

Abstract
Distinct lipid environments, including lipid rafts, are increasingly recognized as a crucial factor affecting membrane protein function in plasma membranes. Unfortunately, an understanding of their role in membrane protein activation and oligomerization has remained elusive due to the challenge of characterizing these often small and transient plasma membrane heterogeneities in live cells. To address this difficulty, we present an experimental model membrane platform based on polymer-supported lipid bilayers containing stable raft-mimicking domains (type I) and homogeneous cholesterol-lipid mixtures (type II) into which transmembrane proteins are incorporated (αvβ3 and α5β1 integrins). These flexible lipid platforms enable the use of confocal fluorescence spectroscopy, including the photon counting histogram method, in tandem with epifluorescence microscopy to quantitatively probe the effect of the binding of native ligands from the extracellular matrix ligands (vitronectin and fibronectin for αvβ3 and α5β1, respectively) on domain-specific protein sequestration and on protein oligomerization state. We found that both αvβ3 and α5β1 sequester preferentially to nonraft domains in the absence of extracellular matrix ligands, but upon ligand addition, αvβ3 sequesters strongly into raft-like domains and α5β1 loses preference for either raft-like or nonraft-like domains. A corresponding photon counting histogram analysis showed that integrins exist predominantly in a monomeric state. No change was detected in oligomerization state upon ligand binding in either type I or type II bilayers, but a moderate increase in oligomerization state was observed for increasing concentrations of cholesterol. The combined findings suggest a mechanism in which changes in integrin sequestering are caused by ligand-induced changes in integrin conformation and/or dynamics that affect integrin-lipid interactions without altering the integrin oligomerization state.
Introduction
The modern view of the plasma membrane is that it is a highly complex fluid system comprised of rapidly changing heterogeneous patches of lipids that may regulate the location and functionality of membrane-associated proteins (1,2). One such class of heterogeneous patches consists of lipid rafts, which are defined as dynamic assemblies enriched in cholesterol (CHOL), sphingolipids, and GPI-anchored proteins (3). Rafts of membrane proteins, in concert with other proteins or lipids, induce a variety of cellular activities, including formation of signaling platforms (4), pathogenesis through endocytosis (5), and changes in cellular adhesion, cell morphology, and angiogenesis (6). An important aspect of lipid rafts is that protein functionality can be regulated by a change in raft association or sequestration (7).
Several possible mechanisms may induce a change in sequestration of receptors. Sequestration of proteins into lipid rafts may arise due to protein acylation (8), receptor clustering (9), ligand addition (10), or other specific protein-protein interactions (11). CHOL concentration is known to be a critical factor in sequestration events, as exemplified by CHOL depletion studies that showed a change in protein activity level that was restored when CHOL returned to normal levels (12,13). However, the underlying mechanisms of protein sequestering, and the interplay among them, are still not well understood because it can be very challenging to distinguish contributions from different factors in plasma membranes. CHOL depletion, for example, may induce artifacts such as cytoskeletal destabilization (14). Furthermore, the common practice of identifying proteins found in detergent-resistant membranes with rafts appears to be, in part, prone to inaccurate analyses (15). Rafts are small in size, and protein-lipid raft associations are dynamic and often short-lived (16,17). One method for increasing raft size and duration in live cells is cross-linking, i.e., through cross-linking antibodies, GM1-cholera toxin B (CTxB) cross-linking, or mechanical cross-linking of ligands (18–21). These methods, however, artificially induce cell responses. Lingwood and Simons (22) called this problem, akin to Heisenberg's uncertainty principle, the observer effect of lipid raft studies: before heterogeneity can be observed in plasma membranes, it must first be induced. Consequently, model studies containing raft-mimicking lipid mixtures have also been pursued. In silico progress has been slow, in part due to the limited availability of complete crystal structures for transmembrane proteins. Synthetic bilayers provide a promising avenue of investigation. If lipid bilayers are composed of ternary mixtures of CHOL, saturated lipids, and unsaturated lipids, they can phase-separate into regions of higher order with more CHOL (lo) and lower order with less CHOL (ld) (23,24). Sequestration of membrane receptors and response of sequestration to cross-linking antibodies have also been confirmed in model membrane environments (25–27).
The elegance of model systems lies in the fact that they enable one to study the sequestering and oligomerization behaviors of receptors in response to biological stimuli without the use of artificial cross-linking agents. Applying this concept, we expanded the model membrane approach to study the effects of ligand addition to the integrins αvβ3 and α5β1 incorporated into ternary phase-separating planar polymer-tethered model lipid bilayers. The polymer-tethered planar system enables stable lipid phase separations (28,29), incorporation of biologically active transmembrane proteins (30–32), and sensitive fluorescence fluctuation spectroscopy investigations into lipid phase partitioning and protein oligomerization state (33). Integrins are well suited for these experiments because they are implicated in many raft-associated activities, including cell adhesion, morphology, motility, and angiogenesis. They also act as bidirectional signaling platforms in which functionality is regulated by different factors, including binding of ligands or other membrane proteins, activation through divalent cations, signals from the cytosolic environment, and microclustering (6). Using confocal spectroscopy XY scans, we show that αvβ3 and α5β1 in a polymer-tethered lipid bilayer in PBS buffer partition preferentially to ld domains, and ligand addition alone causes substantial, quantifiable shifts in protein partitioning to lo domains. Despite these substantial shifts, ligand addition had essentially no impact on receptor oligomerization state, as determined by photon counting histograms (PCHs) for receptors incorporated into binary lipid/CHOL bilayers or ternary raft-mimicking bilayers. By contrast, independently of ligand addition, the concentration of CHOL in binary bilayers affected receptor oligomerization to a moderate degree.
