

Abstract
Nucleoporins (Nups), which are intrinsically disordered, form a selectivity filter inside the nuclear pore complex, taking a central role in the vital nucleocytoplasmic transport mechanism. These Nups display a complex and nonrandom amino-acid architecture of phenylalanine glycine (FG)-repeat clusters and intra-FG linkers. How such heterogeneous sequence composition relates to function and could give rise to a transport mechanism is still unclear. Here we describe a combined chemical biology and single-molecule fluorescence approach to study the large human Nup153 FG-domain. In order to obtain insights into the properties of this domain beyond the average behavior, we probed the end-to-end distance (RE) of several ∼50-residues long FG-repeat clusters in the context of the whole protein domain. Despite the sequence heterogeneity of these FG-clusters, we detected a reoccurring and consistent compaction from a relaxed coil behavior under denaturing conditions (RE/RE,RC = 0.99 ± 0.15 with RE,RC corresponding to ideal relaxed coil behavior) to a collapsed state under native conditions (RE/RE,RC = 0.79 ± 0.09). We then analyzed the properties of this protein on the supramolecular level, and determined that this human FG-domain was in fact able to form a hydrogel with physiological permeability barrier properties.
Introduction
The megaDalton-sized nuclear pore complex (NPC) forms a selective barrier that regulates vital cargo exchange across the nuclear envelope (1,2). It is estimated that NPCs regulate the transport of hundreds of large cargos per second while small molecules (<30 kDa) can pass through unhindered by passive diffusion (3–5). Approximately one-third of the proteins that constitute the NPC have large intrinsically disordered domains (6–8), and due to the existence of several FG-repeats within their amino-acid (AA) sequence, they are frequently termed FG-nucleoporins (FG-Nups). Interaction of FG-repeats with specialized nuclear transport receptors (NTRs) facilitates passage of NTR-cargo complexes through the NPC, thus giving rise to a selective transport mechanism (9). However, the structure of the actual permeability barrier itself is still highly debated, and several transport models have been proposed to explain the sievelike behavior of NPCs (for reviews, see (3,4,10)). Among the most prominent models is the selective phase model which postulates a cohesive meshwork of FG-Nups inside the NPC through which NTRs can efficiently melt (11,12). In fact, it has been shown that some yeast FG-Nups can form a supramolecular hydrogel that has properties reminiscent of the physiological NPC permeability barrier (13–15). In contrast, the entropic barrier model postulates that FG-Nups form a noncohesive brush inside the pore, which can be reversibly collapsed by interactions with NTRs. In support of this model it was found by Lim et al. (16,17) that FG domains of Nup153 immobilized in a dense layer can form a polymer brush.
Precisely how FG-Nup architecture correlates with function is, however, still not very well known. The biological function of FG-Nups is also highly redundant, as several FG-Nups can be deleted without substantial impact on NPC function (18). Furthermore, FG-repeat composition is not well conserved during evolution, and even FG-Nups from related species often display little sequence similarity (19,20). Single FG-Nups themselves do not necessarily exhibit a homogeneous AA composition along their entire sequence, but often display a local bias toward distinct AA distributions (8,21–23). FG-repeats can be grouped into different categories based on neighboring AAs, such as FxFG, PxFG, and GLFG (x = any AA), which are not randomly distributed but seem to occur in patches. For instance, GLFG-repeats are rarely found in humans but frequently in yeast. To what extent different FG-repeat types encode different or similar functionalities is not yet well understood. The AA linkers (spacer regions) between FG-repeats have also been implicated as functionally relevant (21,23–25). A study by Yamada et al. (23) grouped yeast FG-Nups into roughly two categories based on hydrodynamic radii and percentage of charged and hydrophobic residues, and they proposed how different Nup behavior (such as collapsed or extended coil) could give rise to a permeability barrier. The enrichment of particular residues such as N and Q (often found in amyloid-generating proteins (26)) has recently also been linked to function (21,23).
Patently, our understanding of the function of FG-repeat proteins is still limited by an insufficient knowledge of even the basic biophysical properties of these disordered domains. Furthermore, understanding the properties of small fragments is of particular importance because sequence heterogeneities within these FG-repeats occur even within short AA stretches. Here we present an approach to probe the properties of short segments within the large Nup153 FG-domain (Nup153FG). This large domain shows a heterogeneous sequence composition with a spatially clustered occurrence of distinct FG-repeat types. We combined a differential labeling scheme of selected FG-subdomains with single molecule fluorescence detection. Our applied multiparameter technology is particularly well suited to probe biophysical properties of segments of relevant AA lengths in the context of the unperturbed full-length FG-domain. In multiple stretches of the protein with diverse AA and FG-type compositions, we detected a collapsed behavior and a gradual transition to the denatured state. These findings induced us to investigate, on the macromolecular level, whether Nup153FG also belongs to the class of hydrogel-forming proteins. Indeed, the protein was able to form such a state. Thus, our approach provides insights into the behavior of various AA stretches within FG-Nups with implications for nuclear transport mechanisms.
Methods
Protein expression and purification
The codon-optimized (Mr. Gene, Regensburg, Germany) Nup153FG (AA 875–1475 of the human full-length protein) was recombinantly expressed in the Escherichia coli strain BL21(AI) (Invitrogen, Carlsbad, CA). The AA sequence was designed analogously to previous studies to ensure maximal comparability (17). The cysteine-free wild-type (wt) domain and the labeling mutants carried an N-terminal His6 tag followed by a TEV protease cleavage site. On their C-terminus, all protein constructs were fused to an intein followed by a chitin-binding domain. Plasmids from the labeling mutants were cotransformed with pEvol-AcF, which encodes for the p-acetylphenylalanine (AcF) Amber suppressor tRNA/tRNA synthetase pair (27). Cultures were grown in Terrific Broth medium at 37°C, induced with arabinose and IPTG at OD ∼1.0, and harvested by centrifugation after 4 h. Bacterial cultures of Nup153FG labeling mutants additionally contained 2 mM AcF. The protein was purified under mildly denaturing (2 M Urea) conditions on Ni- and chitin-resins following standard protocols. N- and C-terminal purification tags were cleaved by TEV protease and 100 mM β-mercaptoethanol (β-ME), respectively, following standard manufacturer's protocols. Nup153FG wt was further purified, desalted by reversed phase chromatography (SOURCE 15RPC; GE Healthcare, Uppsala, Sweden), and lyophilized.
