEK293T cells, we observe that transient expression of Vav2 or Vav3 together with TrkB results in robust lamellipodia-like protrusions from the transfected cells (C. W. Cowan, unpublished observations), indicative of Rac-induced F-actin assembly in cells. A recent study reported that the inhibitor NSC23766, which is a small-molecule inhibitor of some Rac-GEFs, such as Tiam1 and Trio, did not reduce TBS-LTP in hippocampal slices (Rex et al., 2009). However, this compound exhibits specificity for inhibiting Tiam1-Rac binding and was found to reduce Tiam1-stimulated, but not Vav-stimulated, cell growth, suggesting that the inhibitor does not affect Vav-mediated Rac1 activation (Gao et al., 2004). Consistent with this idea, the NSC compound only reduced TBS-induced Rac-GTP levels by ∼30%, suggesting that a NSC-sensitive GEF, like Tiam1, accounts for only a subset of the Rac activation. Our data would suggest that Vav GEFs account for a majority of the Rac-GTP produced by BDNF/TrkB signaling (Fig. 2).
Despite our initial speculation that Vav GEFs would be recruited to tyrosine-autophosphorylated TrkB via the SH2 phosphotyrosine binding domain, which is how Vav2 interacts with Eph receptors in HEK293T cells (Cowan et al., 2005), our findings in heterologous cells reveal a novel interaction between Vav2 and TrkB involving a TrkB-independent interaction between the Vav DH/PH/ZF core catalytic region and the TrkB domain. A similar interaction mechanism was observed between Ephexin1, a DH family GEF required for axonal outgrowth and repulsion, and two different receptor tyrosine kinases, EphA4 and fibroblast growth factor (FGF) receptors. The DH and PH domains of Ephexin interacted with the kinase domain of the EphA4 receptor (Shamah et al., 2001), and, recently, a similar interaction was described between Ephexin and the FGF receptor (Zhang et al., 2007). In the former case, the addition of ephrinA1 ligand stimulated tyrosine phosphorylation of endogenous Ephexin, which in turn stimulated its RhoA GEF activity and promoted axon growth cone collapse (Sahin et al., 2005).
While our results suggest that Vav-dependent F-actin dynamics in dendritic spines may regulate aspects of postsynaptic structural and functional synapse plasticity, it will be important to determine the downstream signaling events and cellular processes by which Vav GEFs elicit these changes. Our findings suggest a postsynaptic role for Vav GEFs, but as these are total knock-out mice, we cannot rule out a potential presynaptic role that could indirectly mediate TBS-LTP or spine growth. Since Vav2 and Vav3 mRNAs (and Vav2 protein) are expressed at similar levels in both primary neuron and astrocyte cultures (data not shown), we also cannot rule out a possible glial role for Vav GEFs in mediating structural and functional synapse plasticity. Finally, there could also be a developmental role for Vav GEFs that occurs before the structural and functional synapse plasticity events analyzed at P15 (or its rough equivalent in slice culture). Analysis of conditional Vav knock-out mice will be important for resolving these questions in the future. On a mechanistic note, Vav GEFs may be necessary for endocytosis of the BDNF/TrkB receptor complex, which is known to be critical for neuronal survival, as well as for the migration of cerebellar granule cell precursors (Valdez et al., 2005; Zhou et al., 2007). Previously, we found that Vav GEFs are required for endocytosis of the ephrinA1/Eph receptor complex in retinal ganglion cells (Cowan et al., 2005), which led us to speculate that Vav-dependent endocytosis, possibly through a Rho/Rac-dependent process, is a critical step in Eph receptor-mediated forward signaling during axon guidance. Similarly, endocytosis of the BDNF-bound TrkB receptor has been shown to be critical for promoting forward-signaling processes necessary for BDNF-dependent neuronal survival and migration of cerebellar granule cell precursors (Valdez et al., 2005; Zhou et al., 2007); but, in the latter case, this was shown to require Tiam1 (Zhou et al., 2007). In the future, it will be interesting to determine whether Vav GEFs regulate TrkB receptor endocytosis, and whether internalization of the BDNF-activated TrkB receptor is important for TBS-LTP and dendritic spine plasticity.
