

Abstract
Neuroserpin is a regulator of neuronal growth and plasticity. Like other members of the serpin family, neuroserpin undergoes a large conformational change as part of its function. Unlike other serpins such as α1-antitrypsin, wild-type neuroserpin will polymerize under near-physiological conditions, and will spontaneously transition to the latent state. To probe the origins of this conformational lability, we have performed hydrogen exchange measurements and molecular-dynamics simulations on human neuroserpin. Hydrogen exchange indicates that neuroserpin has greater flexibility in the breach region and in β-strand 1C compared with α1-antitrypsin. Molecular-dynamics simulations show that the distance between the top of β-strands 3 and 5A averages 4.6 Å but becomes as large as 7.5 Å in neuroserpin while it remains stable at ∼3.5 Å in α1-antitrypsin. Further simulations show that the stabilizing S340A mutation suppresses these fluctuations in neuroserpin. The first principal component calculated from the simulations shows a movement of helix F away from the face of β-sheet A in neuroserpin while no such movement is evident in α1-antitrypsin. The increased mobility of these regions in neuroserpin relative to α1-antitrypsin provides a basis for neuroserpin's increased tendency toward the formation of polymers and/or the latent state.
Introduction
Neuroserpin is a member of the serpin family of protease inhibitors (1). Inhibitory serpins are unusual in that they do not fold to the lowest free energy conformation, but are instead trapped in a metastable state in which β-strand 4A is exposed to solvent as a flexible loop. Upon cleavage by a target protease, the reactive center loop (RCL) spontaneously inserts into the central β-sheet (sheet A) becoming a sixth strand and in the process translocating the covalently bound protease ∼70 Å and trapping it in an inactive state (2). This transition is accompanied by a large increase in stability (3). Because serpins adopt a strained metastable conformation, they are prone to inappropriate conformational changes that can be triggered both by mutations and by mild environmental perturbations (4). These inappropriate conformational changes frequently lead to polymers, which have been linked to a number of pathologies. In the case of neuroserpin, misfolding and polymerization of pathological mutants has been identified as the molecular basis of familial encephalopathy with neuroserpin inclusion bodies (5). In addition, wild-type neuroserpin is marginally stable and will form polymers under near physiological conditions (6).
The structure of human neuroserpin in the metastable form (3FGQ) (7) is shown in Fig. 1. Neuroserpin has the canonical serpin fold, consisting of three β-sheets, A, B, and C, and nine α-helices, A–I. As in other serpins, the RCL is exposed to solvent and available for cleavage by target proteases. In addition, neuroserpin contains one unique structural feature, the Ω-loop between β-strands 1 and 2 B. The Ω-loop has been shown to contribute to the inhibition of tissue plasminogen activator (tPA) (7).
Figure 1.
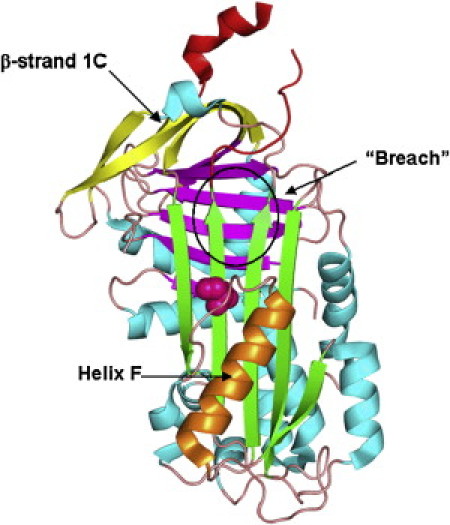
The structure of human neuroserpin (3FGQ). (Green, magenta, and yellow) β-sheets A, B, and C, respectively. (Red) RCL; (orange) helix F. S340 shown as magenta spheres.
Previously, the structural distribution of native state stability and conformational dynamics in the canonical serpin α1-antitrypsin (α1-AT) was probed using hydrogen/deuterium exchange and mass spectrometry (HXMS) (8). HXMS exploits the fact that backbone amide hydrogens in proteins will readily exchange with deuterium when a protein is incubated in D2O. Amide hydrogens that are buried in the protein interior or involved in stable hydrogen bonds are resistant to exchange, and structural fluctuations which transiently expose them to solvent are required in order to allow exchange with deuterium to occur. Thus, amide hydrogens in stable rigid regions will show slow rates of exchange while those located in unstable or flexible regions will exchange rapidly. HXMS thus reveals the distribution of conformational flexibility in proteins through the local rates of H/D exchange (9).
In α1-AT, it was found that β-sheet B, the center of β-sheet A, and helices B and C form the stable slow exchanging core of the molecule. Much of β-sheet C was also found to be stable; in particular, there were stable interactions between β-strands 1 and 2 C. This interaction is critical because it must be broken both to form polymers and to form the latent state (10,11). In contrast, an unexpected degree of flexibility was seen in helices A and F. It was speculated that the flexibility of helix F may contribute to inhibitory efficiency, as this helix must undergo large motions during the inhibitory conformational change.
We employed HXMS and molecular-dynamics (MD) simulations to study the conformational dynamics of human neuroserpin and compare them with α1-AT. One question to be addressed is whether the serpins' metastability and unusual inhibitory mechanism imposes constraints on their conformational flexibility. Conservation of flexibility or rigidity in certain regions may indicate that flexibility/rigidity in these regions is required for function, and would thus shed light on the mechanism of the inhibitory conformational change. We also wish to determine whether differences in local conformational flexibility can explain the differences between the biochemical behavior of neuroserpin and other serpins. Unlike most serpins, wild-type neuroserpin readily form polymers under near-physiological conditions (12). Additionally, unlike α1-AT but similar to plasminogen activator inhibitor (PAI-1), neuroserpin will spontaneously transition to the latent state, in which RCL insertion into β-sheet A occurs without proteolytic cleavage (12).