Importin β was expressed as a C-terminal intein-His6 fusion construct and expressed in E. coli BL21 (AI) (Invitrogen). Cultures were grown in Terrific Broth medium at 37°C and induced at OD 0.4-0.6 with arabinose and IPTG. After induction, temperature was reduced to 30°C and growth was continued for 5–6 h. Bacteria were harvested and purified under native conditions according to standard purification protocols for His-tagged proteins. The C-terminal purification tag was removed by intein cleavage in 100 mM β-ME.
Protein labeling and design of labeling mutants
Single cysteines and Amber stop codons (TAG) were introduced into the Nup153FG wt sequence by site-directed mutagenesis. The purified labeling mutants were site-specifically labeled at their cysteines with Alexa Fluor 594 C5-maleimide (Invitrogen) and the unnatural amino acid AcF was labeled with Alexa Fluor 488 hydroxylamine (Invitrogen) as described previously (28,29). The double-labeled Nup153FG mutants were further purified on a Superdex 200 column (GE Healthcare). Concentrations were determined by UV/Vis spectrometry and typical labeling efficiencies per labeling site were above 70%.
Table S1 in the Supporting Material shows an overview of the labeling sites that were introduced into Nup153FG corresponding to the segments shown in Fig. 1.
Figure 1.
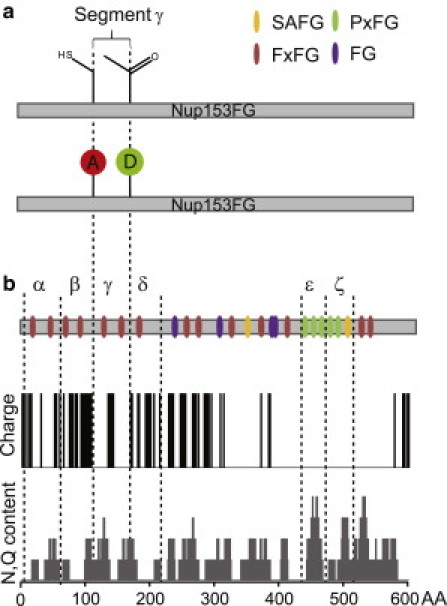
Labeling design and AA composition of Nup153FG. (a) Alexa 594-maleimide (A) attached to a unique cysteine in red and Alexa 488-hydroxylamine (D) coupled to the site-specifically introduced unnatural amino acid with ketone functionality (in green). (b) Different classes of FG-repeats are color-coded and segments analyzed by single-molecule FRET are separated (dashed lines) and labeled α to ζ (upper row). (Middle row) Digital representation of AA charge. (Lower row) Analysis of N and Q content along the sequence with a sliding window of 10 AAs. AA composition was analyzed with the software EMBOSS (57).
Importin β was labeled by cysteine random labeling with Alexa Fluor 594 C5-maleimide. The labeled protein was separated from unreacted dye on a Superdex 200 column; it contained approximately three dye molecules per protein.
Single-molecule fluorescence spectroscopy
Single-molecule fluorescence experiments were performed on a custom-built multiparameter spectrometer centered around a model No. IX81 microscope (Olympus, Hamburg, Germany) equipped with a high-numerical-aperture water-immersion objective (60×, 1.2 NA). A laser diode (LDH 485; Picoquant, Berlin, Germany), pulsed at 27 MHz, was used to excite freely diffusing labeled proteins with linearly polarized light. The fluorescence emission of donor and acceptor dyes was spatially filtered with a 100-μm pinhole, split into parallel and perpendicular polarization directions, separated into green and red fluorescence (emission filters 525/50, 700/75, dichroics 500/660 and 555 from AHF, Tuebingen Germany), and detected by MPDs (Picoquant) in the green channel and APDs (Perkin Elmer, Vaudreuil, Quebec, Canada) in the red channel. Photon signals were counted using a Hydraharp (Picoquant) (see Fig. S1 in the Supporting Material for a scheme of the single-molecule fluorescence setup). Acquired data were subjected to multiparameter fluorescence analysis (30,31) and processed burstwise for fluorescence intensities (I), donor lifetimes (τ), and donor anisotropies (r). Fitting of τ was done according to the procedure from Koshioka et al. (32) and Schaffer et al. (33). Fluorescence lifetimes and Förster resonance energy transfer efficiencies (EFRET) of the individual populations measured during a single-molecule experiment were extracted from two-dimensional Gaussian fits of the two populations in an EFRET-versus-τ histogram, corresponding to donor-only species and FRET species. The Perrin equation
| (1) |
relates the fundamental anisotropy r0 = 0.4, the anisotropy r, the fluorescence lifetime τ, and the rotational correlation coefficient ρ. We fit the two populations in a two-dimensional τ-versus-r histogram with 2D-Gaussians. The value ρ was calculated according to the Perrin equation from τD(0) and rD(0) (the τ and r obtained for the donor-only species from the two-dimensional fit) and Perrin curves were then plotted onto the two-dimensional histograms. The dye was assumed to have free rotation when anisotropies were low (<0.2) and followed the Perrin curve (34). For all analyses, fluorescence intensities of the different polarization directions were corrected by G-factors specifically determined for the setup on the day of the measurement according to
| (2) |
All single-molecule experiments were performed in measurement buffer (phosphate-buffered saline (PBS) pH 7.4, 2 mM dithiothreitol, and 2 mM magnesium acetate) at protein concentrations of 50 pM. Titrations with guanidinium-hydrochloride (GdmCl) contained additional 1 M, 2 M, 3 M, or 4 M GdmCl inside the measurement buffer. In the text, PBS refers to measurement buffer and XM GdmCl to measurement buffer plus XM GdmCl.
All analysis was performed using a custom-written code in IgorPro (Wavemetrics, Lake Oswego, OR).