Materials and Methods
Materials
The lipopolymer 1,2-dioctadecyl-sn-glycero-3-N-poly(2-methyl-2-oxazoline)50 (diC18M50) was synthesized according to previously described procedures (34). Lipopolymer-coated quantum dots were synthesized according to established procedures (35). The phospholipids 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC), 1,2-dioleoyl-sn-glycro-3-phosphocholine (DOPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (DPTE), 1,2-dioleoyl-sn-glycro-3-phosphoethanolamine (DOPE), CHOL,) and ganglioside GM1 (ovine brain) were purchased from Avanti Polar Lipids (Alabaster, AL). The fluorescently labeled phospholipids N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadec-anoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (NBD-DHPE), N-(6-tetramethylrhodamine-thiocarbamoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (TRITC-DHPE), Alexa-555 labeled CTxB (CTxB-555), and kits for fluorescently labeling antibodies with TRITC or Alexa-555 were obtained from Invitrogen (Carlsbad, CA). Chloroform (HPLC grade; Fisher Scientific, Pittsburgh, PA) was used as a spreading solvent for lipid monolayers at the air-water interface, and Milli-Q water (pH = 5.5, 18 MΩ-cm resistivity; Millipore, Billerica, MA) was used as a subphase material for all experiments. Glass coverslips were pretreated by baking for 3 h at 515°C, followed by sonication in a bath sonicator for 45 min first in 1% SDS, then NaOH saturated MeOH, and finally 0.1% HCl (Fisher Scientific), with extensive rinsing between sonication steps and after the final sonication step, and stored in Milli-Q for no more than 1 week. The proteins human integrin αvβ3 and α5β1, octyl-β-D-glucopyranoside (OG) formulation; monoclonal antibodies (MAbs) anti-integrin αvβ3, clone LM609, and anti-integrin α5β1, clone JBS5; vitronectin, human purified (VN); and fibronectin, human purified (FN) were purchased from Millipore (Billerica, MA). Rhodamine 6G was purchased from Sigma-Aldrich (St. Louis, MO). Antibodies were labeled as described in the antibody labeling kits. Efficacy of labeling was checked by determining brightness by fluorescence correlation spectroscopy (FCS).
Construction of polymer-tethered phospholipid bilayers
Polymer-tethered phospholipid bilayers were built with the use of successive Langmuir-Blodgett (LB) and Langmuir-Schaefer (LS) film transfers according to standard procedures (28). To form the first (LB) monolayer, a chloroform solution of a mixture of diC18M50 and lipids was spread at the air-water interface of a film balance with dipper (Labcon, Darlington, UK). The composition was 5 mol % diC18M50, 31.5 mol % DPPC and CHOL, and 32% DOPC for type I bilayers. For type II bilayers, the composition consisted of 5 mol % diC18M50 and 0, 5, or 30 mol % CHOL, with the balance consisting of SOPC. The monolayer was compressed and, after stabilization (30 min for type I bilayers, 20 min for other mixtures), transferred to a glass substrate (28). Next, a chloroform solution containing a lipid mixture (the LS mixture) was spread at the air-water interface and compressed. For type I bilayers, the LS mixture consisted of 1:1:1 DPPC/CHOL/DOPC. For type II bilayers, the LS mixture consisted of 0, 5, or 30 mol % CHOL, with the balance consisting of SOPC. LS transfer was accomplished by stabilizing the LS monolayer with a depression slide positioned underneath the air-water interface, and then carefully pushing the glass substrate containing the LB layer onto the underlying depression slide. With the use of a transfer dish, the depression slide was removed and the bilayer was transferred into a petri dish, where the Milli-Q was replaced by PBS (Fisher Scientific, 10× concentration, diluted in Milli-Q).
Incorporation of proteins into bilayers
Proteins were reconstituted into model bilayers by means of a modified Rigaud technique (36), also known as the direct protein incorporation method (37). All protein studies were carried out in PBS buffer at room temperature. Membrane proteins stabilized in 100 mM OG (1.3 × 10−11 mol proteins leading to bilayer concentrations of at most 10−3 mol %) were added to the PBS solution above the bilayer (6 mL of PBS) for 1.5–2 h, followed by removal of surfactant using a single layer of SM-2 biobeads previously slurried in PBS (Bio-Rad, Hercules, CA) applied for 15 min to solution over bilayers. Incorporation of proteins into type I bilayers was assisted by initially diluting the proteins into 1 mL of 250 μM OG before addition to the PBS solution above the bilayer, leading to a detergent concentration in the presence of the bilayer of ∼0.002 cmc, but this step was not necessary for type II bilayers. Presoftening of the bilayer by addition of OG or other detergent before protein incorporation (37–39) was not required at these low protein concentrations (250 pM) and long incubation times. Extensive rinsing in PBS ensured efficient removal of surfactant, as confirmed by analysis of domain shapes (see Fig. S1 in the Supporting Material) and lipid fluidity as discussed below. Fluorescently labeled MAbs were then added for 2–4 h and excess antibodies were removed by rinsing. Antibody binding assays were used to confirm the functional reconstitution of integrins, and fluidity was also checked. After data were acquired on these systems, FN or VN was added in a 1:1 ratio to membrane proteins and permitted to equilibrate 3–4 h, and data were taken on the same substrates. Several experiments were conducted at 12 h after ligand addition, and no differences were observed. Unbound FN and VN were removed by rinsing before imaging. MAbs were added to bilayers in the absence of integrins as a control to check for nonspecific binding of antibodies.
