

Abstract
We investigated the influence of cardiac myosin binding protein-C (cMyBP-C) and its constitutively unphosphorylated status on the radial and longitudinal stiffnesses of the myofilament lattice in chemically skinned myocardial strips of the following mouse models: nontransgenic (NTG), effective null for cMyBP-C (t/t), wild-type cMyBP-C expressed into t/t (WTt/t), and constitutively unphosphorylated cMyBP-C (AllP-t/t). We found that the absence of cMyBP-C in the t/t and the unphosphorylated cMyBP-C in the AllP-t/t resulted in a compressible cardiac myofilament lattice induced by rigor not observed in the NTG and WTt/t. These results suggest that the presence and phosphorylation of the N-terminus of cMyBP-C provides structural support and radial rigidity to the myofilament lattice. Examination of myofilament longitudinal stiffness under rigor conditions demonstrated a significant reduction in cross-bridge-dependent stiffness in the t/t compared with NTG controls, but not in the AllP-t/t compared with WTt/t controls. The absence of cMyBP-C in the t/t and the unphosphorylated cMyBP-C in the AllP-t/t both resulted in a shorter myosin cross-bridge lifetime when myosin isoform was controlled. These data collectively suggest that cMyBP-C provides radial rigidity to the myofilament lattice through the N-terminus, and that disruption of the phosphorylation of cMyBP-C is sufficient to abolish this structural role of the N-terminus and shorten cross-bridge lifetime. Although the presence of cMyBP-C also provides longitudinal rigidity, phosphorylation of the N-terminus is not necessary to maintain longitudinal rigidity of the lattice, in contrast to radial rigidity.
Introduction
The cardiac isoform of myosin binding protein-C (cMyBP-C) has drawn considerable attention since it was first recognized that mutations in the human MYBPC3 gene underlie the development of a high percentage of cases of familial cardiomyopathy (1–4). A significant reduction in cMyBP-C density has now been observed in some humans carrying mutant alleles for MYBPC3 (5,6). These findings heighten the importance of understanding the functional and structural effects of a reduced content of cMyBP-C while we continue to discover the physiological function of cMyBP-C and its phosphorylation.
cMyBP-C is normally incorporated into the thick filament at regular 43 nm intervals (7,8), and its complete absence from the sarcomere leads to an atypical dilated phenotype in three independently developed mouse models lacking cMyBP-C, labeled −/− for the genetic null and t/t for the effective null (9–11). Compared with nontransgenic (NTG) controls, the absence of cMyBP-C in structurally intact cardiac myofilaments leads to reduced rigor cross-bridge-dependent stiffness and thick filament stiffness (12,13). Furthermore, the absence of cMyBP-C leads to greater myofilament disorder (14,15) and an expanded (14) or possibly unchanged (12) myofilament lattice spacing.
The C-terminus of cMyBP-C binds to the myosin rod, titin, and other cMyBP-Cs within the thick filament backbone (16–18). These binding properties of the C-terminus appear to underlie a structural role in stabilizing and stiffening the thick filament, thereby facilitating force transmission between the acto-myosin cross-bridge and the M-line (13). In contrast, the N-terminus of cMyBP-C is not incorporated as part of the thick filament backbone and possesses multiple phosphorylation sites as well as binding sites to myosin S2 and actin (17,19,20). The arrangement of myosin heads along the thick filament and the kinetics of acto-myosin cross-bridges are sensitive to cMyBP-C phosphorylation (19–21), and evidence suggests that cMyBP-C phosphorylation modulates sarcomere function via its direct influence on myosin head position, myofilament lattice spacing, and/or thin filament activation (4,15,20–26).
Using the t/t mouse as a background, we developed a transgenic mouse line to test the effects of unphosphorylated cMyBP-C on sarcomere mechanical properties related to the possible structural role of cMyBP-C in the context of an intact myofilament lattice. The transgenic mouse expresses a cMyBP-C, termed AllP-t/t, that has five possible phosphorylation sites (Thr-272, Ser-273, Thr-281, Ser-282, and Ser-302) exchanged with Ala to mimic a nonphosphorylated cMyBP-C (27). Compared with control mice expressing the wild-type cMyBP-C in the t/t background (WTt/t), AllP-t/t hearts develop an atypical dilated cardiomyopathy (9–11,27).