Determination of fluorophore distances through FRET
For every detected FRET population, EFRET was calculated either from the donor lifetimes according to
| (3) |
where τD(0) and τDA are the fluorescence lifetimes of the donor only population and of the FRET-species, or from the intensity measured in the green and the red channels
| (4) |
with correction factor γ, where IA and ID correspond to the acceptor and donor fluorescence intensities, respectively. Intensity-based FRET efficiencies were plotted in the 2D EFRET-versus-τ histograms. The end-to-end-distances RE = used throughout the text were determined from EFRET according to
| (5) |
with the radial probability distribution P(r) for a Gaussian chain (see (35–38) and Supporting Material) and
| (6) |
The Förster distance (R0) was determined to be 53 Å in 4 M GdmCl and 54 Å in PBS from ensemble absorbance and fluorescence data. The value R0 contains the orientation factor κ2 between donor emission dipole and acceptor excitation dipole, the overlap integral J(λ) between the donor emission spectrum and the acceptor absorbance spectrum, the refractive index n of the buffer, the Avogadro number NA, and the fluorescence quantum yield of the donor in the absence of acceptor (ΦD(0)). The value κ2 is 2/3 under the assumption of free dye rotation, which is valid based on the low anisotropies measured (39). J(λ) was calculated using a donor-only labeled Nup153FG mutant for the donor emission spectrum and the same mutant carrying both labels for the acceptor absorbance spectrum. The value ΦD(0) was determined to be 0.85 ± 0.03 and 0.91 ± 0.03 in PBS and 4 M GdmCl, respectively, based on the average quantum yield of all donor-only labeled mutants. Quantum yields were determined by the comparative method (40) using Rhodamine 123 (33) and Fluorescein (41) as quantum yield standards.
When necessary, the contribution of dye size and linker length to the protein backbone was approximated under consideration of bond lengths of the energy-minimized dye-linker-conformation using ChemBioOffice (CambridgeSoft, Cambridge, MA).
Based on Tcherkasskaya et al. (42) and Teraoka (43), the end-to-end distance RE, RC is given by
| (7) |
with b = 0.255 in good solvent and M = molecular weight, describing the distances of a relaxed coil behavior, which we use for normalization throughout the text.
We note that the center of the FRET histogram is determined by a Gaussian fit over the signal from several hundred single molecules, and thus very precise and reproducible. However, the conversion into distance is affected by multiple factors (quantum yield measurements, orientation of the dyes, etc.). Because the exact error contribution of, e.g., κ2 cannot be determined, the systematic uncertainty of the absolute distance lies in the few Å regime (31,39). To account for this, we performed our statistical analysis over all studied Nup153FG mutants.
Hydrogel formation and transport assays
Lyophilized Nup153FG wt was taken up in an aqueous solution of 0.1% trifluoroacetic acid to a final concentration of ∼200 mg/mL and placed onto a coverslip while still liquid. Gels were allowed to solidify overnight at room temperature and were equilibrated in measurement buffer (as above) for at least 2 h. Nup153FG hydrogels were imaged on a Leica SP5 microscope (Mannheim, Germany) with a 63× 1.2 NA water objective and localized by laser reflection in an xz-scan. Images of 512 × 512 pixels and a zoom factor 3 yielded a pixel size of 160 × 160 nm. The hydrogel/buffer interface was brought into focus and fluorescence images were taken with an xy-scan setting. Samples were excited at 488 nm with an Argon laser and at 561 nm with a DPSS laser. Fluorescence images were recorded in two channels, collecting light from 500 to 550 nm (green channel) and from 580 to 650 nm (red channel). Alexa 594-labeled Importin β and 70 kDa dextran (FITC-labeled) were added to the sample to a final concentration of 1 μM.
Results
Experimental strategy to study human Nup153
The Nup153 FG-domain is 601 AAs long and has an N-terminal part enriched in FxFG-repeats. The C-terminal domain largely consists of PxFG-repeats, while the middle domain has a less regular FG-repeat pattern. Fig. 1 b shows the overall FG composition of the Nup153 transport domain (Nup153FG) with color-coded FG-repeats and the distribution of AA charge, and N+Q residues along the sequence, revealing that the C-terminal PxFG domain is essentially uncharged but N+Q-rich. It is evident that the whole protein domain displays a heterogeneous, nonrandom AA composition. Common methods such as size exclusion chromatography (23), light scattering, and pulling on the whole domain by force microscopy techniques (17) typically yield the behavior of the whole domain. To overcome this limitation, we aimed to probe the behavior of different segments within the full-length transport domain, avoiding physical fragmentation of the protein chain that would preclude observing long distance effects, which can influence intrinsically disordered protein (IDP) function (44).
In the last few years single-molecule FRET (smFRET) has been demonstrated to be an extremely powerful tool to provide insights into the structure and architecture of IDPs (35–38,45,46) and to reliably report the length (RE) of an AA chain (for review, see (31,47)). A prerequisite to determine RE using smFRET is the introduction of a FRET dye pair site-specifically into the protein chain. The fluorescence donor (D) and acceptor (A) dyes need to be spaced at an appropriate distance because smFRET reports with sufficient sensitivity and precision on distances between 30 and 80 Å. This converts into suitable distances of ∼40–70 AA. Because FG-repeats are typically 10–20 AAs apart, this distance range allows us to zoom into small segments of the FG domain and to take a closer look at the subdomain structure (containing 1–3 FG-repeats, Fig. 1).
Labeling of Nup153 segments
Commonly, dual labeling of a protein is done by placing two cysteines into an otherwise cysteine-free protein. This only allows random labeling with D and A. To ensure maximal reproducibility and specificity in sample preparation we adapted a recently presented strategy by Brustad et al. (28). In this technology, one artificially mutated cysteine serves as a unique handle to install Alexa 594 (A) selectively via maleimide chemistry. For the other labeling site, we genetically encoded the unnatural amino-acid AcF, which serves as a unique linker to attach an Alexa 488 (D) hydroxylamine dye. We note that for all mutants planned for this work, this method successfully and reliably generated dual-labeled species, minimizing concerns from heterogeneous sample preparations or random labeling. We deliberately chose the Alexa 488/Alexa 594 dye pair, as it is a well-established dye pair in smFRET protein folding studies (35–38,46,48,49). The Förster distance for this dye pair is 54 Å (in measurement buffer), which corresponds to high sensitivity in the distance regime relevant for this study.