Materials and Methods
Expression and purification of human neuroserpin
The purification of the wild-type neuroserpin was done following the previous protocol reported by Belorgey et al. (12) with some modifications. In brief, the plasmid containing the wild-type neuroserpin gene was kindly provided by Dr. David A. Lomas (Respiratory Medicine Unit, Department of Medicine, University of Cambridge, Cambridge, UK). Escherichia coli BL21 (DE3) were used for expression. All chemicals were obtained from Sigma (St. Louis, MO) unless otherwise specified. Initially, cells were cultured at 37°C for ∼3 h until the A600 reached to 0.7–1.2, followed by induction with 1 mM isopropyl-β-D-1-thiogalactopyranoside and the culture was transferred to 30°C for further 16–18 h. The cells were harvested by centrifugation, resuspended in cold Buffer A (50 mM Na2HPO4, 10 mM imidazole, 500 mM NaCl, pH 7.8) with protease inhibitor cocktail and 0.5 mM phenylmethylsulfonyl fluoride and disrupted by sonication. The disrupted cell suspension was further centrifuged at 12,000g for 15 min and the supernatant was collected. The pellet was again suspended in Buffer A and centrifuged again, which was repeated twice and then the pooled supernatants were incubated with Ni-NTA agarose (Qiagen, Germantown, MD) at 4°C for 60 min.
The protein-bound Ni-NTA agarose was washed thoroughly with Buffer A and packed in column to perform the fast protein liquid chromatography purification. The column was washed three times with 20 mM Na2HPO4, pH 7.8. Finally, the protein was eluted with the 20 mM Na2HPO4, pH 7.8 buffer containing 250 mM imidazole. The fractions containing recombinant neuroserpin were pooled, diluted fourfold and the fractions were buffer-exchanged into Buffer B (20 mM Tris-HCl, pH 7.4, 20 mM NaCl) to remove the imidazole. This fraction was loaded on Hitrap Q HP (GE Healthcare Life Sciences, Waukesha, WI) and washed with Buffer B and bound neuroserpin was eluted with the linear gradient (0–1) M of Buffer C (Buffer B with 1 M NaCl). At the end, the eluted fraction was buffer-exchanged with Buffer D (10 mM Sodium Phosphate, 50 mM NaCl, pH 7.5) and concentrated using Amicon Ultra-15 (Millipore, Billerica, MA) and Amicon Ultra-0.5 (Millipore). Activity assay was performed using the previously reported protocol by Belorgey et al. (12). All inhibitory reactions were performed at 25°C in the inhibition buffer (50 mM HEPES, 150 mM NaCl, 0.01% w/v dodecyl-maltoside (Sigma), pH 7.4) by adding 20 nM tPA (Calbiochem, San Diego, CA) to a mixture of neuroserpin of different concentrations (from 200 to 1000 nM) and 1 mM substrate S-2288 (Chromogenix, Milano, Italy). The release of p-nitroanilide in every reaction was recorded at 410 nm as a function of time and the inhibitory activity of the purified neuroserpin was verified.
Hydrogen/deuterium exchange and mass spectrometry
Neuroserpin was diluted 25-fold into D2O buffer (10 mM sodium phosphate, 50 mM NaCl, pD 7.8, and incubated at 25°C for 10, 50, 100, 500, 1000, 2000, or 3000 s. Exchange was then quenched by adding an equal volume of ice-cold 100 mM sodium phosphate, pH 2.5 and placing the samples on ice. Porcine pepsin was added to a 1:1 (m/m) ratio and digestion proceeded for 5 min. The digest was loaded on to a microbore C18 column and eluted at 50 μL/min directly into an LTQ XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) equipped with an ESI course. A linear acetonitrile-water gradient of 10–45% over 15 min was employed and the column, injector and connection lines were immersed in an ice bath throughout. Peaks were identified manually and centroid masses were calculated using the software MagTran (AmGen, Thousand Oaks, CA). A fully deuterated reference sample was prepared and used to correct for deuterium loss during sample handling as described previously (8). Three repeat measurements of the fully deuterated reference sample indicate that the average experimental variability is ±0.5 deuteria. Before performing H/D exchange measurements, the peptides resulting from peptic digestion were sequenced using tandem mass spectrometry. A digest of undeuterated neuroserpin was eluted into an LTQ XL mass spectrometer using an acetonitrile-water gradient of 10–45% over 60 min and 45–98% over 30 min and peptides were subjected to collision-induced dissociation. The sequencing data was analyzed using the SEQUEST algorithm in the BioWorks software package (Thermo Electron, Waltham, MA).
MD simulations and analysis
MD simulations for neuroserpin and α1-AT were conducted using NAMD v 2.7 (13) and the CHARMM27 force field (14) with the CMAP correction (15,16) and TIP3P water (17). Simulations were performed using particle-mesh Ewald electrostatics and periodic boundary conditions. The starting structures were 1QLP (α1-AT) and 3FGQ (neuroserpin). The missing residues in the RCL in 3FGQ were modeled in using the ModLoop server (18). Starting structures for the S340A and S49P mutants of neuroserpin were generated by in silico mutagenesis. For each protein the solvated, energy-minimized system was heated to 310 K in 10° steps, simulations were run for 50 ns and the last 45 ns were used for analysis. In the case of S49P, simulations were run for 35 ns and the last 30 ns were used for analysis. Root mean-square fluctuations (RMSF), residue interaction energy networks, and distance measurements were performed using the software WORDOM (19). Hydrogen-bond occupancies were calculated using VMD (20). Principal component analysis (PCA) was performed and PCA projections and animations were prepared using the software package ProDy (21). Images of protein structures were generated using PyMol (22).