In this study, we examined t/t, AllP-t/t, and control mice expressing WT cMyBP-C to elucidate the structural role of cMyBP-C in providing radial and longitudinal stiffness to the myofilament lattice. We report that the maintenance of myofilament order and also possibly interfilament lattice rigidity are dependent on the presence of the N-terminus of cMyBP-C and/or its phosphorylation capacity. We also find that the absence of cMyBP-C or its effective dephosphorylation leads to a shorter myosin cross-bridge lifetime (ton).
Materials and Methods
Mouse models
All procedures were reviewed and approved by the institutional animal care and use committees of the Cincinnati Children's Hospital Medical Center and University of Vermont Medical School, and were in compliance with the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health. The mice used in this study were generated as previously described (27,28). A cohort of WTt/t and AllP-t/t mice received 3 μg/g body weight L-thyroxine (T4, T-2501; Sigma-Aldrich, St. Louis, MO) by six consecutive daily intraperitoneal injections of 200 μL of 0.9% saline + 10 mM NaOH. This was done to mimic hyperthyroidism and elicit expression of cardiac α-MyHC. A similar T4 treatment in the t/t mice led to >50% mortality, which led us to choose another treatment in these mice. A cohort of NTG and t/t mice were fed an iodine-deficient, 0.15% 6-n-propyl-2-thiouracil (PTU) diet (No. TD97061; Harlan-Teklad) for at least 10 weeks, which induced hypothyroidism and a downregulation of cardiac α-MyHC expression.
Isolated cardiomyocytes
Mouse left ventricle (LV) cardiomyocytes were isolated by retrograde perfusion with collagenase solution as described elsewhere (13), but without blebbistatin. Cardiomyocytes were observed with a 40× objective on an inverted microscope (Nikon Diaphot) while bathed in normal Tyrode's solution containing (in mmol/L) 137 NaCl, 5.4 KCl, 0.5 MgCl2, 10 HEPES, 5.5 glucose, 0.5 Probenecid at pH 7.4. We detected the resting sarcomere length at 37°C using Fourier transforms of the digital image (IonOptix, Milton, MA). At least 10 cardiomyocytes were used to detect the average sarcomere length for nine NTG, six t/t, seven WTt/t, and 10 AllP-t/t hearts.
Quantification of site-specific phosphorylation
Phosphorylated amino acids were identified at four specific sites within cMyBP-C isolated from NTG expressing WT MyBP-C, and the degree of phosphorylation was quantified by label-free liquid chromatography-mass spectrometry. A thorough explanation of these methods is provided in the Supporting Material and elsewhere (29).
Solutions for skinned strips
Chemicals and reagents were obtained from Sigma (St. Louis, MO) unless otherwise noted. We formulated the solution concentrations (expressed in mmol/L unless otherwise noted) by solving equations describing ionic equilibria according to Godt and Lindley (30). The relaxing solution consisted of pCa 8.0, 5 EGTA, 5 MgATP, 1 Mg2+, 0 Pi, 40 phosphocreatine (PCr), 240 U/mL creatine kinase (CK), ionic strength 190, pH 7.0. The activation solution was the same as the relaxing solution but with pCa 4.5. The rigor solution was the same as the activating solution but with no ATP. The skinning solution was the same as the relaxing solution but without PCr or CK and with 10 μg/mL leupeptin, 1% Triton-X100 wt/vol, and subsequently 50% glycerol wt/vol. The storage solution was the same as the relaxing solution but without PCr or CK and with 10 μg/mL leupeptin and 50% glycerol wt/vol.
X-ray diffraction
Papillary muscles were dissected to no less than 1 mm diameter and chemically skinned for 2 h at room temperature, stored at −20°C for no more than 4 days, and stretched to 2.2–2.4 μm sarcomere length. We obtained equatorial x-ray diffraction patterns at room temperature using the BioCAT small-angle x-ray diffraction facility on beamline 18ID at the Advanced Photon Source (Argonne, IL) as previously described (31,32). The separations of the 1,0 equatorial reflections were measured by two independent observers and then converted into the mean distance between planes of thick filaments (d1,0) (31).