RE determination of C-terminal repeats under native conditions
First, the two PxFG containing double mutants (termed ε and ζ) were analyzed using multiparameter fluorescence detection (MFD) on the single molecule level. The simultaneous readout of D and A intensities, anisotropies (r), and donor dye lifetimes (τ) allows us to measure distances with high precision while circumventing potential sources of artifacts, such as from dye sticking or nonspecific quenching of the dyes (30,31). Fig. 2, a and b, shows the result for segment ε, while the raw data for segment ζ is shown in Fig. S2. In the τ-versus-EFRET histogram, two populations are clearly visible. The so-called zero-peak population centered at τD(0) = 3.5 ns and EFRET = 0 originates from those species where only donor signal was collected. This phenomenon is typical in single molecule measurements and has origins in photophysically inactive acceptor dye or imperfect labeling (29). The other species (centered at τDA = 1.44 ns, EFRET = 0.75) is due to resonance energy transfer from D to A, reducing the donor lifetime. These two populations are also resolved in the r-versus-τ histogram. The black line shows that the slightly increased anisotropy for the short lifetime species is in agreement with predictions from the Perrin equation. The Perrin equation relates the expected fluorescence lifetime to the rotational motion of the dyes, and deviations from the predicted line would reveal potential artifacts in the measurement (31).
Figure 2.
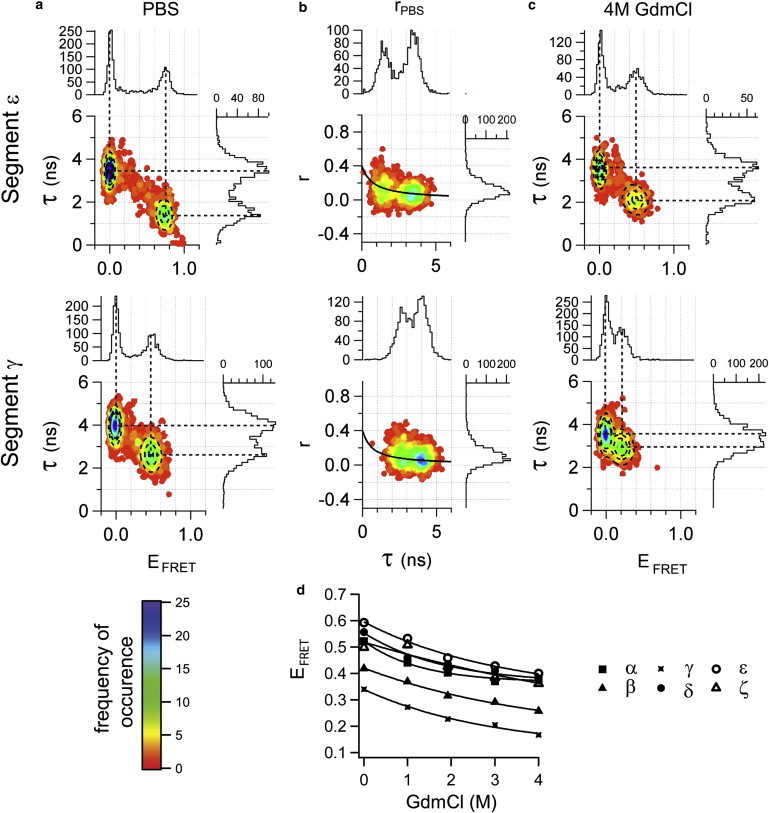
MFD smFRET analysis of the Nup153FG segments γ and ε. (a) Burst-integrated fluorescence-lifetime analysis of segment ε (upper row) and segment γ (lower row) under native conditions (PBS). The two-dimensional plots are color-coded for frequency of occurrence. (Top and right histograms) Maximum projections lifetime (τ) and EFRET. (Dashed circles) Result from two-dimensional Gaussian fits of both the 0-peak and the FRET-peak population. (Dashed lines) Centers of the fits to their position in the one-dimensional representation of EFRET and τ data. (b) Relationship between donor fluorescence anisotropy (r) and corresponding τ. (Solid line) Expected trend according to the Perrin equation. (c) Burst-integrated fluorescence-lifetime histograms analogous to (a) for segment ε and γ under unfolding conditions (4 M GdmCl). (d) EFRET from all experiments plotted as a function of GdmCl concentration for all mutants α to ζ. FRET efficiencies (from lifetime) in native conditions range from EFRET = 0.34 (segment γ) to 0.59 (segment ε) and decrease during denaturation to a final value of EFRET = 0.14 (segment γ) to 0.40 (segment ε) in 4 M GdmCl. (Solid lines) Result of monoexponential fits.
The simultaneous recording of fluorescence lifetimes and intensities also allows us to estimate the approximate end-to-end distances (RE) between the dye pairs and thus the approximate extension of the enclosed AA stretch. RE, RG (radius of gyration), and RH (hydrodynamic radius) are related measures for protein shape, and can be interconverted (43). We can thus compare our results to the prediction based on previous work by Tcherkasskaya et al. (42), who determined the dependence of RH on M for unfolded proteins that exhibit almost exclusively a relaxed coil behavior. As shown in Fig. 3 a, our measured distances are much shorter than expected for an IDP behaving like a relaxed coil (42,50). In fact, the distances are reminiscent of molten-globule or collapsed-coil behavior (23,42,51), showing that the protein stretches are compact in their native state.
Figure 3.