Microscopy techniques
A commercial ConfoCor 2 (Zeiss, Jena, Germany) was used for epifluorescence (EPI) microscopy and fluorescence fluctuation spectroscopy (FFS). EPI was conducted with the use of an inverted optical microscope (Axiovert 200M, Zeiss, Oberkochen, Germany) in which the beam was focused to the sample by a microscopy objective (C-Apochromat, water immersion, 40× NA = 1.2; Zeiss). EPI studies were undertaken with a Zeiss AxioCam MRm monochrome digital camera and Axiovision 4.8 software or a CoolSNAPfx camera (Roper Scientific, Princeton, NJ) and Roper Scientific imaging software. FFS data were acquired using a 1.8 mW HeNe laser (543 nm) with a 560–605 nm emission filter (red channel) or a 30 mW Argon laser (514 nm) with a 500–530 emission filter (green channel). We performed confocal spectroscopic XY (CS-XY) scans (10 × 10 μm, 0.5 μm steps) of both channels after taking EPI micrographs of the same location. Control CS-XY scans were taken of bilayers with NBD-PE and no proteins to determine the amount of bleed-through from the green channel to the red channel. FFS data for FCS and PCH analyses were acquired for 50 s runs on the bilayer or in solution, using the same pinhole size, with focusing of planar systems accomplished by maximizing peak count rates from single fluorescent molecules diffusing through the confocal volume. Ternary lipid mixtures (type I) induced phase separations into lo (liquid-ordered) and ld (liquid-disordered) domains, which were visualized by EPI through one of three NBD-PE labeling strategies: 1), addition of 0.5 mol % NBD-PE directly to the bilayer with a correction for background bleed-through in the CS-XY scans; 2), addition of 0.2 mol % NBD-PE (sufficiently low to avoid the need for background correction); or 3), addition of NBD-PE only after completion of all protein data through the use of fusogenic vesicles (35). All three strategies provided similarly accurate quantitative analyses of protein distributions. We investigated protein diffusion using wide-field, single-molecule fluorescence microscopy of membrane proteins in dilute (10−8) concentrations after fluorescent MAb labeling on an inverted microscope (Zeiss Axiovert S100TV) as previously described (40). Further details about this methodology are provided in the Supporting Material. We investigated lipid diffusion for the purpose of analyzing bilayer fluidity by incorporating 2 × 10−3 mol % TRITC-DHPE into type I and type II bilayers, and using FCS to determine characteristic diffusion times for the lipids before and after addition of detergent and rinsing with biobeads.
Data analysis
We analyzed integrin sequestering by identifying partition coefficients (Kp (lo/ld)), defined as Ilo/Ild (41) from CS-XY scans. Raw scans were corrected for NBD and background contributions to determine protein signal average intensities in lo phase (Ilo) and ld phase (Ild). To quantify changes in Kp, we introduce Eraft, defined as the difference in signal intensities between lo and ld phases normalized by the sum of the signal intensities.
| (1) |
For example, for a system perturbed by the addition of ligands, the change in raftophilic excess, divided by two, equals the fraction of proteins Xmigrate that have migrated from ld to lo:
| (2) |
We monitored integrin oligomerization behavior using PCHs of avalanche photodiode photon counts (42,43). To ensure that the data were sufficiently robust above background for the low-concentration diffusing proteins on the bilayer, we also analyzed the intensity trace. We found that when the average signal for the top 1% of the counts collected by intensity trace were at least 20-fold above the average count rate, sufficient data were available to fit the PCH data to a PCH algorithm-generated curve. After finding the average number and brightness of the fluorescent particles (Navg, ε), as described more fully in the Supporting Material, we refitted the data to look for dimers by assuming a second fluorescent species of twice the brightness and finding the average number for both the primary species and secondary species (Navg, ε, Navgdimer, εdimer (= 2ε)). Navg and Navgdimer were then used to determine Xdimer (mole fraction dimers). We tested the PCH model using fluorescent particles known to be monomers, such as Rhodamine 6G, MAbs in solution, and TRITC-DHPE in a bilayer, and found that these species had Xdimer < 1% (data not shown). The data were also refit with a model encompassing tetramers, which showed correspondingly similar results for both the control systems and the membrane proteins of interest (data not shown). It was also necessary to calibrate the PCH algorithm for brightness of particles in solution relative to particles in a thin slab, such as a planar bilayer (44). This was done by constructing a bilayer with SOPC and 2 × 10−2 mol % DPTE and adding maleimide functionalized quantum dots (35). We compared the PCH calculations for brightness of the QDs on the bilayer with the brightness of dilute QDs in solution, and determined that the brightness of the species in solution and the brightness of the species on the bilayer were within 5% of each other (Fig. S2).
Results and Discussion
Determination of fluidity of αvβ3 and α5β1 incorporated into model bilayers
Type I and type II lipid bilayers with 5 mol % DiC18M50 in the LB layer were constructed, and integrins were incorporated therein and subsequently labeled with MAbs as described above. We checked the integrity of the bilayers before protein addition and after detergent extraction by EPI analysis of the bilayers and by determining characteristic diffusion times for lipid probes using FCS. These experiments showed no change in bilayer domain structure (Fig. S1) or lipid fluidity for both type I and type II (0% CHOL) bilayers within the experimental uncertainty of 5% (data not shown). The EPI analysis showed no statistically significant aggregation of integrins on the bilayer. To quantify the functional reconstitution and lateral fluidity of integrins in planar membrane systems, we determined the lateral diffusion of αvβ3 and α5β1 reconstituted into type II (0% CHOL) bilayers before (−VN/FN) and after (+VN/FN) ligand binding. The calculated diffusion coefficients were as follows: for αvβ3, D = 0.39 ± 0.04 (−VN), 0.42 ± 0.04 (+VN); for α5β1 D = 0.71 ± 0.07 (−FN), 0.87 ± 0.08 (+FN; all values μm2s−1), immobile fraction (IF) < 10% by inspection of cumulative distribution functions). These diffusion data are in good agreement with data for αIIbβ3 integrins reconstituted onto other planar model membranes, but differ in terms of IF (45,46). The differences in IF can be attributed to the different protein labeling and reconstitution strategies used. (In the αIIbβ3 system, integrins are fluorescently labeled using 5-(and-6)-carboxy-tetra-methylrhodamine succinimidyl ester and then are reconstituted via proteosomes, in which both membrane protein orientations in the planar membrane system are likely to occur with similar probabilities. In this case, a substantial IF can be expected because integrins pointing with their ectodomains toward the solid substrate are immobile. The DPI reconstitution method, as used in the current study, has been shown to lead to a more unidirectional incorporation of transmembrane proteins (37). By monitoring proteins through the use of fluorescent antibodies that only bind to the protein's extracellular domain, this work also assured that only the properties of correctly oriented proteins (extracellular domain points to water phase) are analyzed, which are less prone to immobilization in the planar bilayer.)