Length perturbation analysis
LV skinned myocardial strips were studied as previously described (33,34). Papillary muscles were dissected to yield at least two thin strips (∼140 μm diameter, ∼800 μm length) with longitudinally oriented parallel fibers, skinned for 2 h at room temperature, and stored at –20°C for no more than 4 days. At the time of study, aluminum T-clips were attached to the ends of a strip ∼300 μm apart. The strip was mounted between a piezoelectric motor (Physik Instrumente, Auburn, MA) and a strain gauge (SensorNor, Horten, Norway), lowered into a 30 μL droplet of relaxing solution (pCa 8.0) maintained at 17°C, and incrementally stretched to 2.2 μm sarcomere length as measured by digital Fourier transform. Sinusoidal length perturbations of amplitude 0.125% strip length were applied at 0.125–250 Hz as previously described (33,34). The elastic and viscous moduli were measured from the in-phase and out-of-phase portions of the tension response to length perturbation. The fitting of the frequency dependence of the elastic and viscous moduli to a mathematical model provided a measure of myosin ton as demonstrated previously (35).
Analysis
Multiple measurements from the same heart were averaged to provide a single measure for that heart. All data are presented as the mean ± SE, and significance by statistical tests are reported at the p < 0.05 and p < 0.01 levels.
Results
Characteristics of mouse hearts
The LVs of the t/t and AllP-t/t mice were significantly larger than those of their respective NTG and WTt/t controls (Table 1). Despite a normal abundance of cMyBP-C in the AllP-t/t, these hearts developed an atypical dilated cardiomyopathy similar to that developed in the −/− and t/t mice lacking cMyBP-C, as reported previously (9–11,27).
Table 1.
Mouse characteristics, cardiomyocyte unloaded sarcomere length, activated myofilament tension, and myosin lifetime (ton)
| NTG (n = 7) | t/t (n = 9) | WTt/t (n = 8) | AllP-t/t (n = 9) | |
|---|---|---|---|---|
| Age (weeks) | 28.7 ± 4.3 | 24.3 ± 3.5 | 22.7 ± 2.3 | 29.9 ± 4.0 |
| Body mass (g) | 30.0 ± 1.5 | 31.1 ± 0.7 | 31.5 ± 0.8 | 32.4 ± 1.3 |
| LV/body (mg/g) | 3.57 ± 0.26 | 6.65 ± 0.32∗ | 3.70 ± 0.19 | 5.01 ± 0.26∗ |
| RV+LV/body (mg/g) | 4.58 ± 0.17 | 8.48 ± 0.47∗ | 4.81 ± 0.20 | 6.24 ± 0.32∗ |
| Sarcomere length (μm) | 1.71 ± 0.02 | 1.60 ± 0.05∗ | 1.94 ± 0.03 | 1.74 ± 0.02∗∗ |
| Tmin (mN.mm−2) | 2.24 ± 0.25 | 2.72 ± 0.32 | 2.36 ± 0.50 | 1.98 ± 0.33 |
| Tdev (mN.mm−2) | 13.6 ± 1.8 | 11.9 ± 1.2 | 16.0 ± 1.3 | 15.3 ± 1.5 |
| ton (ms) | 11.0 ± 0.8 | 14.7 ± 1.0∗ | 11.5 ± 0.6 | 10.5 ± 0.5 |
| NTGPTU (n = 5) | t/tPTU (n = 6) | WTt/tT4 (n = 4) | AllP-t/tT4 (n = 5) | |
| ton (ms) | 60.6 ± 3.3 | 39.3 ± 2.6∗ | 10.5 ± 1.8 | 7.6 ± 0.6† |
LV and added right ventricle (RV) weights were greater in the t/t compared with NTG and in the AllP-t/t compared with WTt/t. ∗Different from respective control (p < 0.05); ∗∗p < 0.01; †different from WTt/tT4 controls at p = 0.085 and from AllP-t/t at p < 0.05.