Results
Hydrogen/deuterium exchange
Deuterium uptake versus time curves for two representative peptides covering residues 202–211, located in β-strand 4C, and residues 299–314, encompassing helix I and a portion of β-strand 6A, are shown in Fig. 2. A map of our sequence coverage and normalized deuterium versus time curves for all 26 peptic fragments employed in this study are given in Fig. S1 and Fig. S2 (see the Supporting Material). Residues 202–211 contain five residues in β-strands and four residues in a surface-exposed loop. At 3000 s, ∼4 amide hydrogens (∼47% of the total) have exchanged with deuterium. These likely correspond to the four residues in the loop. The β-strands in this region are therefore highly stable and resistant to exchange. The fact that all four loop residues do not exchange immediately suggests that the two intramain chain hydrogen bonds seen in the crystal structure are moderately stable in solution. In contrast, of the 15 exchangeable amide hydrogens in residues 299–314, only two remain unexchanged after 3000 s despite the fact that the majority of residues in this region are located in regular secondary structure. In fact, six residues undergo exchange with 10 s (Fig. 2), indicating that at least some of the secondary structure seen in the crystal structure is unfolded in solution. This region is therefore unstable and highly dynamic.
Figure 2.
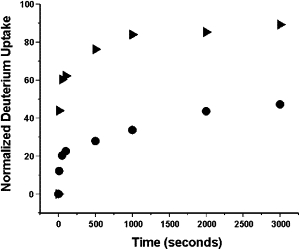
Normalized deuterium versus time curves for two representative peptic fragments of neuroserpin covering residues 202–211 (circles) and 299–314 (triangles).
Results for all peptic fragments identified in this study are mapped onto the crystal structure of human neuroserpin (3FGQ) in Fig. 3. As in α1-AT, β-sheets B and C and α-helices G and H are slow-exchanging, indicating structural rigidity. Also as in α1-AT, α-helix D and much of helix A are highly flexible. There are, however, several notable differences in the H/D exchange patterns of neuroserpin and α1-AT. Two regions of particular interest are the breach region at the top of β-strands 3 and 5A and β-strand 1C. The peptide containing β-strand 1C includes residues 359–378 and contains three residues in β-strand 1C, three residues in β-strand 4B, and 13 residues in loops and turns. Eight of these residues exchange immediately and are likely located in the solvent-exposed RCL. A further seven exchange during the subsequent incubation in D2O, and by 3000 s only three amide hydrogens remain unexchanged. While it is not possible to identify the location of these three stable amide hydrogens, it is reasonable to argue that they are much more likely to be located in the deeply buried and fully hydrogen-bonded β-strand 4B than in the surface-exposed β-strand 1C. Thus, H/D exchange suggests that β-strand 1C is labile to exchange on timescales of thousands of seconds, in contrast to α1-AT (8). Residues 185–201 include four residues in β-strand 3A, two residues in β-strand 4C along with a single turn of helix, and the remaining residues in loops and turns. Despite the six residues involved in β-sheets, this peptide reaches ∼100% exchange at 3000 s. This indicates that both β-strands spanned by these residues are flexible, in contrast to the rigidity seen in α1-AT (8).
Figure 3.
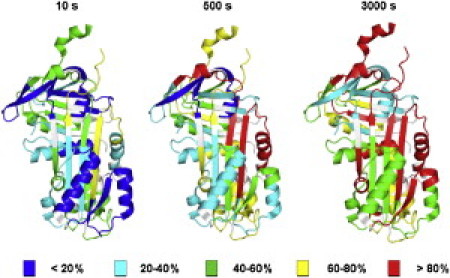
Distribution of H/D exchange rates in neuroserpin. Normalized deuterium levels at 10, 500, and 3000 s mapped onto the structure of neuroserpin.
MD simulations
Residue fluctuations
All-atom MD simulations on neuroserpin and α1-AT were run for 50 ns and the last 45 ns were used for analysis. RMSF per residue for both proteins are shown in Fig. 4. Helices C and D are both more mobile in neuroserpin than in α1-AT, as is the top of helix F. The RCL of neuroserpin is less mobile than that of α1-AT and in fact the Ω-loop is more mobile than the RCL in neuroserpin. The helical stretch in the RCL that is present in the crystal structure of neuroserpin unravels early in the simulations, consistent with the supposition that it is stabilized by crystal contacts (7). While most serpins have an alanine residue at position 340, neuroserpin has a serine, and it has been shown that mutation of the serine to the more common alanine results in both increased thermal stability and resistance to polymerization. To further explore the connection between local conformational lability and polymerization, we generated the S340A mutant of neuroserpin in silico and performed MD simulations. In terms of RMSF, S340A shows relatively minor differences from wild-type. S340A has reduced fluctuations in helices C and D and increased fluctuations in the loop between helix A and β-strand 6B, the Ω-loop, and the C-terminal portion of the loop connecting helix F to β-strand 3A. The S49P mutation likewise has minor effects on the average RMSF, although there is a significant increase in the RMSF of residues 170–176 in the loop connecting the F-helix to β-strand 3A.