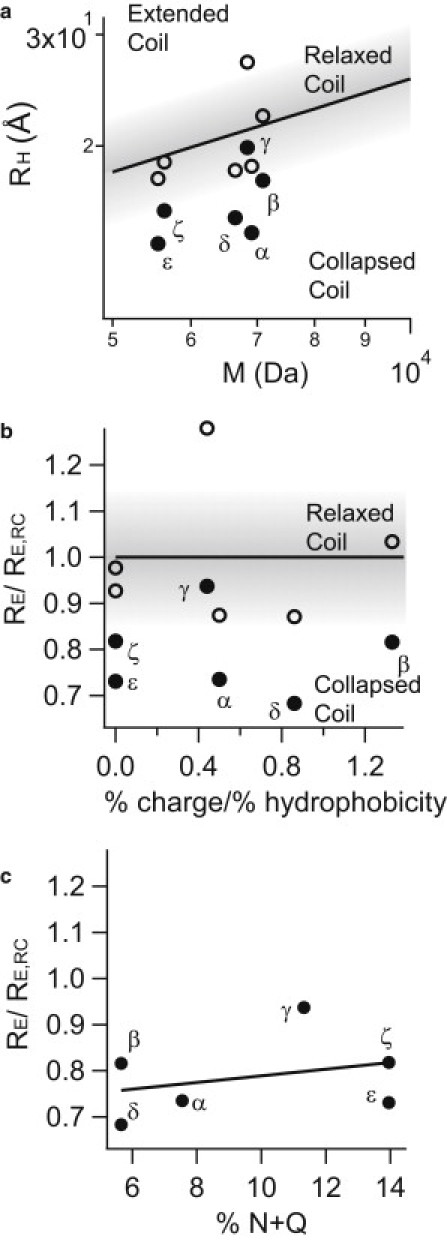
Comparison and classification of Nup153FG segments. (a) Dependence of RH on molecular weight (segment length). The predicted curve for a relaxed behavior of a polypeptide chain (solid representation) (42). The previously determined confidence interval for this relaxed coil behavior and thus the boundaries of collapsed and extended coil behavior are visualized by the shaded gradient. (b) Relation of RE to % charge/% hydrophobicity (analog to Yamada et al. (23)). Measured RE were related to the end-to-end distance predicted according to the relaxed coil model (RE,RC) to provide a segment-length-independent measure of distances. (Solid line) Position where a segment with ideal relaxed coil behavior would occur. (Shaded) Relaxed coil regions. (c) Dependence of RE/RE,RC from the content of N and Q within the probed segment. (Solid line) Linear fit through all data-points. (Solid circles) Distances measured under native conditions. (Open circles) Distances under unfolding conditions (4 M GdmCl).
RE determination of N-terminal repeats under native conditions
It is interesting to compare the RE results of the C-terminal region with other regions of the protein, a task our method is particularly suited to, as we can vary the labeling site while keeping all other experimental parameters (dyes, instrumentation, analysis) constant. We therefore designed four additional mutants following similar design criteria as before and measured their shape, i.e., the RE, using MFD smFRET. We focused on the N-terminal domain, which does not contain any PxFG-repeats but exclusively FxFG. The four tested mutants (Fig. 2, a and b, Fig. 3 a, and Fig. S2) show a strong collapsed state under native conditions with RE/RE,RC between 0.68 and 0.94. We further analyzed the data (FxFG and PxFG segments) with respect to % charged versus % hydrophobic AAs. High charge content could (but does not have to; see (36)) favor repulsive interactions within the protein sequence and thus a more extended conformation, while hydrophobicity could facilitate attractive interactions and therefore favor a more condensed conformation (23). As shown in Fig. 3 b, no substantial correlation between the measured RE/RE,RC and the % charge/% hydrophobicity ratio could be found (Pearson correlation coefficient, RP = −0.04), while only a minor trend depending on N+Q enrichment was found (RP = 0.31, Fig. 3 c). Despite the different characteristics of the N-terminal stretch, a similar phenotypic property (RE) of both the N-terminal and the C-terminal stretches was observed.
Relaxing the collapsed-coil behavior of Nup153
If an IDP is indeed natively collapsed, it must be possible to further denature it and relax its protein chain (38). Accordingly, we performed a GdmCl titration for all labeling mutants (see Fig. 2 c for mutant γ and ε and Fig. S2 for all other mutants). As shown in Fig. 2, c and d, EFRET substantially decreases with increasing GdmCl concentration. We note that MFD smFRET analysis allows us to correct photophysical effects from changing the environment (buffer) of the dyes and to exclude artifacts (31). Fig. 2 d shows that all segments undergo a similarly drastic expansion under unfolding conditions. A t-test analysis of the two populations including all our mutants (compact state versus extended state) reveals this effect to be significant with p < 0.0005. This also verifies that our technology has sufficient sensitivity to easily distinguish between collapsed and relaxed coil behavior. As another internal validation, we determined that the relaxed state in a high concentration of denaturant has the expected RH for a fully denatured protein (42,51) (Fig. 3 a). Within the resolution of the performed titration, the denaturation transition itself was continuous and absent of a multistate transition.
Nup153 FG can form a porelike permeability barrier
As IDPs lack strong structured elements and are intrinsically hard to study due to their high flexibility and dynamics, studying the volume occupied by the polypeptide chain has been a powerful tool to describe the basic properties of the protein/domain. However, in many IDP studies it is challenging and often impossible to link the observed behavior (collapsed coil, premolten globule, etc. (42,51)) to actual function. In the case of FG-Nups, Yamada et al. (23) recently suggested in a systematic analysis that yeast FG-Nups can be grouped into collapsed and extended FG-Nups. Notably, the N-terminal domain of Nsp1 was found to be natively collapsed while its C-terminal domain showed extended behavior (23). In a separate study by Ader et al. (21), a similar Nsp1 N-terminal domain, which is enriched in uncharged and N+Q residues, formed a cohesive hydrogel while the C-terminal domain did not. Also, the recent observation that yeast Nup49 can form a hydrogel (15) is in line with a potential link between collapsed-coil behavior and gel-forming propensity of yeast Nups (23). While yeast FG-Nups share little sequence similarity with human FG-Nups, it is fascinating to follow this logic for a moment. We observed that most segments we investigated in this study exist in a collapsed state. Furthermore, Nup153 is also N+Q-rich (Fig. 1, see Discussion and Conclusion).
We therefore tested the gel-forming propensity of Nup153FG following procedures that have previously been used to form yeast FG-repeat hydrogels (13–15). As shown in Fig. 4 a, Nup153FG indeed formed a hydrogel. The next step was to test whether this gel is functional in the sense that it mimics basic properties of the physiological NPC permeability barrier (14,15,52). A gel was mounted on a microscope and in Fig. 4 b, we show the results of a dual color fluorescence experiment. The large transport receptor Importin β (97 kDa) added to the surrounding buffer could easily enter the gel. The smaller dextran (70 kDa) added simultaneously did not considerably penetrate the gel. These findings demonstrate that highly concentrated human Nup153FG can also form a functional hydrogel in vitro.
Figure 4.