Regulation of local protein synthesis in dendrites plays a critical role in stabilizing long-lasting changes in dendritic spine morphology and functional synapse plasticity (Tanaka et al., 2008; Yang et al., 2008). However, the mechanisms by which de novo protein synthesis regulates spine and synapse plasticity are poorly understood. Based on the transient nature of BDNF-induced Vav tyrosine phosphorylation and Rac-GTP production, we speculate that Vav GEF signaling stimulates transient F-actin assembly, but an independent, protein synthesis-dependent molecular process likely stabilizes the F-actin cytoskeleton and the maintenance of LTP. Consistent with this idea, we find that addition of exogenous BDNF to hippocampal slices is sufficient to induce rapid spine head growth (Fig. 4). However, addition of exogenous BDNF, in the absence of presynaptic stimulation, does not by itself produce LTP, but coupling exogenous BDNF preincubation with TBS does enhance CA3-CA1 LTP (L. Leverich, unpublished observations). This suggests that BDNF-induced spine head growth may be permissive for functional synapse plasticity, but additional activity-dependent processes are required to induce and stabilize LTP. It is also important to note that the addition of 20 ng/ml BDNF to hippocampal slices did not alter spine head size unless coupled with glutamate uncaging (Tanaka et al., 2008). In contrast, we found that the addition of 250 ng/ml BDNF was sufficient for spine head growth (Fig. 4A,B), suggesting that there is a threshold BDNF concentration required to stimulate spine growth without strong, coupled glutamate release, which appears to stimulate endogenous BDNF release. Glutamate is also known to stimulate other Rac-GEFs, such as Tiam1 and Kalirin-7, via activation of ionotropic AMPA or NMDA glutamate receptors (Tolias et al., 2005; Xie et al., 2007), which may synergize with BDNF/TrkB signaling mechanisms to facilitate spine growth.
Together, the findings described here introduce a novel role for Vav GEFs in BDNF-induced Rac activation and dendritic spine head growth in neurons, as well as in TBS-induced hippocampal LTP. By regulating Rho GTPase activity in response to BDNF/TrkB signaling, Vav proteins may modulate F-actin remodeling in dendritic spines to facilitate dendritic structural changes and activity-dependent synapse plasticity.
Footnotes
Purified GST-PBD protein was provided by the Recombinant Protein Biochemistry Core (Grant P30 EY020799 to C.W.C.). C.F.H. was supported by an institutional fellowship from NIDA (T32 DA07290). C.B.W. was supported in part by the Summer Undergraduate Research Fellowship program at The University of Texas Southwestern Medical Center. We acknowledge the generous support of the Whitehall Foundation (C.W.C.), the National Eye Institute (Grants RO1 EY018207 to C.W.C.), and the National Institute on Drug Abuse (Grants P01 DA008277 to C.W.C.). We thank Wojciech Swat for providing Vav2 and Vav3 knock-out mice, and Caroline Hamilton-Smith for assistance with hippocampal field recordings. Hippocampal dendritic spine imaging was performed on a two-photon confocal microscope system in the The University of Texas Southwestern Medical Center Live Cell Imaging Core facility.
The authors declare no competing financial interests.
References
- Abe K, Rossman KL, Liu B, Ritola KD, Chiang D, Campbell SL, Burridge K, Der CJ. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275:10141–10149. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- Aghazadeh B, Lowry WE, Huang XY, Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–633. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Bramham CR. Local protein synthesis, actin dynamics, and LTP consolidation. Curr Opin Neurobiol. 2008;18:524–531. doi: 10.1016/j.conb.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR. Vav proteins, adaptors and cell signaling. Oncogene. 2001;20:6372–6381. doi: 10.1038/sj.onc.1204780. [DOI] [PubMed] [Google Scholar]
- Cerione RA, Zheng Y. The Dbl family of oncogenes. Curr Opin Cell Biol. 1996;8:216–222. doi: 10.1016/s0955-0674(96)80068-8. [DOI] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Cowan CW, Shao YR, Sahin M, Shamah SM, Lin MZ, Greer PL, Gao S, Griffith EC, Brugge JS, Greenberg ME. Vav family GEFs link activated Ephs to endocytosis and axon guidance. Neuron. 2005;46:205–217. doi: 10.1016/j.neuron.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Crespo P, Schuebel KE, Ostrom AA, Gutkind JS, Bustelo XR. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Ehler E, van Leeuwen F, Collard JG, Salinas PC. Expression of Tiam-1 in the developing brain suggests a role for the Tiam-1-Rac signaling pathway in cell migration and neurite outgrowth. Mol Cell Neurosci. 1997;9:1–12. doi: 10.1006/mcne.1997.0602. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett TA, Van Buul JD, Burridge K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp Cell Res. 2007;313:3285–3297. doi: 10.1016/j.yexcr.