MyHC isoform profiles from these LVs are illustrated in Fig. 1 A and demonstrated significant concentrations of β-MyHC in the t/t (∼40%) and AllP-t/t (∼40%). PTU treatment in the NTG and t/t led to near 100% incorporation of β-MyHC (Fig. 1 B). Thyroxine (T4) treatment in the t/t and AllP-t/t induced a greater expression of α-MyHC compared with β-MyHC (Fig. 1 B). The concentrations of α-MyHC in the AllP-t/tT4 (∼90%) were comparable to WTt/tT4 (∼90%) with T4 treatment and WTt/t (∼95%) without T4 treatment.
Figure 1.
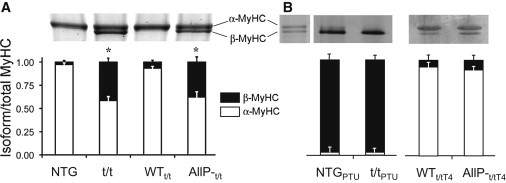
MyHC isoform profiles in mouse LV. (A) Gel electrophoresis stained with Coomassie Blue reveals the relative contents of α-MyHC and β-MyHC in NTG, t/t, WTt/t, and AllP-t/t genotypes (27). LVs from NTG and WTt/t contained an ∼90/10 ratio of α/β-MyHC, and LVs of t/t and AllP-t/t contained an ∼60/40 ratio of α/β-MyHC. (B) NTG and t/t mice made hypothyroid by a PTU diet expressed predominantly (nearly 100%) β-MyHC. WTt/t and AllP-t/t mice made hyperthyroid by thyroxine (T4) treatment expressed predominantly (nearly 100%) α-MyHC.
The degree of phosphorylation of Ser-273, -282, -302, and -307 in the endogenous protein of the NTG, presented in Table 2, was determined by mass spectroscopy (see Supporting Material for raw data). Assuming that these four serines constitute the phosphorylatable sites of cMyBP-C, the percentage of cMyBP-C that is totally nonphosphorylated and singly, doubly, triply, and quadruply phosphorylated would be 0%, ∼15%, ∼55%, ∼27.5%, and ∼2.5%, respectively. This phosphorylation profile is consistent with that previously observed for the NTG and WTt/t using isoelectric focusing gels (27).
Table 2.
Phosphorylated peptides observed and degree of phosphorylation determined for endogenous cMyBP-C isolated for NTG mice
| Phosphorylated amino acid | Phosphopeptide observed | Phosphorylation (% ± SD) |
|---|---|---|
| Ser-273 | 272TSpLAGAGR279 | 76 ± 11 |
| 271RTSpLAGAGR279 | ||
| Ser-282 | 281TSpDSHEDAGTLDFSSLLK298 | 100 ± 0 |
| 280RTSpDSHEDAGTLDFSSLLK298 | ||
| Ser-302 | 299KRDSpFR304 | 31 ± 12 |
| Ser-307 | 305RDSpKLEAPAEEDVWEILR322 | 11 ± 3 |
Ser-273, -282, and -302 are homologous to three of the five sites exchanged with Ala in the AllP-t/t. Although the probability of having Ser-273, -282, and -302 phosphorylated is low (∼24%), the majority of the endogenous protein (∼76%) contains phosphorylation on Ser-273 and -282. We infer this phosphorylation profile to be that of the NTG and WTt/t, in contrast to the fully dephosphorylated profile of the AllP-t/t.
Myofilament lattice spacing
Small-angle x-ray diffraction was used to detect the mean distance between planes of thick filaments (Fig. 2 A), which served as a quantitative index of myofilament lattice spacing, d10. Raw x-ray intensity profiles under relaxed conditions (Fig. 2 B) demonstrated qualitatively a greater degree of myofilament lattice disorder in the t/t and AllP-t/t, in similarity to that quantified and reported previously (14,15). Example x-ray intensity profiles for all four populations under the three conditions examined are presented in the Supporting Material. As can be seen in Fig. 2, Fig. S3, and Fig. S4, the d1,0 and d1,1 peaks are less prominent and broader in the t/t and AllP-t/t compared with the NTG and WTt/t. These features are indicative of greater myofilament disorder reported previously for the sarcomere of the t/t (9) and possibly in concert with myosin head disorder as occurs with no MyBP-C or dephosphorylation of MyBP-C detected by electron microscopy (21). At a sarcomere length of 2.2–2.4 μm and under maximum calcium-activated conditions (pCa 4.5) or rigor conditions, the myofilament lattice spacing of the t/t and AllP-t/t was less (p < 0.05 by two-way repeated-measures analysis of variance) than their respective NTG and WTt/t controls (Fig. 2 C). Of interest, both the t/t and AllP-t/t showed shorter sarcomere lengths in unloaded cardiomyocytes (Table 1), as reported previously for −/− and t/t (13,36), and also more compressed myofilament lattices when sarcomeres were set to 2.2–2.4 μm length and calcium activated (Fig. 2). These data are internally consistent with an inverse relationship between myofilament lattice spacing and sarcomere length in these populations (31,37,38). Specifically, resting sarcomere length in unloaded single cardiomyocytes of the t/t and AllP-t/t was significantly shorter than that of NTG and WTt/t controls (Table 1). We would therefore expect a more compressed lattice from a sarcomere that is stretched to 2.2–2.4 μm when its unloaded sarcomere length is relatively shorter, as was the case for the t/t and AllP-t/t.