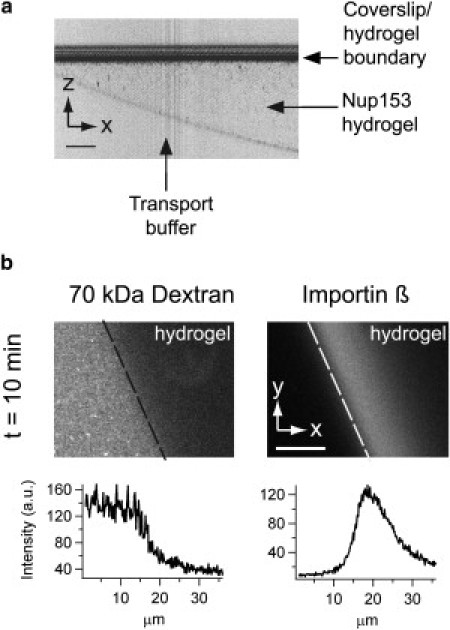
Functionality of Nup153FG hydrogels by fluorescence microscopy. (a) Laser-reflection image of a Nup153FG hydrogel allows positioning the gel before fluorescence image acquisition (inverted colors). (b) Selectivity of Nup153FG hydrogels. Spectrally separated confocal images of a 70 kDa dextran labeled with FITC and Importin β labeled with Alexa 594 were taken simultaneously. The hydrogel (located on the right side of the dashed line) shows Alexa 594 fluorescence (Importin β) in the hydrogel after 10 min, while FITC fluorescence (dextran) remains outside the hydrogel. (Lower panel) Corresponding fluorescence intensity analysis of FITC and Alexa 594 fluorescence. Scale bar is 10 μm.
Discussion and Conclusion
The high flexibility of FG-repeat nucleoporins make them challenging proteins to study with conventional methods. Most previously established techniques only provide a read-out of the behavior of the entire protein and do not provide direct insights into the behavior of individual regions within the context of the whole protein. Such sequence heterogeneity is particularly important for repeat proteins like Nup153, which, despite their lack of apparent structure, do not display completely random sequence architecture. We have used a labeling scheme that makes it possible to unambiguously study segments of individual proteins while leaving the whole protein intact. This also eliminates concerns that potential long-range effects are not preserved in our experiments. Although our method reliably produces site-specifically double-labeled samples, it does not yet fulfill the requirements of a high throughput method. We consequently limited our analysis to selected segments in the N- and C-terminal stretch of the domain, which mainly contain FxFG and PxFG-repeats, respectively. In the middle region, the FG-repeat composition is more complex and thus beyond the scope of this investigation.
Because IDPs cannot be described in terms of folded secondary structure, a major discriminator is polypeptide shape/extension and its relation to the primary AA sequence. In contrast to our initial expectation, and even though the overall AA composition in the N- and C-terminus differ substantially, we observed a similar collapsed coil behavior for all mutants under our measurement conditions. An analysis of RE (Fig. 3 b) with respect to % charge/% hydrophobicity did not reveal substantial differences between the segments (RP = −0.04), and the N-terminal segments of the protein showed collapsed behavior despite their charge content (23). Analysis of RE with respect to enrichment of N and Q in the segments (Fig. 3 c) revealed a minor trend (RP = 0.31). N and Q residues are enriched in many other IDPs, particularly those involved in aggregation via β-sheet formation (21,26), and they are possibly important for supramolecular hydrogel formation (21).
A computational study showed that high proline content in IDPs can be correlated with increased hydrodynamic radii (53). As shown in the Supporting Material we find that collapsed-coil behavior does not depend substantially (RP = 0.13) on proline content for our tested segments (Fig. S3). Recently, Cheng et al. (25) have shown by molecular dynamics (also explicitly for FG-repeat Nups) that proline content is not necessarily the only discriminating factor for chain extension, in agreement with our experimental observation. Prolines can facilitate sharp turns in secondary structure elements like β-hairpins. Stable secondary structure is often evidenced by a two- or multistate transition in the protein denaturation curve (28). Within the resolution of the experiment, we found a continuous decay from the natively collapsed to the denatured state (Fig. 2 d) for all our labeling mutants, indicating that stable secondary structure is absent or too transient to be detected.
Taken together, our results indicate that similar properties (dimension/shape/RE) can be achieved despite different AA composition or FG-type. Our denaturation experiments unambiguously verify that differences between relaxed and collapsed coil behavior (p < 0.0005) can be easily distinguished with our method and that in the native state the dominant property of all tested segments is the collapsed state. For IDPs, no general functional description between shape and function has yet been made, and it is a long term goal of the whole field to establish such concepts (50). Already taking the results from the very few different studies together, there seems to be overlap between those yeast FG-Nups (approximately three) that are natively collapsed and can form a hydrogel (13,15,23). Stimulated by this idea, and by the fact that the tested Nup153 segments showed some form of collapsed-coil behavior, we investigated whether the whole Nup153FG could also form a hydrogel, despite its central role for the entropic barrier model (16,17). Indeed, we were able to successfully verify that Nup153FG can form hydrogels with physiological transport properties (i.e., exclusion of cargo but easy penetration by large transport proteins). This furthermore demonstrates that not only yeast, but also human FG-Nups can have functional gel-forming propensities, indicating a conservation of biophysical properties of at least some FG-Nups across distant species.
While our study does not yet allow us to weigh the relative importance of N- versus C-terminal fragments for the gelation properties of the full-length domain, further experiments using this technology—ideally nuclear-pore proteome-wide—can help to establish relationships between supramolecular and molecular properties of FG-repeat proteins. Taking the high sequence variability of FG-Nups (which is typical for IDPs (50)) into account, studying segment properties within full-length proteins can also assist to decode the high redundancies of FG-Nups in the transport pathway (18).
Currently, a comparable study to the one by Yamada et al. (23) is absent for human nucleoporins, which have very little sequence similarity with yeast nucleoporins, and vary dramatically in the occurrence of FG-repeat types (e.g., GFLG is less frequent in humans). It is important to note that supramolecular hydrogel formation of proteins in general is still a poorly understood process, as for example made clear by a systematic study of aggregation properties of short 3–10-mer polypeptides (54). Thus, strict design rules even for general gel formation of proteins do not yet exist (and certainly not for functional hydrogels). Taken together, the few studies—including this one—on FG-repeat IDPs clearly indicate that a single parameter is unlikely to be the only discriminating factor to determine whether functional supramolecular hydrogel formation can occur, and that this is a much higher complexity issue (21,23,24). Indeed, it is evident that straightforward relationships between segment composition and general polymer properties are already difficult to establish (25,36,53,55). However, our study indicates that the presented technology can assist in probing properties beyond the average behavior of the protein and in linking the results to macromolecular properties. As there is compelling evidence that Nup153 can also contribute to an entropic brush under different experimental conditions (immobilization of densely packed proteins on a surface (16,17)), we believe that our data revealed additional properties of the protein. Rather than exclusively exhibiting entropic brush or gel-behavior, proteins like Nup153 can show a more multifaceted character.