Figure 4.
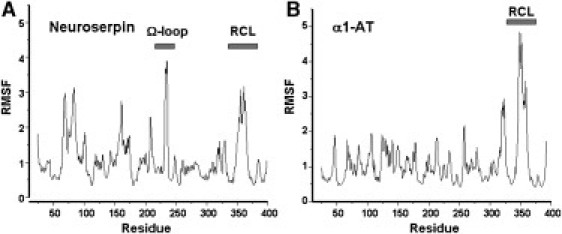
Fluctuations from MD simulations. Alpha-carbon RMSFs for (A) neuroserpin and (B) α1-antitrypsin calculated from MD simulations. For each protein the two N-terminal and C-terminal residues, which are highly flexible, have been omitted for ease of visualization.
Interaction energy networks
Residue interaction energy networks were calculated according to the method of Vijayabaskar and Vishveshwara (23) as implemented in the program WORDOM (19). It has been argued that identifying networks of strongly interacting residues as a function of an interaction cutoff energy can reveal stabilization centers in proteins that may be related to folding or function (23). The residue interaction energy networks calculated from the simulations of neuroserpin and α1-AT reveal striking differences. Fig. 5 shows interaction energy networks for both molecules identified using a cutoff of −2.5 kcal/mol (results are qualitatively similar for somewhat higher or lower cutoffs). The clusters represent connected networks of residues that interact with a favorable energy of at least −2.5 kcal/mol during at least 50% of the simulation.
Figure 5.
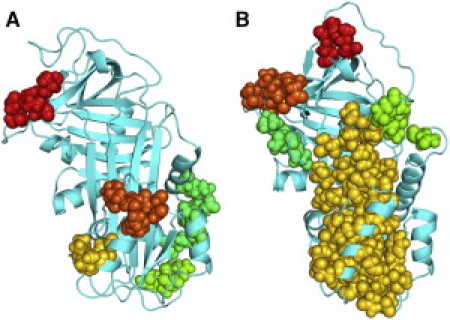
Clusters of strongly interacting residues identified from MD simulations. Residue interaction energy networks (see text) determined from MD simulations for (A) neuroserpin and (B) α1-antitrypsin. (Colored spheres) Networks of strongly interacting residues.
The most notable difference is that α1-AT has a large network of strongly interacting residues that encompasses nearly the whole of β-sheet A and much of helix F. Such a network is completely absent in neuroserpin, although neuroserpin does show two clusters of strongly interacting residues within helix F. Another noteworthy difference is that in α1-AT, two residues flanking β-strand 1C, Pro361 and Asn367, belong to strongly interacting clusters of residues located in β-strands 2C and 4C, respectively. Again, no such networks of strongly interacting residues are seen in neuroserpin. The S340A mutation does not result in a network of strongly interacting residues in β-sheet A but, interestingly, it does result in a strongly interacting network of residues in β-sheet C including interactions of residues Gln369 and Ile371 on β-strand 1C with Glu289 and Tyr291 on β-strand 2C. No significant changes in interaction networks are seen in the S49P mutant.
Fluctuations in the breach and shutter regions
To compare the relative mobilities of the breach regions in both neuroserpin and α1-AT, the distance between the α-carbon of residue 189 and the oxygen of residue 337 in α1-AT and between the α-carbon of residue 187 and the oxygen of residue 343 in neuroserpin were calculated over the entire simulation. These pairs of residues occupy analogous positions near the top of β-strands 3 and 5A in both proteins. In Fig. 6 a it is clear that while in α1-AT the distance between the two strands remains stable at ∼3.5 Å, in neuroserpin there is considerably greater mobility. Not only is the distance between the tops of β-strands 3 and 5A larger on average, but there are large fluctuations during which the distance becomes as much as 7.5 Å. As an additional test of the linkage between fluctuations in the breach region and polymerization, the distance between the α-carbon of residue 187 and the oxygen of residue 343 of the polymerization resistant neuroserpin mutant S340A was also calculated. S340A does not exhibit the large fluctuations in distance between the tops of β-strands 3 and 5A that are seen in neuroserpin (Fig. 6 b). The S49P mutation, which promotes polymerization, does not appreciably alter fluctuations in the breach region but it does increase the distance between β-sheets A and B, particularly in the so-called shutter region. Fig. 6 c shows the distance between the α-carbons of F48 in β-strand 6B and S339 in β-strand 5A. A clear increase in the interstrand distance is evident in S49P (average distance of 7.7 ± 0.3 for wild-type versus 9.3 ± 0.3 for S49P).
Figure 6.
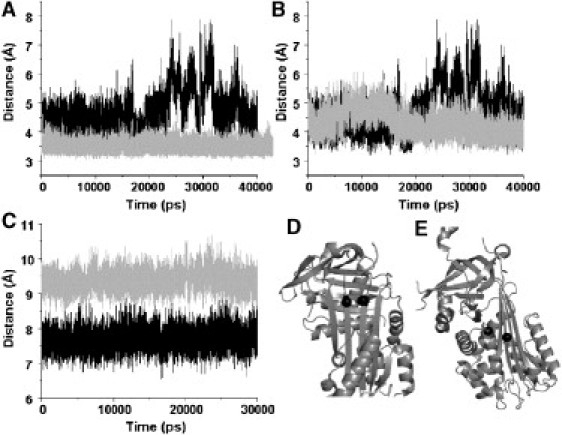
Distance fluctuations between selected residues in α1-antitrypsin, native neuroserpin, and neuroserpin mutants. (A) Distance between α-carbon of residue 189 and the oxygen of residue 337 in α1-AT (gray lines) and between the α-carbon of residue 187 and the oxygen of residue 343 in neuroserpin (black lines). (B) Distance between the α-carbon of residue 187 and the oxygen of residue 343 in wild-type (black) and S340A (gray) neuroserpin. (C) Distance between the α-carbons of residues 48 and 339 in wild-type (black) and S49P (gray) neuroserpin. (D) Positions of residues 187 and 343 (solid spheres). (E) Positions of residues 48 and 339 (solid spheres).