Figure 2.
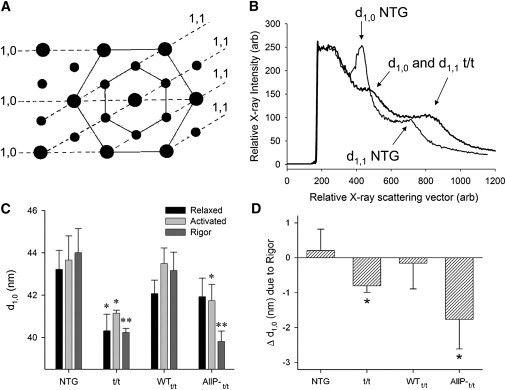
Small-angle x-ray diffraction detection of myofilament lattice spacing. (A) This cartoon of a myofilament lattice cross section illustrates the planes of thick filaments and thin filaments separated by distances termed d1,0 and d1,1, respectively. (B) The distance from the origin to peaks in the x-ray intensity profile along the equator provides a measure of the mean distance between planes of thick filaments. Under relaxed conditions, the diffraction peaks indicating d1,0 and d1,1 profiles of t/t and AllP-t/t tended to be broader and weaker, which are characteristics consistent with increased paracrystalline disorder in the myofilament lattice. (C) Myofilament lattice spacing was compressed in t/t and AllP-t/t compared with respective controls. ∗Different from respective control under the same conditions (p < 0.05), ∗∗p < 0.01. (D) Rigor induced significant compression of the myofilament lattice in the t/t and AllP-t/t compared with activated conditions. ∗Different from no change by paired t-test (p < 0.01); n = 7 NTG, 5 t/t, 8 WTt/t, and 6 AllP-t/t.
Rigor compressed the lattice to a greater extent (p < 0.05 by two-way repeated-measures analysis of variance) than did calcium activation (Fig. 2 D). Rigor conditions in skeletal muscle have been shown to impose a radial compressive force on the myofilament lattice, presumably due to a radial component of the rigor cross-bridge force as it adopts a post-power-stroke state while bound to actin (39,40). We found that the skinned mouse cardiac myofilament lattice is not compressed under rigor versus calcium-activated conditions in the NTG and WTt/t controls, as was reported previously for skinned rat myocardium (41). These findings imply that the presence of WT cMyBP-C and its in vivo phosphorylation in the NTG and WTt/t provide mechanical support for the myofilament lattice of cardiac muscle, and that the N-terminus of cMyBP-C may effectively provide a structural strut, either directly or indirectly through the myosin head, between myofilaments.
Effects of cMyBP-C on cross-bridge lifetime
We performed small length perturbation analysis on skinned myocardial strips of NTG versus t/t, and WTt/t versus AllP-t/t to examine the effects of MyBP-C and its unphosphorylated state on myofilament mechanical characteristics under activated and rigor conditions. The elastic and viscous moduli recorded at maximum calcium activation are presented in Fig. 3. Over several frequencies examined, the elastic and viscous moduli were significantly lower in the t/t compared with NTG controls (Fig. 3, A and B). The cross-bridge ton, which qualitatively is reciprocally related to the frequency of peak viscous modulus (35), was significantly prolonged in the t/t compared with NTG, but was similar between AllP-t/t and WTt/t (Table 1).