Determination of raft sequestration of proteins before and after ligand binding
To test the sensitivity of the experimental EPI/FFS setup, we constructed type I bilayers with registered lo and ld domains with an addition to the LS mixture of 2 × 10−3 mol % GM1 and 0.5 mol % of the lipid raft marker NBD-PE. CTxB-555 was subsequently added and CS-XY scans were performed. As expected, the raftophilic CTxB colocalized with the NBD-PE: Eraft for CTxB = 0.68 ± 0.07. This corresponds to a Kp of 5.2 ± 1.2, verifying the sensitivity of the type I bilayers to induce sequestration in the GM1/CTxB system. Next, type I bilayers with registered lo and ld domains were constructed with 0.5 mol % NBD-PE added to the LS mixture, and αvβ3 was incorporated as described above. After EPI micrographs were taken (Fig. 1, C and F), CS-XY scans were performed, representatives of which are shown in Fig. 1, where the same area is scanned by the red (protein) channel (A and D) and the green (NBD) channel (B and E). A comparison of the CS-XY scans shows that before addition of VN, αvβ3 displays a marked preference for the nonraft ld phase (Fig. 1, A and B). As illustrated in Fig. 1, D–F, adding VN to the same substrate induced a dramatic change in raft preference. The αvβ3 proteins switched from an ld preference to an lo preference (Fig. 1 D). The NBD-PE distribution was not perturbed by the addition of VN (Fig. 1 F). Motivated by the question of whether this large change in raftophilicity was specific to the αvβ3 integrins or was more generally applicable to other integrins, we performed the experiment again, this time using α5β1 integrins and FN as the ECM ligand; the results are shown in Fig. 2. In similarity to the αvβ3 integrins, before addition of ligand, the α5β1 partitioned preferentially to the ld phase (Fig. 2 A). Subsequent to FN addition, however, the α5β1 switched to displaying no strong preference for either the ld or the lo phase (Fig. 2 D). The results from the CS-XY scans are quantified and tabulated, along with the partitioning behavior of the GM1-CTxB system, in Fig. 3, which shows Eraft for αvβ3 and α5β1 before and after ligand binding (VN and FN, respectively), and Eraft for CTxB-555 linked to GM1. The fraction of receptors that translocated from the ld domains to lo domains can be quantified as Xmigrate, as discussed above (Eq. 2). The CS-XY scans showed Xmigrate = 53 ± 6% for αvβ3 integrins, and Xmigrate = 27 ± 3% for α5β1 integrins.
Figure 1.
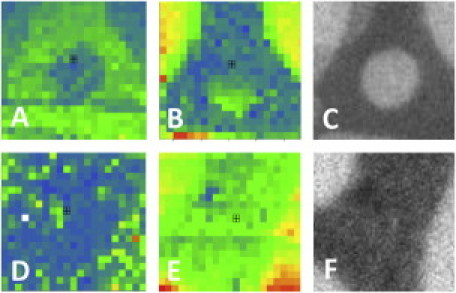
(A) CS-XY scan of αvβ3 − VN. (B) CS-XY scan of NBD-PE in the same location as A. (C) EPI micrograph of NBD-PE, same location as A. (D) CS-XY scan of αvβ3 + VN. (E) CS-XY scan of NBD-PE, same location as D. (F) EPI micrograph of NBD-PE, same location as D. (A–F) Box = 10 × 10 μm2.
Figure 2.
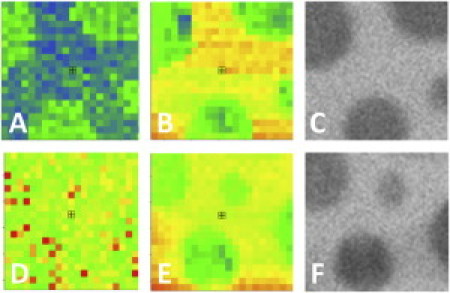
(A) CS-XY scan of α5β1 − FN. (B) CS-XY scan of NBD-PE same location as A. (C) EPI micrograph of NBD-PE in the same location as A. (D) CS-XY scan of α5β1 + FN. (E) CS-XY scan of NBD-PE, same location as D. (F) EPI micrograph of NBD-PE, same location as D. (A–F) Box = 10 × 10 μm2.
Figure 3.
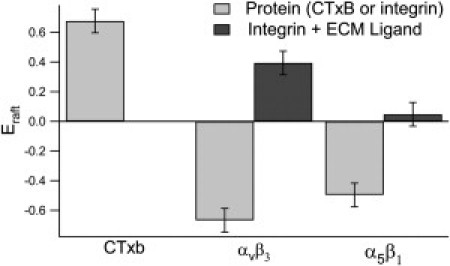
Normalized difference in intensity between lo phase and ld phase (Eraft) shown for GM1-CTxB and αvβ3 and α5β1 before and after ligand addition (light bars: − ECM ligand; dark bars: + ECM ligand). Negative values of Eraft correspond to Kp < 1.
Cell studies have shown that inactive and unbound integrins, similarly to the proteins reconstituted into the model bilayers in PBS buffer in our experiment, are nonraft-associated (19,48). By contrast, integrin signaling and involvement in cell adhesion, motility, and angiogenesis are all raft-related activities, as demonstrated by studies involving alteration of plasma membrane CHOL levels or association with other raft-associated proteins (49–52). It appears that in the model system presented here, binding to ECM ligands alone suffices to increase a preference in αvβ3 and α5β1 for the CHOL-rich (lo) phase, even in the absence of other protein cofactors, activating cations, or known cross-linking agents such as CTxB or cross-linking MAbs. It is notable that αvβ3 is more sensitive to raft sequestration than α5β1. This finding is intriguing in light of the observation that αvβ3 regulates the adhesive and phagocytic activity of α5β1 (53). Several research groups have investigated the partitioning of raft-associated proteins using giant unilamellar vesicles and giant plasma membrane vesicles. Results show that the relative preference for ordered phases is higher for these proteins than for nonraft-associated proteins, and this preference increases upon addition of cross-linking agents (25–27,54). Our system differs from those approaches in that it is planar and investigates the oligomerization state along with sequestration in the absence of artificial cross-linking.