This study was limited to Nup153 on the single molecule level, thus providing an unperturbed view on the biophysical properties of segments of a single nucleoporin. In the pore, the sum and potentially even geometrical constraints of FG-repeat Nups give rise to the permeability barrier. It is particularly important to understand the properties of Nup153 ranging from the single molecule level to the network level, as it is known that Nup153 also shuttles away from the pore to perform unknown functions (56). Therefore, within the cell, Nup153 experiences a large difference in FG-concentrations ranging from very low away from the NPC to higher, depending on its location within the pore (center, rim, etc.) (4). One can easily imagine that these extremely varied conditions trigger fundamentally different behavior of one and the same protein. Our segmental analysis within the context of the full-length domain permits unique insights into the subdomain properties of this protein, and thus opens a new strategy to study nuclear pore architecture.
Acknowledgments
We are grateful for the help of C. Koehler in sample preparation and N. Banterle with programming. We very much appreciate the helpful discussion and critical proofreading by Drs. Beck and VanDelinder.
This work was funded by the Emmy Noether program of the Deutsche Forschungsgemeinschaft and a fellowship of the Boehringer Ingelheim Fonds to S.M.. We also acknowledge the support granted by the European Molecular Biology Laboratory Advanced Light Microscopy Facility and Proteomics core facilities.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Alber F., Dokudovskaya S., Rout M.P. The molecular architecture of the nuclear pore complex. Nature. 2007;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- 2.Beck M., Lucić V., Medalia O. Snapshots of nuclear pore complexes in action captured by cryo-electron tomography. Nature. 2007;449:611–615. doi: 10.1038/nature06170. [DOI] [PubMed] [Google Scholar]
- 3.D'Angelo M.A., Hetzer M.W. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol. 2008;18:456–466. doi: 10.1016/j.tcb.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim R.Y., Ullman K.S., Fahrenkrog B. Biology and biophysics of the nuclear pore complex and its components. Int .Rev. Cell Mol. Biol. 2008;267:299–342. doi: 10.1016/S1937-6448(08)00632-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wente S.R., Rout M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010;2:a000562. doi: 10.1101/cshperspect.a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denning D.P., Patel S.S., Rexach M. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc. Natl. Acad. Sci. USA. 2003;100:2450–2455. doi: 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen N.P., Patel S.S., Rexach M. Deciphering networks of protein interactions at the nuclear pore complex. Mol. Cell. Proteomics. 2002;1:930–946. doi: 10.1074/mcp.t200012-mcp200. [DOI] [PubMed] [Google Scholar]
- 8.Patel S.S., Belmont B.J., Rexach M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell. 2007;129:83–96. doi: 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 9.Cook A., Bono F., Conti E. Structural biology of nucleocytoplasmic transport. Annu. Rev. Biochem. 2007;76:647–671. doi: 10.1146/annurev.biochem.76.052705.161529. [DOI] [PubMed] [Google Scholar]
- 10.Wälde S., Kehlenbach R.H. The part and the whole: functions of nucleoporins in nucleocytoplasmic transport. Trends Cell Biol. 2010;20:461–469. doi: 10.1016/j.tcb.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Ribbeck K., Görlich D. Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 2001;20:1320–1330. doi: 10.1093/emboj/20.6.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ribbeck K., Görlich D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J. 2002;21:2664–2671. doi: 10.1093/emboj/21.11.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frey S., Richter R.P., Görlich D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science. 2006;314:815–817. doi: 10.1126/science.1132516. [DOI] [PubMed] [Google Scholar]
- 14.Frey S., Görlich D. A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell. 2007;130:512–523. doi: 10.1016/j.cell.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 15.Frey S., Görlich D. FG/FxFG as well as GLFG repeats form a selective permeability barrier with self-healing properties. EMBO J. 2009;28:2554–2567. doi: 10.1038/emboj.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim R.Y., Fahrenkrog B., Aebi U. Nanomechanical basis of selective gating by the nuclear pore complex. Science. 2007;318:640–643. doi: 10.1126/science.1145980. [DOI] [PubMed] [Google Scholar]
- 17.Lim R.Y., Huang N.P., Aebi U. Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA. 2006;103:9512–9517. doi: 10.1073/pnas.0603521103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strawn L.A., Shen T., Wente S.R. Minimal nuclear pore complexes define FG repeat domains essential for transport. Nat. Cell Biol. 2004;6:197–206. doi: 10.1038/ncb1097. [DOI] [PubMed] [Google Scholar]
- 19.Denning D.P., Rexach M.F. Rapid evolution exposes the boundaries of domain structure and function in natively unfolded FG nucleoporins. Mol. Cell. Proteomics. 2007;6:272–282. doi: 10.1074/mcp.M600309-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.DeGrasse J.A., DuBois K.N., Chait B.T. Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol. Cell. Proteomics. 2009;8:2119–2130. doi: 10.1074/mcp.M900038-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ader C., Frey S., Baldus M. Amyloid-like interactions within nucleoporin FG hydrogels. Proc. Natl. Acad. Sci. USA. 2010;107:6281–6285. doi: 10.1073/pnas.0910163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnan V.V., Lau E.Y., Rexach M.F. Intramolecular cohesion of coils mediated by phenylalanine-glycine motifs in the natively unfolded domain of a nucleoporin. PLOS Comput. Biol. 2008;4:e1000145. doi: 10.1371/journal.pcbi.1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamada J., Phillips J.L., Rexach M.F. A bimodal distribution of two distinct categories of intrinsically disordered structures with separate functions in FG nucleoporins. Mol. Cell. Proteomics. 2010;9:2205–2224. doi: 10.1074/mcp.M000035-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dölker N., Zachariae U., Grubmüller H. Hydrophilic linkers and polar contacts affect aggregation of FG repeat peptides. Biophys. J. 2010;98:2653–2661. doi: 10.1016/j.bpj.2010.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng S., Cetinkaya M., Gräter F. How sequence determines elasticity of disordered proteins. Biophys. J. 2010;99:3863–3869. doi: 10.1016/j.bpj.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alberti S., Halfmann R., Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemke E.A. Site-specific labeling of proteins for single-molecule FRET measurements using genetically encoded ketone functionalities. Methods Mol. Biol. 2011;751:3–15. doi: 10.1007/978-1-61779-151-2_1. [DOI] [PubMed] [Google Scholar]