Principal component analysis
Principal component analysis (PCA) has proven an effective method for extracting the large-scale collective motions from MD trajectories when they might otherwise be masked by the many small-scale rapid fluctuations (24). Two frames projected along the first principal component for both neuroserpin and α1-AT are shown in Fig. 7, A and B, respectively, and animations are given in Movie S1 and Movie S2 (see the Supporting Material), respectively. The first principal component for both proteins is dominated by a bending and twisting motion around an axis that cuts horizontally through the center of β-sheet A. However, on a more local scale the two proteins clearly differ, with neuroserpin displaying greater overall mobility. In neuroserpin, there is a significant movement of helix F away from the face of β-sheet A that is not evident in α1-AT. Neuroserpin also shows greater mobility at the top of β-sheet A, particularly in β-strands 3 and 6A. Helices C and D are much more mobile in neuroserpin compared with α1-AT, and in fact the first few turns of helix D in neuroserpin unravel during the simulation.
Figure 7.
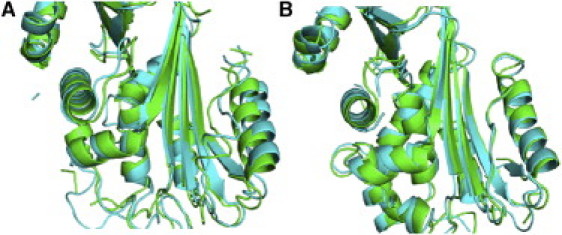
Movement of the F-helix. Snapshots of projections along the first principal component extracted from MD simulations for (A) neuroserpin and (B) α1-antitrypsin. (Green) First frame; (cyan) last frame. A movement of the F-helix away from β-sheet A is clearly evident in neuroserpin but absent in α1-antitrypsin. See also Movie S1 (neuroserpin) and Movie S2 (α1-antitrypsin) in the Supporting Material.
Hydrogen-bond occupancy
Specific hydrogen bonds have been posited to play important roles in regulating serpin conformational change. In particular, a hydrogen-bond network centered on residues 49, 52, 182, and 338 is believed to play a role in the opening of β-sheet A (7). We calculated the hydrogen-bond occupancy over the entire simulation time for every donor-acceptor pair in both neuroserpin and α1-AT. All of the four critical residues referenced above participate in high occupancy hydrogen bonds. The occupancy is 37% for the Ser49 sidechain-Leu389 mainchain bond; 41% for the Ser52 sidechain-His338 sidechain bond; 21% for the Asn182 mainchain-Ile337 mainchain bond; and 45% for the Ile301 mainchain-His338 mainchain bond. The three strongest (most occupied) hydrogen bonds are between the side chains of Arg36 in helix A and Asp305 in helix I, Asp373 in the RCL and Arg362 located immediately after β-strand 1C, and Asp230 at the beginning of the Ω-loop and Arg259 in the loop immediately preceding helix G (Fig. 8). All three of these hydrogen bonds are present during >99% of the simulation. The most highly occupied hydrogen bond in α1-AT (>99% occupancy) is between the side chains of Glu354 in the RCL and Arg223 in β-strand 3C. The fact the strongest hydrogen bonds in both neuroserpin and α1-AT involve anchoring functional loops to the body of the protein suggests that they might play a role by maintaining these loops in conformations that are optimal for function. The S340A mutation does not significantly alter the occupancies of these high-occupancy hydrogen bonds. However, the polymerization promoting S49P mutation does significantly lower the occupancy of the bond between the side chains of Ser52 in helix B and His338 in β-strand 5A from 41% to 31%.
Figure 8.
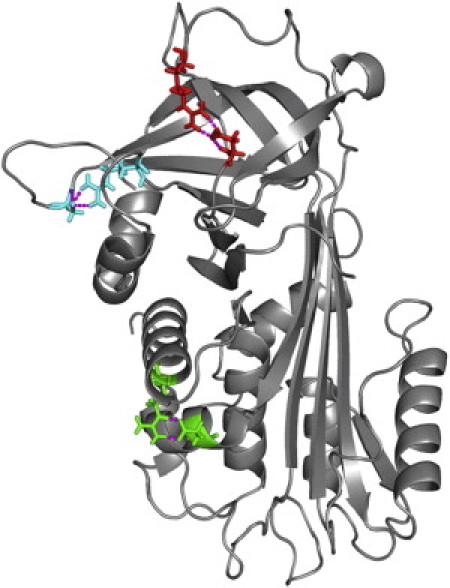
The three most highly occupied hydrogen bonds in neuroserpin shown on the average structure calculated from MD simulations. (Green) Arg36 and Asp305. (Red) Asp373 and Arg362. (Cyan) Asp230 and Arg259.