Determination of degree of oligomerization
Our previous data showed that ligand addition causes substantial changes in Eraft, which raises the possibility that these changes are accompanied by similar substantial changes in integrin oligomerization state. Ligands are known to induce clustering in membrane proteins other than integrins (55–57). Moreover, clustering (through agents such as GM1 or a cross-linking antibody) is known to induce raftophilicity in integrins (20,58,59). To investigate whether ligand addition by itself induces a change in oligomerization state separately from any change that might be induced by a change in lipid phase, we constructed a series of type II bilayers, which do not phase-separate, with 0, 5, and 30 mol % CHOL. We probed the oligomerization state of αvβ3 and α5β1 by analyzing the PCH data acquired in these six systems before and after ligand binding. The PCH curves and best fits for species brightness are provided in Fig. S3 and Fig. S4. They show that the integrins were primarily monomers both before and after ligand binding, and the best fits for brightness for these systems in each case were within 15% of the best fit for the brightness of the MAbs in solution (Fig. S4). The results for oligomerization state as deduced from solutions to the PCH algorithm are shown in Fig. 4. For the αvβ3 system, Xdimer is below 5% for the CHOL free bilayer, but increases to 5–10% for 5 mol % CHOL and then to ∼12% for the 30 mol % CHOL bilayer. The α5β1 system shows the same trend, with possibly higher Xdimer (2–7%) found for 0% CHOL. This finding is interesting in light of the observation that reduction of CHOL levels leads to a reduction in integrin functioning as observed by reduced cellular adhesion capabilities (13). Of more importance, there is no statistical difference in oligomerization state before (light bars) and after (dark bars) ligand binding, with the possible exception of a moderate increase in oligomerization after ligand binding in the 5 mol % CHOL αvβ3 system.
Figure 4.
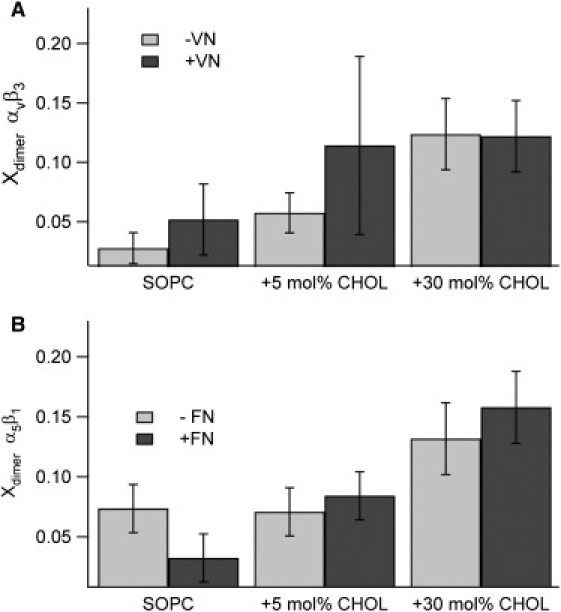
Fraction of dimers found through PCH analysis for αvβ3 (A) and α5β1 (B) before (light bars) and after (dark bars) ligand binding in type II bilayers. These data show that increasing CHOL increases oligomerization for both αvβ3 and α5β1, but only to moderate levels, and ligand binding has no statistically measurable effect on dimerization.
After characterizing integrin oligomerization in type II bilayers, we conducted corresponding experiments on raft-mimicking type I bilayers, utilizing the dual EPI/FFS setup to acquire FFS data in lo and ld phases for both αvβ3 and α5β1 before and after ligand addition for the purpose of analyzing oligomerization state in this system. The results of these studies are shown in Fig. 5, which include the PCH data sets as well as the findings for brightness relative to MAbs in solution and Xdimer. PCH analysis shows that the primary brightness of the integrins relative to the fluorescent MAbs in solution was 77 ± 9% before ligand binding and 81 ± 9% after ligand binding, and thus was essentially the same before and after ligand binding (Fig. 5 E). The rate of dimerization was moderate, between 5 and 20 mol % for both αvβ3 and α5β1 (Fig. 5 F), thus mirroring the results found in the type II bilayers. Using our system and the PCH algorithm, we were able to sensitively identify the oligomerization state, and found that raft sequestration due to monomeric, nonclustered ligands does not induce oligomerization in either αvβ3 or α5β1 integrins.
Figure 5.
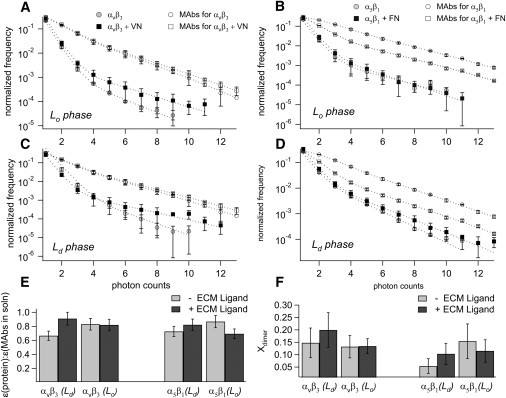
(A–D) PCH curves for αvβ3 (A and C) and α5β1 (B and D) before (light markers) and after (dark markers) ligand binding in both lo phase (A and B) and ld phase (C and D), along with PCH curves for MAbs for integrins in solution (open markers). MAbs data were acquired twice: at the time of initial PCH acquisition (before ligand binding) and at the time of subsequent PCH addition (after ligand binding). Dotted lines are best-fit curves from the PCH algorithm. (E) Brightness compared with MAbs in solution. (F) Fraction of dimers found through PCH analysis of αvβ3 (left) and α5β1 (right) before (light bars) and after (dark bars) ligand binding in ld and lo phases.
Interestingly, our findings are in good agreement with an early study involving octyl-glucoside, in which Hantgan et al. (60) quantified the average molecular weight of the integrin αIIbβ3 through centrifugation before and after addition of an RGD peptide ligand-mimetic. Their study showed only a 10% increase in molecular weight after ligand addition, and thus did not detect significant integrin oligomerization upon ligand addition either. Our PCH data are also supported by an elegant cell study that revealed that ligand addition to αvβ3 or other forms of αvβ3 activation was not capable of inducing clustering in the absence of cytosolic-integrin linkages (58). These findings, as well as our data, imply that αvβ3 integrin-ligand binding alone is insufficient for integrin clustering or oligomerization. Investigators have studied α5β1 clustering by monitoring the difference in strength of cellular adhesion (a proxy for integrin clustering) of magnetic beads coated with different concentrations of FN (21) or polymers linked with 1.7, 3.6, or 5.4 RGD peptides (20). In both cases, cellular adhesion was significantly stronger (per ligand attached) for clustered ligands than for monovalent ligands, indicating that single ligands were not spontaneously forming clusters.