Figure 3.
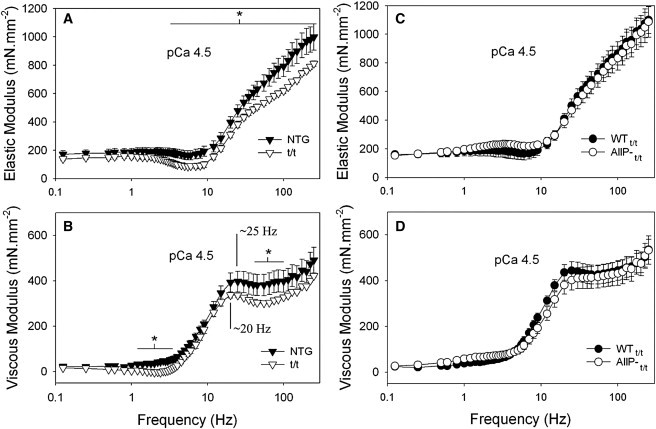
Frequency characteristics of the elastic and viscous moduli at maximum calcium activation and 17°C. (A and B) Values for the elastic and viscous moduli were significantly reduced in the t/t myofilaments over several frequencies compared with NTG. The frequency characteristics of the moduli were also shifted to lower frequencies in the t/t, as indicated qualitatively by the position of the peak in the viscous modulus. (C and D) Elastic and viscous moduli in AllP-t/t are similar to those of WTt/t. ∗Different from control population by t-test (p < 0.05); n = 8 NTG, 8 t/t, 8 WTt/t, and 8 AllP-t/t.
The myosin cross-bridge lifetime, ton, is known to be significantly longer in β-MyHC compared with the α-MyHC normally expressed in adult mouse hearts (42–44). However, it is difficult to detect and compare ton for mouse myosin isoform using the laser trap unless MgATP is reduced substantially below saturating concentrations to prolong the ton. Using laser trap techniques at saturating levels of MgATP, Palmiter et al. (45) observed ton in rabbit β-MyHC to be double that of α-MyHC. We previously estimated ton in mouse β-MyHC (∼43 ms) to be four times longer than that of α-MyHC (∼11 ms) using conditions, including a saturating MgATP concentration, similar to those used in this study (35). Because both the t/t and AllP-t/t express ∼40% β-MyHC (compared with nearly 95–100% α-MyHC in the NTG and WTt/t), and because of the linear relationship between α/β-MyHC ratio and myosin kinetics (46,47), we would have expected to find similarly prolonged ton at ∼24 ms for both the t/t and AllP-t/t when compared with their respective controls with ton ∼11 ms. Instead, we found ton ∼15 ms for t/t and ∼11 ms AllP-t/t, which are much shorter than the 24 ms predicted based on the MyHC isoform. These results suggest that cross-bridge-dependent mechanics, which dominate the mechanical response to length perturbations at maximum calcium activation, do not strictly depend on the MyHC isoform, and that ton may be shortened by the absence of cMyBP-C in the t/t and by the unphosphorylated state of cMyBP-C in the AllP-t/t.
To further examine the influence of MyBP-C on myosin cross-bridge ton without the complication of differential MyHC isoform profiles, we calculated myosin cross-bridge ton in NTG and t/t mice that were made hypothyroid by the PTU diet and thereafter expressed predominantly β-MyHC (Fig. 1 B). We performed small length perturbation analysis under the same conditions described above. The mean cross-bridge ton was significantly shorter (i.e., shortened by 1/3) in the t/tPTU compared with the NTGPTU (Table 1). Because both the NTGPTU and t/tPTU express 90–100% β-MyHC, we attribute the shorter ton in the t/tPTU to its lack of cMyBP-C. We also estimated myosin cross-bridge ton in the WTt/t and AllP-t/t that had been treated with T4, which induced an increased expression of α-MyHC (Fig. 1 B). Myosin ton was shorter in the AllP-t/tT4 compared with WTt/tT4 and compared with AllP-t/t without T4 treatment (Table 1). These data confirm that myosin ton is shortened by the absence of the cMyBP-C in the t/t and by the unphosphorylated state of cMyBP-C in the AllP-t/t.