Discussion
The polymerization of neuroserpin mutants has been identified as the molecular basis of familial encephalopathy with neuroserpin inclusion bodies, and this linkage between polymerization prone mutants and pathology is similar to what is seen in other serpinopathies such as α1-AT deficiency (25). However, in the case of human neuroserpin the wild-type protein is also highly prone to form polymers under near-physiological conditions, and in addition will spontaneously transition to the latent state. Crystal structures and mutagenesis data provide some clues to this unusually high degree of conformational lability (7). However, in providing insight into molecular mobility, static structures have inherent limitations. Additionally, while instability has been proposed to make contributions to both function and dysfunction in neuroserpin (7), global stability is, structurally speaking, a low-resolution measure. Additional insight can be gained by examining the distribution of local stability and flexibility of neuroserpin.
Direct comparison of H/D exchange rates in neuroserpin and α1-AT is problematic because the different sequences give rise to distinct peptic cleavage patterns and, as a result, the solvent exposure, hydrogen bonding, and loop/secondary structure content of peptic fragments from the two proteins differ. However, we can still make some informative comparisons by examining the number of protected amide hydrogens remaining in each peptide at 3000 s of incubation and comparing this with the amount of secondary structure the peptide contains. For example, it was noted above that in peptide 359–378, two amide hydrogens are hydrogen-bonded in β-strand 1C and three are hydrogen-bonded in the portion of β-strand 4B that the peptide covers. Only three amide hydrogens remain protected at 3000 s, and it is reasonable to ascribe these to the buried β-strand 4B than to the much more exposed β-strand 1C. Thus, β-strand 1C's hydrogen bonds are labile on the ∼1 h timescale. In contrast, peptide 352–372 in α1-AT also contains two amide hydrogen bonds in β-strand 1C and three in β-strand 4B. This peptide shows five protected amide hydrogens at 3000 s, suggesting stability of hydrogen bonds of both β-strands 1C and 4B on this timescale (8). The comparison allows us to conclude that β-strand 1C is more stably hydrogen-bonded to β-strand 2C in α1-AT than in neuroserpin.
Because the removal of strand 1C from 2C is required both for the active → latent transition and for polymer formation, this observation has clear implications for the increased tendency of neuroserpin toward latency and polymerization. As another example, in both neuroserpin and α1-AT we identify peptic fragments (of different lengths) that cover portions of β-strands 3A and 4C along with a single turn of helix. In α1-AT this peptide contains 10 amide hydrogens that are hydrogen-bonded in elements of secondary structure, and shows seven protected amide hydrogens after 3000 s (8). In neuroserpin, the corresponding peptide (185–201) contains five amide hydrogens hydrogen bonded in secondary structure and shows only one protected hydrogen after 3000 s (Fig. 9). Even lacking the ability to identify the locations of the protected amide hydrogens, we can conclude that the top of β-strand 3A, one-half of the breach region critical for RCL insertion, is more labile in neuroserpin than in α1-AT. This conclusion is supported by the MD simulations, which show that distance fluctuations between the tops of strands 3 and 5A are significantly larger in neuroserpin than in α1-AT.
Figure 9.
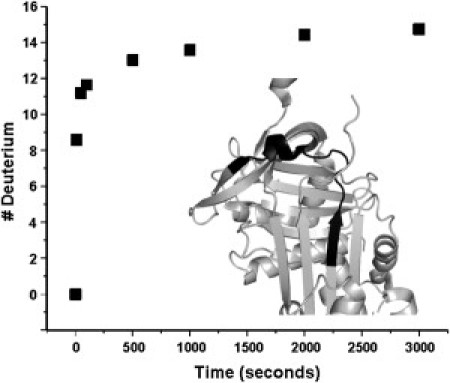
Instability in the breach region of neuroserpin. Deuterium uptake versus time for residues 185–201 in neuroserpin. (Inset, in black) Location of these residues in the three-dimensional structure.
Neuroserpin lacks a highly conserved sequence motif in the loop at the C-terminal end of helix F, and substitution of these residues back into neuroserpin results in variants that are fully functional but both more stable and resistant to polymerization (7). Somewhat surprisingly, H/D exchange indicates that amide hydrogen bonds in helix F are more stable in neuroserpin than in α1-AT. This observation is supported by the simulations. The third, fourth, seventh, and eighth helix F hydrogen bonds in α1-AT have very low occupancies compared to the corresponding hydrogen bonds in neuroserpin (7%, 6%, 3%, and 11% in α1-AT compared with 28%, 49%, 21%, and 26% in neuroserpin). However, while hydrogen-bonding within helix F is stronger in neuroserpin, interactions between helix F and sheet A are stronger in α1-AT. This is clear qualitatively from the presence of a large network of strongly interacting residues spanning both β-sheet A and helix F in α1-AT that is absent in neuroserpin. More quantitatively, we calculated the electrostatic and van der Waals energies between Lys168 and Phe189 in α1-AT and between the corresponding residues Leu162 and Tyr185 in neuroserpin.
The average total interaction energy is 1.4 kcal/mol more favorable in α1-AT, with the major contribution coming from improved electrostatic interactions, although improved van der Waals interactions also contribute. PCA also indicates weakened helix F-sheet A interactions in neuroserpin, as the first principal component involves helix F moving away from the face of sheet A (such a motion is not seen in α1-AT). Weakened hydrogen-bonding within helix F in α1-AT can be explained by positing that optimal interactions with sheet A require small distortions in the helix geometry that make it less optimal for amide hydrogen-bond formation. Neither polymerization nor the transition to the latent state can proceed without temporary displacement of helix F from the front of sheet A. The weaker helix F-sheet A interactions and the tendency of helix F to move away from sheet A in neuroserpin offer additional clues to its propensity for polymerization.