The combined integrin sequestering and PCH data clearly indicate that ligand binding does not affect Xdimer for either αvβ3 or α5β1 in phase-separating type I lipid mixtures. In other words, the observed ligand-mediated changes of αvβ3 and α5β1 sequestering reported in Figs. 1–3 are not caused by changes in receptor oligomerization state. Moreover, our study demonstrates that the change in the fraction of proteins that dimerize before and after ligand binding (≤5% difference in Xdimer on ligand addition) is far less than the fraction of proteins that migrated from disordered to ordered lipid phases for both the α5β1 system, where 27% migrated to induce an even distribution, and the αvβ3 system, where 54% migrated to the lo phase to induce a clear preference for the lo phase. This platform therefore gives us the ability to sensitively distinguish two separate aspects (i.e., raft-association and oligomerization state) and to conclude that although ligand binding is sufficient to induce raft association, it is not directly implicated in oligomerization.
Our data suggest that the observed protein sequestering is due to ligand-induced conformational changes of integrins influencing integrin-lipid interactions. It is well known that ligand addition causes substantial structural changes to both the ectodomains and transmembrane domains of integrins. Electron microscopy studies have analyzed the headgroup conformation of the EC domains of both αvβ3 and α5β1 in solution before and after quantitative exposure to RGD peptides designed to mimic ECM ligands (61,62). Both αvβ3 and α5β1 integrin fragments started in the resting, bent conformations before they were exposed to ligands. After ligand exposure, 98% of the αvβ3 headgroups adopted an open conformation, even in the absence of the known activating factor, Mn2+ (61), and 25% of the α5β1 protein extracellular domains switched to the open conformation (62). The position of the headgroups in relation to the membrane also changes: integrins in the bent, resting position have the RGD binding pocket near the plasma membrane, but when activated, the RGD binding pocket is thought to straighten out and pull far away from the membrane (63). These changes in the ectodomain may influence the observed change in sequestration directly by altering the ectodomain-membrane interface, or indirectly by inducing changes in the integrin transmembrane domains. Furthermore, studies on mutations of integrins indicate that the transmembrane domains of the α and β subunits are associated with substantial conformational changes in response to external stimuli during processes associated with integrin outside-in and inside-out signaling (64). Such conformational changes likely expose different residues and increase the number of residues in the β subunit that reside within the lipid membrane, thus changing the tilt angle of the β subunit (65). This process may affect the mismatch between the integrin transmembrane domain and the hydrophobic region of the lipid bilayer (66).
Conclusion
This study demonstrates that polymer-tethered planar model membranes are sensitive tools for studying transmembrane proteins in different lipid environments and acquiring quantitative data regarding the partitioning preference and oligomerization state of these systems. In this study, we investigated integrin preferences for raft-like and nonraft-like lipid environments using a bilayer phase-separated into lo and ld phases. We found that two members of the integrin family, αvβ3 and α5β1, partition preferentially into the ld phase in their native state. However, addition of ECM ligands induced a quantifiable change in the partition preferences: FN caused α5β1 to lose preference and partition equally between ordered and disordered states, and VN caused αvβ3 to partition as strongly in the lo phase as it did in the ld phase. In addition, using PCH, we were able to quantify the degree of oligomerization of the proteins, and found that most of the proteins, for both systems, were monomers, and addition of ligands did not change the oligomerization state. We further investigated the effect of CHOL on the oligomerization state of the proteins, and found for both systems that increasing CHOL levels increased the oligomerization state, but only moderately, and again, addition of ECM ligands had no effect on the oligomerization state. Our data show that the experimental platform presented here enables the isolation of different factors that describe the effect of lipid rafts in regulating membrane proteins without the use of confounding cross-linking agents. This methodology can be expanded to study the incorporation of other membrane proteins and cofactors to the model system, as well as to study the effect of asymmetric bilayers with lipid rafts contained in only the inner or outer leaflet. Recent work showing that α5β1 appears to recruit CHOL to the outer leaflet in plasma membranes (67) makes this a fascinating avenue for investigation. The methodology can further be combined with other approaches involving the use of polymer-tethered lipid bilayers, such as that used by Purrucker et al. (30) to investigate the cell adhesion strength of integrins incorporated in polymer-tethered model systems. Finally, we hope that this work will further membrane protein molecular-dynamics simulations incorporating CHOL.
Acknowledgments
This research was supported in part by the National Science Foundation (grants MCB-0416779 and MCB-0920134), the Indiana University Purdue University Indianapolis Center for Membrane Biosciences, and the Indiana University Purdue University Indianapolis Nanoscale Imaging Center.
Footnotes
Ann Kimble-Hill's present address is Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN.
Sumit Garg's present address is Materials Science Division, Argonne National Laboratories, Lemont, IL.