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruart A, Sciarretta C, Valenzuela-Harrington M, Delgado-García JM, Minichiello L. Mutation at the TrkB PLCγ-docking site affects hippocampal LTP and associative learning in conscious mice. Learn Mem. 2007;14:54–62. doi: 10.1101/lm.428307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Das B, Wei W, Van Aelst L, Mosteller RD, Khosravi-Far R, Westwick JK, Der CJ, Broek D. Lck regulates Vav activation of members of the Rho family of GTPases. Mol Cell Biol. 1997;17:1346–1353. doi: 10.1128/mcb.17.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- Heo J, Thapar R, Campbell SL. Recognition and activation of Rho GTPases by Vav1 and Vav2 guanine nucleotide exchange factors. Biochemistry. 2005;44:6573–6585. doi: 10.1021/bi047443q. [DOI] [PubMed] [Google Scholar]
- Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron. 2008;57:719–729. doi: 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Hunter SG, Zhuang G, Brantley-Sieders D, Swat W, Cowan CW, Chen J. Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol Cell Biol. 2006;26:4830–4842. doi: 10.1128/MCB.02215-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossel AH, Cambridge SB, Wagner U, Bonhoeffer T. A caged Ab reveals an immediate/instructive effect of BDNF during hippocampal synaptic potentiation. Proc Natl Acad Sci U S A. 2001;98:14702–14707. doi: 10.1073/pnas.251326998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Zhang W, Wayman GA, Kwon CH, Westbrook GL, Parada LF. Neurotrophin-dependent dendritic filopodial motility: a convergence on PI3K signaling. J Neurosci. 2008;28:7006–7012. doi: 10.1523/JNEUROSCI.0195-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Yamauchi J, Tanoue A, Wu C, Mobley WC. TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc Natl Acad Sci U S A. 2006;103:10444–10449. doi: 10.1073/pnas.0603914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Pandey A, Podtelejnikov AV, Blagoev B, Bustelo XR, Mann M, Lodish HF. Analysis of receptor signaling pathways by mass spectrometry: identification of vav-2 as a substrate of the epidermal and platelet-derived growth factor receptors. Proc Natl Acad Sci U S A. 2000;97:179–184. doi: 10.1073/pnas.97.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, Greengard P, Nairn AC, Nestler EJ, Cowan CW. Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron. 2008;59:621–633. doi: 10.1016/j.neuron.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Steward O. Evidence that protein constituents of postsynaptic membrane specializations are locally synthesized: analysis of proteins synthesized within synaptosomes. J Neurosci. 1991;11:2881–2895. doi: 10.1523/JNEUROSCI.11-09-02881.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramár EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, Wright TM, Shamah SM, O'connell S, Cowan CW, Hu L, Goldberg JL, Debant A, Corfas G, Krull CE, Greenberg ME. Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron. 2005;46:191–204. doi: 10.1016/j.neuron.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Schuebel KE, Movilla N, Rosa JL, Bustelo XR. Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 1998;17:6608–6621. doi: 10.1093/emboj/17.22.6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Bazalakova M, Neve RL, Corfas G, Debant A, Greenberg ME. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–244. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- Shen K, Cowan CW. Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol. 2010;2:a001842. doi: 10.1101/cshperspect.a001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J Physiol. 2003;553:497–509. doi: 10.1113/jphysiol.2003.052639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Akmentin W, Philippidou P, Kuruvilla R, Ginty DD, Halegoua S. Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J Neurosci. 2005;25:5236–5247. doi: 10.1523/JNEUROSCI.5104-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–5751. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zeng L, Sachdev P, Yan L, Chan JL, Trenkle T, McClelland M, Welsh J, Wang LH. Vav3 mediates receptor protein tyrosine kinase signaling, regulates GTPase activity, modulates cell morphology, and induces cell transformation. Mol Cell Biol. 2000;20:9212–9224. doi: 10.1128/mcb.20.24.9212-9224.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sawada T, Jing X, Yokote H, Yan X, Sakaguchi K. Regulation of ephexin1, a guanine nucleotide exchange factor of Rho family GTPases, by fibroblast growth factor receptor-mediated tyrosine phosphorylation. J Biol Chem. 2007;282:31103–31112. doi: 10.1074/jbc.M704430200. [DOI] [PubMed] [Google Scholar]
- Zhou P, Porcionatto M, Pilapil M, Chen Y, Choi Y, Tolias KF, Bikoff JB, Hong EJ, Greenberg ME, Segal RA. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55:53–68. doi: 10.1016/j.neuron.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]