- 28.Brustad E.M., Lemke E.A., Deniz A.A. A general and efficient method for the site-specific dual-labeling of proteins for single molecule fluorescence resonance energy transfer. J. Am. Chem. Soc. 2008;130:17664–17665. doi: 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemke E.A., Gambin Y., Deniz A.A. Microfluidic device for single-molecule experiments with enhanced photostability. J. Am. Chem. Soc. 2009;131:13610–13612. doi: 10.1021/ja9027023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eggeling C., Berger S., Seidel C.A. Data registration and selective single-molecule analysis using multi-parameter fluorescence detection. J. Biotechnol. 2001;86:163–180. doi: 10.1016/s0168-1656(00)00412-0. [DOI] [PubMed] [Google Scholar]
- 31.Sisamakis E., Valeri A., Seidel C.A. Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 2010;475:455–514. doi: 10.1016/S0076-6879(10)75018-7. [DOI] [PubMed] [Google Scholar]
- 32.Koshioka M., Sasaki K., Masuhara H. Time-dependent fluorescence depolarization analysis in three-dimensional microspectroscopy. Appl. Spectrosc. 1995;49:224–228. [Google Scholar]
- 33.Schaffer J., Volkmer A., Seidel C.A.M. Identification of single molecules in aqueous solution by time-resolved fluorescence anisotropy. J. Phys. Chem. A. 1999;103:331–336. [Google Scholar]
- 34.Roy R., Hohng S., Ha T. A practical guide to single-molecule FRET. Nat. Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffmann A., Kane A., Schuler B. Mapping protein collapse with single-molecule fluorescence and kinetic synchrotron radiation circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA. 2007;104:105–110. doi: 10.1073/pnas.0604353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller-Späth S., Soranno A., Schuler B. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA. 2010;107:14609–14614. doi: 10.1073/pnas.1001743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nettels D., Müller-Späth S., Schuler B. Single-molecule spectroscopy of the temperature-induced collapse of unfolded proteins. Proc. Natl. Acad. Sci. USA. 2009;106:20740–20745. doi: 10.1073/pnas.0900622106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyay S., Krishnan R., Deniz A.A. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. USA. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iqbal A., Arslan S., Lilley D.M. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nucleic acids. Proc. Natl. Acad. Sci. USA. 2008;105:11176–11181. doi: 10.1073/pnas.0801707105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams A.T.R., Winfield S.A., Miller J.N. Relative fluorescence quantum yields using a computer-controlled luminescence spectrometer. Analyst (Lond.) 1983;108:1067–1071. [Google Scholar]
- 41.Brannon J.H., Magde D. Absolute quantum yield determination by thermal blooming fluorescein. J. Phys. Chem. 1978;82:705–709. [Google Scholar]
- 42.Tcherkasskaya O., Davidson E.A., Uversky V.N. Biophysical constraints for protein structure prediction. J. Proteome Res. 2003;2:37–42. doi: 10.1021/pr025552q. [DOI] [PubMed] [Google Scholar]
- 43.Teraoka I. Wiley; New York: 2002. Polymer Solutions: An Introduction to Physical Properties. 338. [Google Scholar]
- 44.Brocca S., Testa L., Grandori R. Compaction properties of an intrinsically disordered protein: Sic1 and its kinase-inhibitor domain. Biophys. J. 2011;100:2243–2252. doi: 10.1016/j.bpj.2011.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Best R.B., Merchant K.A., Eaton W.A. Effect of flexibility and cis residues in single-molecule FRET studies of polyproline. Proc. Natl. Acad. Sci. USA. 2007;104:18964–18969. doi: 10.1073/pnas.0709567104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferreon A.C., Gambin Y., Deniz A.A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deniz A.A., Mukhopadhyay S., Lemke E.A. Single-molecule biophysics: at the interface of biology, physics and chemistry. J. R. Soc. Interface. 2008;5:15–45. doi: 10.1098/rsif.2007.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung H.S., Gopich I.V., Eaton W.A. Extracting rate coefficients from single-molecule photon trajectories and FRET efficiency histograms for a fast-folding protein. J. Phys. Chem. A. 2011;115:3642–3656. doi: 10.1021/jp1009669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merchant K.A., Best R.B., Eaton W.A. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. USA. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tompa P. Chapman & Hall/CRC Press; Boca Raton, FL: 2009. Structure and Function of Intrinsically Disordered Proteins. 331. [Google Scholar]
- 51.Kohn J.E., Millett I.S., Plaxco K.W. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mohr D., Frey S., Görlich D. Characterization of the passive permeability barrier of nuclear pore complexes. EMBO J. 2009;28:2541–2553. doi: 10.1038/emboj.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marsh J.A., Forman-Kay J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010;98:2383–2390. doi: 10.1016/j.bpj.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hauser C.A., Deng R., Hauser U.A. Natural tri- to hexapeptides self-assemble in water to amyloid β-type fiber aggregates by unexpected α-helical intermediate structures. Proc. Natl. Acad. Sci. USA. 2011;108:1361–1366. doi: 10.1073/pnas.1014796108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mao A.H., Crick S.L., Pappu R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA. 2010;107:8183–8188. doi: 10.1073/pnas.0911107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rabut G., Doye V., Ellenberg J. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat. Cell Biol. 2004;6:1114–1121. doi: 10.1038/ncb1184. [DOI] [PubMed] [Google Scholar]
- 57.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 58.Gopich I.V., Szabo A. Single-macromolecule fluorescence resonance energy transfer and free-energy profiles. J. Phys. Chem. B. 2003;107:5058–5063. [Google Scholar]
- 59.Laurence T.A., Kong X., Weiss S. Probing structural heterogeneities and fluctuations of nucleic acids and denatured proteins. Proc. Natl. Acad. Sci. USA. 2005;102:17348–17353. doi: 10.1073/pnas.0508584102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.