Supporting Material
References
- 1.Edidin M. Shrinking patches and slippery rafts: scales of domains in the plasma membrane. Trends Cell Biol. 2001;11:492–496. doi: 10.1016/s0962-8924(01)02139-0. [DOI] [PubMed] [Google Scholar]
- 2.Welti R., Glaser M. Lipid domains in model and biological membranes. Chem. Phys. Lipids. 1994;73:121–137. doi: 10.1016/0009-3084(94)90178-3. [DOI] [PubMed] [Google Scholar]
- 3.Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 4.Holowka D., Gosse J.A., Baird B. Lipid segregation and IgE receptor signaling: a decade of progress. Biochim. Biophys. Acta. 2005;1746:252–259. doi: 10.1016/j.bbamcr.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Pelkmans L. Secrets of caveolae-and lipid raft-mediated endocytosis revealed by mammalian viruses. Biochim. Biophys. Acta. 2005;1746:295–304. doi: 10.1016/j.bbamcr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Carman C.V., Springer T.A. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin. Cell Biol. 2003;15:547–556. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Brown D.A., London E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 8.Zacharias D.A., Violin J.D., Tsien R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 9.Gulbins E., Grassmé H. Ceramide and cell death receptor clustering. Biochim. Biophys. Acta. 2002;1585:139–145. doi: 10.1016/s1388-1981(02)00334-7. [DOI] [PubMed] [Google Scholar]
- 10.Fallahi-Sichani M., Linderman J.J. Lipid raft-mediated regulation of G-protein coupled receptor signaling by ligands which influence receptor dimerization: a computational study. PLoS ONE. 2009;4:e6604. doi: 10.1371/journal.pone.0006604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura A., Baumann C.A., Saltiel A.R. The sorbin homology domain: a motif for the targeting of proteins to lipid rafts. Proc. Natl. Acad. Sci. USA. 2001;98:9098–9103. doi: 10.1073/pnas.151252898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Depry C., Allen M.D., Zhang J. Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst. 2011;7 doi: 10.1039/c0mb00079e. 52–8. [DOI] [PubMed] [Google Scholar]
- 13.Dibya D., Arora N., Smith E.A. Noninvasive measurements of integrin microclustering under altered membrane cholesterol levels. Biophys. J. 2010;99:853–861. doi: 10.1016/j.bpj.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ganguly S., Chattopadhyay A. Cholesterol depletion mimics the effect of cytoskeletal destabilization on membrane dynamics of the serotonin1A receptor: A zFCS study. Biophys. J. 2010;99:1397–1407. doi: 10.1016/j.bpj.2010.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lichtenberg D., Goñi F.M., Heerklotz H. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem. Sci. 2005;30:430–436. doi: 10.1016/j.tibs.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Murase K., Fujiwara T., Kusumi A. Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys. J. 2004;86:4075–4093. doi: 10.1529/biophysj.103.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plowman S.J., Muncke C., Hancock J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA. 2005;102:15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng P.C., Dykstra M.L., Pierce S.K. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J. Exp. Med. 1999;190:1549–1560. doi: 10.1084/jem.190.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leitinger B., Hogg N. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 2002;115:963–972. doi: 10.1242/jcs.115.5.963. [DOI] [PubMed] [Google Scholar]
- 20.Koo L.Y., Irvine D.J., Griffith L.G. Co-regulation of cell adhesion by nanoscale RGD organization and mechanical stimulus. J. Cell Sci. 2002;115:1423–1433. doi: 10.1242/jcs.115.7.1423. [DOI] [PubMed] [Google Scholar]
- 21.Roca-Cusachs P., Gauthier N.C., Sheetz M.P. Clustering of α(5)β(1) integrins determines adhesion strength whereas α(v)β(3) and talin enable mechanotransduction. Proc. Natl. Acad. Sci. USA. 2009;106:16245. doi: 10.1073/pnas.0902818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lingwood D., Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 23.Dietrich C., Bagatolli L.A., Gratton E. Lipid rafts reconstituted in model membranes. Biophys. J. 2001;80:1417–1428. doi: 10.1016/S0006-3495(01)76114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaiser H.-J., Lingwood D., Simons K. Order of lipid phases in model and plasma membranes. Proc. Natl. Acad. Sci. USA. 2009;106:16645–16650. doi: 10.1073/pnas.0908987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumgart T., Hammond A.T., Webb W.W. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc. Natl. Acad. Sci. USA. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kahya N., Brown D.A., Schwille P. Raft partitioning and dynamic behavior of human placental alkaline phosphatase in giant unilamellar vesicles. Biochemistry. 2005;44:7479–7489. doi: 10.1021/bi047429d. [DOI] [PubMed] [Google Scholar]
- 27.Sengupta P., Hammond A., Baird B. Structural determinants for partitioning of lipids and proteins between coexisting fluid phases in giant plasma membrane vesicles. Biochim. Biophys. Acta. 2008;1778:20–32. doi: 10.1016/j.bbamem.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garg S., Rühe J., Naumann C.A. Domain registration in raft-mimicking lipid mixtures studied using polymer-tethered lipid bilayers. Biophys. J. 2007;92:1263–1270. doi: 10.1529/biophysj.106.091082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kiessling V., Crane J.M., Tamm L.K. Transbilayer effects of raft-like lipid domains in asymmetric planar bilayers measured by single molecule tracking. Biophys. J. 2006;91:3313–3326. doi: 10.1529/biophysj.106.091421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purrucker O., Gönnenwein S., Tanaka M. Polymer-tethered membranes as quantitative models for the study of integrin-mediated cell adhesion. Soft Matter. 2007;3:333–336. doi: 10.1039/b612069e. [DOI] [PubMed] [Google Scholar]
- 31.Deverall M.A., Gindl E., Naumann C.A. Membrane lateral mobility obstructed by polymer-tethered lipids studied at the single molecule level. Biophys. J. 2005;88:1875–1886. doi: 10.1529/biophysj.104.050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner M.L., Tamm L.K. Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys. J. 2000;79:1400–1414. doi: 10.1016/S0006-3495(00)76392-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kahya N., Schwille P. Fluorescence correlation studies of lipid domains in model membranes. Mol. Membr. Biol. 2006;23:29–39. doi: 10.1080/09687860500489099. (Review) [DOI] [PubMed] [Google Scholar]
- 34.Lüdtke K., Jordan R., Naumann C.A. Lipopolymers from new 2-substituted-2-oxazolines for artificial cell membrane constructs. Macromol. Biosci. 2005;5:384–393. doi: 10.1002/mabi.200500004. [DOI] [PubMed] [Google Scholar]