Single-molecule spectroscopic experiments indicate that neuroserpin polymerization is initiated by the formation of an activated monomer that is capable of interacting with other copies of neuroserpin to form dimers and higher-order oligomers (26). This activated species, often termed M∗, is also implicated in the polymerization of other serpins such as α1-AT. Increased conformational lability in the breach region and β-strand 1C, as well as increased mobility of the F helix relative to β-sheet A, rationalize the enhanced propensity of neuroserpin to form this activated species compared with other serpins. It should be noted that neuroserpin is not less stable than α1-AT throughout the structure. β-sheets B and C are quite stable in neuroserpin and interactions within helix F are actually stronger in neuroserpin than in α1-AT, although interactions between helix F and β-sheet A are weaker. Therefore, local loss of stability and increased flexibility appear to underlie neuroserpin's propensity for spontaneous conformational changes. There is evidence that temperature can significantly effect the conformational rearrangements of neuroserpin, with distinct latent and polymeric species forming at moderate and high temperatures (27). Further studies of the effect of temperature on the conformational ensemble of neuroserpin will be needed to elucidate the role of local flexibility in these conformational transitions.
Neuroserpin has a serine at position 340 in place of the more common alanine that typically occupies this position in serpins. The additional oxygen significantly perturbs the stability of β-sheet A and mutation of the serine to alanine both increases thermal stability and diminishes neuroserpin's propensity to polymerize (7). Simulations of the S340A mutant indicate that substitution of alanine for serine results in decreased fluctuations in the distance between β-strand 3 and 5 A in the breach region, which supports a link between local conformational flexibility and polymerization. The polymerization promoting mutation S49P does not result in increased fluctuations in the breach region on the timescale of our simulations. However, it does disrupt packing and hydrogen bonding in the shutter region, as evidenced by the increased distance between β-sheets A and B and the weakening of a conserved hydrogen bond between helix B and β-strand 5A. This disruption of core interactions may contribute S49P's enhanced propensity to form polymers.
Neuroserpin is closely related to PAI-1, which is also more prone to spontaneous conformational changes than other serpins (2). Like neuroserpin, PAI-1 will spontaneously transition to the latent state, although in PAI-1 this transition occurs much more rapidly, with a half-time of ∼120 min as opposed to >24 h for neuroserpin (6). In the case of PAI-1, this excess conformational lability provides an additional level of functional regulation, as the rate of the active → latent transition can be controlled by ligands such as vitronectin (28). It is unclear whether there exists a direct connection between the increased local flexibility in neuroserpin and its functional regulation. Neuroserpin-tPA complexes are short lived compared to most other serpin-target protease complexes (29). It has been suggested that covalent serpin-protease complexes exist in an equilibrium between a fully inhibited state and a partially active state from which the protease can escape through cleavage of the protease-P1 bond (30). The fully inhibited form is that seen in crystal structures in which the protease is located at the bottom of the serpin and the F-helix lies against the face of β-sheet A, while it is suggested that in the partially active form the protease interacts with the lower portion of β-sheet A, displacing the F-helix. If displacement of the F-helix from β-sheet A is required for formation of the partially active state, then weakened sheet A-helix F interactions as well as the intrinsic propensity of the F-helix to move away from sheet A (as seen in the lowest PCAs derived from MD simulations) in neuroserpin could explain the relatively short lifetimes of neuroserpin-protease complexes.
Unlike serpins such as α1-antitrypsin, neuroserpin is internalized by LRP receptors in its active form, indicating that part of its regulation involves being continuously turned over (31). The basis for LRP recognition of noncomplexed neuroserpin is poorly understood, but it has been suggested that the process is mediated by neuroserpin polymerization (31). If that is the case, then increased local flexibility in the breach region of neuroserpin may play a role in regulation by promoting polymerization leading to internalization. Neuroserpin is neuroprotective in a mouse model of Alzheimer's disease and has been shown to form stable complexes with amyloid Aβ in vitro (32). The model proposed for the neuroserpin-Aβ complex has the Aβ peptide inserted between β-strands 3 and 5A in a manner analogous to the RCL. If this model is correct, then weakened interactions between strands 3 and 5A and the resulting increased fluctuations may be important in allowing for the efficient binding of Aβ by neuroserpin. Instability of the active state relative to the cleaved state is critical for the serpin inhibitory mechanism. If the dynamic nature of β-sheet A and the breach region in neuroserpin do in fact contribute either to turnover (through facilitating polymerization) or to neuroprotective binding of Aβ, then this local lack of conformational stability has presumably been selected-for during neuroserpin evolution and represents additional example of functional instability in serpins.
Acknowledgments
We thank Dr. David A. Lomas (University of Cambridge, UK) for providing plasmid coding human neuroserpin.
This work was supported by National Institutes of Health grant No. RO1-HL085469.