Under rigor conditions, the t/t mice demonstrated significantly reduced elastic and viscous moduli compared with NTG controls over the majority of frequencies examined (Fig. 4 A), as reported previously (12,34). The elastic and viscous moduli were not different between the AllP-t/t and WTt/t (Fig. 4, C and D), which would indicate that the mutations in the cMyBP-C N-terminus did not significantly influence the longitudinal viscoelastic properties of the sarcomere.
Figure 4.
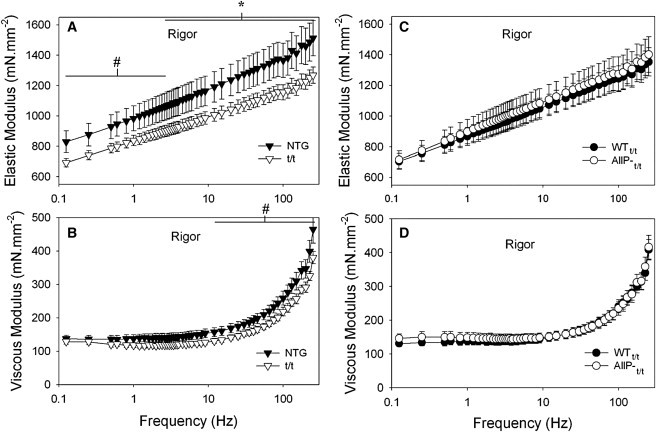
Frequency characteristics of the elastic and viscous moduli under rigor conditions and 17°C. (A and B) Values for the elastic and viscous moduli were significantly reduced in the t/t myofilaments over several frequencies compared with NTG. The frequency characteristics of the moduli were also shifted to lower frequencies in the t/t. (C and D) Elastic and viscous moduli in AllP-t/t are similar to those of WTt/t. ∗Different from control population by t-test (p < 0.05); n = 8 NTG, 8 t/t, 8 WTt/t, and 8 AllP-t/t.
Discussion
There are three important findings of this study: 1), Both the t/t and AllP-t/t demonstrated significant myofilament lattice compression due to rigor compared with controls. This result may be due to the lack of cMyBP-C (t/t) and the unphosphorylated state of cMyBP-C (AllP-t/t) independently leading to an abnormally high radial compressibility of the cardiac myofilament lattice by rigor cross-bridges, although it is also possible that the ∼40% density of β-MyHC in these populations may underlie the higher compressibility compared with the controls, which comprise nearly 100% α-MyHC. 2), The absence of cMyBP-C led to abnormally lower longitudinal stiffness of the sarcomere. 3), The absence and unphosphorylated state of cMyBP-C also led to significantly shorter cross-bridge ton independently of the MyHC isoform.
N-terminus, lattice structure, and compressibility
Previous findings from skinned skeletal muscle suggest that calcium activation and rigor conditions compress the myofilament lattice due to acto-myosin cross-bridge formation (39,40,48,49), whereas observations from skinned or intact cardiac muscle suggest no differences in interfilament spacing under relaxed, activated, or rigor conditions (38,41). The greater relative compressibility of the skeletal myofilament lattice compared with the cardiac myofilament lattice due to cross-bridge formation may reflect the greater tendency for cardiac myosin to bind weakly to the actin filament under relaxed and partially activated conditions (41). Other structural differences between skeletal and cardiac myofilaments that could be responsible for these disparate observations in myofilament lattice compressibility are the smaller size, smaller number, and possibly dissimilar function of myosin-binding protein-C in the skeletal muscle sarcomere compared with cardiac muscle (8).