- 35.Murcia M.J., Minner D.E., Naumann C.A. Design of quantum dot-conjugated lipids for long-term, high-speed tracking experiments on cell surfaces. J. Am. Chem. Soc. 2008;130:15054–15062. doi: 10.1021/ja803325b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rigaud J.-L., Levy D. Reconstitution of membrane proteins into liposomes. Methods Enzymol. 2003 doi: 10.1016/S0076-6879(03)72004-7. 2003;372:65–86. [DOI] [PubMed] [Google Scholar]
- 37.Milhiet P.E., Gubellini F., Lévy D. High-resolution AFM of membrane proteins directly incorporated at high density in planar lipid bilayer. Biophys. J. 2006;91:3268–3275. doi: 10.1529/biophysj.106.087791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renner L., Pompe T., Werner C. Controlled enhancement of transmembrane enzyme activity in polymer cushioned supported bilayer membranes. Soft Matter. 2010;6:5382–5389. [Google Scholar]
- 39.Picas L., Carretero-Genevrier A., Hernández-Borrell J. Preferential insertion of lactose permease in phospholipid domains: AFM observations. Biochim. Biophys. Acta. 2010;1798:1014–1019. doi: 10.1016/j.bbamem.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 40.Deverall M., Garg S., Naumann C. Transbilayer coupling of obstructed lipid diffusion in polymer-tethered phospholipid bilayers. Soft Matter. 2008;4:1899–1908. [Google Scholar]
- 41.Silvius J.R. Partitioning of membrane molecules between raft and non-raft domains: insights from model-membrane studies. Biochim. Biophys. Acta. 2005;1746:193–202. doi: 10.1016/j.bbamcr.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y., Müller J.D., Gratton E. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 1999;77:553–567. doi: 10.1016/S0006-3495(99)76912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang B., Perroud T.D., Zare R.N. Photon counting histogram: one-photon excitation. ChemPhysChem. 2004;5:1523–1531. doi: 10.1002/cphc.200400176. [DOI] [PubMed] [Google Scholar]
- 44.Macdonald P.J., Chen Y., Mueller J.D. Brightness analysis by Z-scan fluorescence fluctuation spectroscopy for the study of protein interactions within living cells. Biophys. J. 2010;99:979–988. doi: 10.1016/j.bpj.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erb E.M., Tangemann K., Engel J. Integrin αIIb β3 reconstituted into lipid bilayers is nonclustered in its activated state but clusters after fibrinogen binding. Biochemistry. 1997;36:7395–7402. doi: 10.1021/bi9702187. [DOI] [PubMed] [Google Scholar]
- 46.Goennenwein S., Tanaka M., Sackmann E. Functional incorporation of integrins into solid supported membranes on ultrathin films of cellulose: impact on adhesion. Biophys. J. 2003;85:646–655. doi: 10.1016/S0006-3495(03)74508-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reference deleted in proof.
- 48.Baron W., Decker L., ffrench-Constant C. Regulation of integrin growth factor interactions in oligodendrocytes by lipid raft microdomains. Curr. Biol. 2003;13:151–155. doi: 10.1016/s0960-9822(02)01437-9. [DOI] [PubMed] [Google Scholar]
- 49.Hogg N., Laschinger M., McDowall A. T-cell integrins: more than just sticking points. J. Cell Sci. 2003;116:4695–4705. doi: 10.1242/jcs.00876. [DOI] [PubMed] [Google Scholar]
- 50.Gopalakrishna P., Chaubey S.K., Pande G. Modulation of α5β1 integrin functions by the phospholipid and cholesterol contents of cell membranes. J. Cell. Biochem. 2000;77:517–528. [PubMed] [Google Scholar]
- 51.Gopalakrishna P., Rangaraj N., Pande G. Cholesterol alters the interaction of glycosphingolipid GM3 with α5β1 integrin and increases integrin-mediated cell adhesion to fibronectin. Exp. Cell Res. 2004;300:43–53. doi: 10.1016/j.yexcr.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Ramprasad O.G., Srinivas G., Pande G. Changes in cholesterol levels in the plasma membrane modulate cell signaling and regulate cell adhesion and migration on fibronectin. Cell Motil. Cytoskeleton. 2007;64:199–216. doi: 10.1002/cm.20176. [DOI] [PubMed] [Google Scholar]
- 53.Blystone S.D., Graham I.L., Brown E.J. Integrin α v β 3 differentially regulates adhesive and phagocytic functions of the fibronectin receptor α 5 β 1. J. Cell Biol. 1994;127:1129–1137. doi: 10.1083/jcb.127.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalvodova L., Kahya N., Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005;280:36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- 55.Cunningham O., Andolfo A., Sidenius N. Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. EMBO J. 2003;22:5994–6003. doi: 10.1093/emboj/cdg588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong S.W., Kwon M.J., Hwang D.H. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J. Biol. Chem. 2009;284:27384–27392. doi: 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y., Wei L.N., Müller J.D. Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc. Natl. Acad. Sci. USA. 2003;100:15492–15497. doi: 10.1073/pnas.2533045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cluzel C., Saltel F., Wehrle-Haller B. The mechanisms and dynamics of (α)v(β)3 integrin clustering in living cells. J. Cell Biol. 2005;171:383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiseman P.W., Brown C.M., Horwitz A.F. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J. Cell Sci. 2004;117:5521–5534. doi: 10.1242/jcs.01416. [DOI] [PubMed] [Google Scholar]
- 60.Hantgan R.R., Paumi C., Weisel J.W. Effects of ligand-mimetic peptides Arg-Gly-Asp-X (X = Phe, Trp, Ser) on αIIbβ3 integrin conformation and oligomerization. Biochemistry. 1999;38:14461–14474. doi: 10.1021/bi9907680. [DOI] [PubMed] [Google Scholar]
- 61.Takagi J., Petre B.M., Springer T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 62.Takagi J., Strokovich K., Walz T. Structure of integrin α5β1 in complex with fibronectin. EMBO J. 2003;22:4607–4615. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo B.H., Carman C.V., Springer T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim M., Carman C.V., Springer T.A. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 65.Wang W., Luo B.-H. Structural basis of integrin transmembrane activation. J. Cell. Biochem. 2010;109:447–452. doi: 10.1002/jcb.22427. [DOI] [PubMed] [Google Scholar]
- 66.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. Biomembranes. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 67.Pankov R., Markovska T., Momchilova A. Cholesterol distribution in plasma membranes of β1 integrin-expressing and β1 integrin-deficient fibroblasts. Arch. Biochem. Biophys. 2005;442:160–168. doi: 10.1016/j.abb.2005.08.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.