Supporting Material
References
- 1.Miranda E., Lomas D.A. Neuroserpin: a serpin to think about. Cell. Mol. Life Sci. 2006;63:709–722. doi: 10.1007/s00018-005-5077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gettins P.G. Serpin structure, mechanism, and function. Chem. Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 3.Kaslik G., Kardos J., Gráf L. Effects of serpin binding on the target proteinase: global stabilization, localized increased structural flexibility, and conserved hydrogen bonding at the active site. Biochemistry. 1997;36:5455–5464. doi: 10.1021/bi962931m. [DOI] [PubMed] [Google Scholar]
- 4.Belorgey D., Irving J.A., Lomas D.A. Characterization of serpin polymers in vitro and in vivo. Methods. 2011;53:255–266. doi: 10.1016/j.ymeth.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Davis R.L., Shrimpton A.E., Lomas D.A. Familial dementia caused by polymerization of mutant neuroserpin. Nature. 1999;401:376–379. doi: 10.1038/43894. [DOI] [PubMed] [Google Scholar]
- 6.Onda M., Belorgey D., Lomas D.A. Latent S49P neuroserpin forms polymers in the dementia familial encephalopathy with neuroserpin inclusion bodies. J. Biol. Chem. 2005;280:13735–13741. doi: 10.1074/jbc.M413282200. [DOI] [PubMed] [Google Scholar]
- 7.Takehara S., Onda M., Lomas D.A. The 2.1-Å crystal structure of native neuroserpin reveals unique structural elements that contribute to conformational instability. J. Mol. Biol. 2009;388:11–20. doi: 10.1016/j.jmb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Tsutsui Y., Liu L., Wintrode P.L. The conformational dynamics of a metastable serpin studied by hydrogen exchange and mass spectrometry. Biochemistry. 2006;45:6561–6569. doi: 10.1021/bi060431f. [DOI] [PubMed] [Google Scholar]
- 9.Wales T.E., Engen J.R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 10.Bottomley S.P., Lawrenson I.D., Pike R.N. The role of strand 1 of the C β-sheet in the structure and function of α1-antitrypsin. Protein Sci. 2001;10:2518–2524. doi: 10.1110/ps.ps.24101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang W.S.W., Whisstock J.C., Wardell M.R. Importance of the release of strand 1C to the polymerization mechanism of inhibitory serpins. Protein Sci. 1997;6:89–98. doi: 10.1002/pro.5560060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belorgey D., Crowther D.C., Lomas D.A. Mutant neuroserpin (S49P) that causes familial encephalopathy with neuroserpin inclusion bodies is a poor proteinase inhibitor and readily forms polymers in vitro. J. Biol. Chem. 2002;277:17367–17373. doi: 10.1074/jbc.M200680200. [DOI] [PubMed] [Google Scholar]
- 13.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 15.Buck M., Bouguet-Bonnet S., MacKerell A.D., Jr. Importance of the CMAP correction to the CHARMM22 protein force field: dynamics of hen lysozyme. Biophys. J. 2006;90:L36–L38. doi: 10.1529/biophysj.105.078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackerell A.D., Jr. Empirical force fields for biological macromolecules: overview and issues. J. Comput. Chem. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 17.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 18.Fiser A., Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 19.Seeber M., Cecchini M., Caflisch A. WORDOM: a program for efficient analysis of molecular dynamics simulations. Bioinformatics. 2007;23:2625–2627. doi: 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
- 20.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 21.Bakan A., Meireles L.M., Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 1575–1577;27:1575–1577. doi: 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.2002. The PyMOL Molecular Graphic System. DeLano Scientific, San Carlos, CA.
- 23.Vijayabaskar M.S., Vishveshwara S. Interaction energy based protein structure networks. Biophys. J. 2010;99:3704–3715. doi: 10.1016/j.bpj.2010.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitao A., Gō N. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 1999;9:164–169. doi: 10.1016/S0959-440X(99)80023-2. [DOI] [PubMed] [Google Scholar]
- 25.Gooptu B., Lomas D.A. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu. Rev. Biochem. 2009;78:147–176. doi: 10.1146/annurev.biochem.78.082107.133320. [DOI] [PubMed] [Google Scholar]
- 26.Chiou A., Hägglöf P., Klenerman D. Probing neuroserpin polymerization and interaction with amyloid-β peptides using single molecule fluorescence. Biophys. J. 2009;97:2306–2315. doi: 10.1016/j.bpj.2009.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricagno S., Pezzullo M., Bolognesi M. Two latent and two hyperstable polymeric form of human neuroserpin. Biophys. J. 2010;99:3401–3411. doi: 10.1016/j.bpj.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindahl T.L., Sigurdardottir O., Wiman B. Stability of plasminogen activator inhibitor 1 (PAI-1) Thromb. Haemost. 1989;62:748–751. [PubMed] [Google Scholar]
- 29.Barker-Carlson K., Lawrence D.A., Schwartz B.S. Acyl-enzyme complexes between tissue-type plasminogen activator and neuroserpin are short-lived in vitro. J. Biol. Chem. 2002;277:46852–46857. doi: 10.1074/jbc.M207740200. [DOI] [PubMed] [Google Scholar]
- 30.Gettins P.G. The F-helix of serpins plays an essential, active role in the proteinase inhibition mechanism. FEBS Lett. 2002;523:2–6. doi: 10.1016/s0014-5793(02)02924-1. [DOI] [PubMed] [Google Scholar]
- 31.Makarova A., Mikhailenko I., Strickland D.K. The low density lipoprotein receptor-related protein modulates protease activity in the brain by mediating the cellular internalization of both neuroserpin and neuroserpin-tissue-type plasminogen activator complexes. J. Biol. Chem. 2003;278:50250–50258. doi: 10.1074/jbc.M309150200. [DOI] [PubMed] [Google Scholar]
- 32.Kinghorn K.J., Crowther D.C., Lomas D.A. Neuroserpin binds Aβ and is a neuroprotective component of amyloid plaques in Alzheimer disease. J. Biol. Chem. 2006;281:29268–29277. doi: 10.1074/jbc.M600690200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.