The x-ray data presented here suggest a more compressible myofilament lattice in the t/t and AllP-t/t under rigor conditions, which suggests that the presence of cMyBP-C or the phosphorylation of Ser-273 and -282 contribute significantly to the radial stiffness of the cardiac myofilament lattice. However, we cannot exclude the possibility that the incorporation of β-MyHC in the t/t and AllP-t/t results in greater radial compression during rigor induction or leads to greater radial compliance of the lattice, although there is no evidence for or against either possibility at this time. There is evidence for similar myofilament lattice spacing for α-MyHC versus β-MyHC in intact ferret ventricular muscle under relaxed conditions (50). In addition, we would not expect the incorporation of β-MyHC to lead to the greater myofilament disorder we observed in the t/t and AllP-t/t. We are therefore biased toward the cMyBP-C explanation regarding increased lattice compliance in the t/t and AllP-t/t, although further studies are clearly warranted. Based on our current results, one might expect that a fully phosphorylated cMyBP-C, such as that provided by the AllP+t/t mouse bearing aspartic acid residues instead of alanines at the sites targeted in the AllP-t/t, would lead to a more rigid myofilament lattice and greater myofilament order like that observed in the NTG and WTt/t (28). This possible structural result for AllP+t/t mice could underlie in part the rescue of t/t with the fully phosphorylated cMyBP-C (28).
In another study of whole-heart function in the same populations used here (51), both the t/t and AllP-t/t demonstrated blunted responses to isoproterenol, which initiates protein kinase-A (PKA)-mediated phosphorylation of several myofibrillar proteins, including cMyBP-C. Other studies have also demonstrated blunted cardiac myofibrillar response to PKA in the cMyBP-C knockout mouse (23,26). Our data correlate with these findings and suggest the possibility that reduced PKA responsiveness in the t/t and AllP-t/t are due to a loss of myofilament lattice radial stiffness that occurs with missing or unphosphorylatable Ser-273 and -282 of cMyBP-C.
C-terminus, longitudinal stiffness, and cross-bridge lifetime
Previous studies into the functional effects of an undetectable amount (t/t) or a true knockout (−/−) of cMyBP-C in the mouse revealed the development of a cardiomyopathy characterized in part by reduced rates of relaxation (9–11). A shorter myosin ton due to the absence of cMyBP-C would contribute to a faster rate of relaxation in the −/− (10,11,36) and t/t (9,33,34). Our measure of a shorter myosin ton is therefore not consistent with slowed relaxation rates due to a lack of MyBP-C. However, the enhanced rate of force redevelopment in myofilaments of the −/− (15,52,53), unloaded shortening velocity in the −/− (54) and t/t (33), and frequency of oscillatory work in the t/t (34) are consistent with our finding of a shorter ton due to a lack of cMyBP-C. We propose that cMyBP-C normally provides a significant elastic and viscous load, which would inhibit contractile performance and yet provide a means of storing elastic recoil energy to facilitate subsequent relaxation function. In other words, we believe that the presence of cMyBP-C allows for storage of mechanical recoil energy in the sarcomere. The greater recoil energy stored during systole in the stiffer cMyBP-C-containing sarcomere would in turn drive the relaxation function more so than in the compliant cMyBP-C-lacking sarcomere.
A diminished and abbreviated systolic elastance has been observed in −/− and t/t mice but not in AllP-t/t or other mice with clearly developed cardiomyopathies (33,51). The abbreviated systolic elastance characteristic appears to be due to the missing C-terminus, which is normally incorporated into the thick filament and provides significant structural support to account for ∼40% of thick filament stiffness (13). We report here that myofilament stiffness, measured as the elastic moduli at very high frequencies, is significantly lower in the t/t but not different in AllP-t/t compared with controls expressing WT cMyBP-C. One important structural dissimilarity between the t/t and the NTG, AllP-t/t, and WTt/t is again the missing C-terminus in the t/t. A more compliant thick filament, as observed in the t/t, would theoretically reduce the backward-going or resistive strain on the myosin head in its post-power-stroke confirmation. The result at the single-molecule level (55,56) would be a faster ADP off-rate and a shorter ton, as we observed here in the t/t and AllP-t/t. In this case, we might conclude that the more compliant thick filament in the t/t, and possibly the greater radial compliance in the t/t and AllP-t/t, would shorten or abbreviate myosin cross-bridge ton.
Conclusion
Collectively, our data suggest a structural role for cMyBP-C that provides mechanical rigidity to the myofilament lattice as well as the thick filament (13). We propose that the cMyBP-C N-terminus must be structurally intact and phosphorylated at Ser-273 and -282 in the mouse cMyPB-C to maintain normal myofilament order and possibly also lattice spacing. We propose that the increased stiffness of the thick filament and possibly also the lattice provided by cMyBP-C lead to a greater amount of stored recoil energy available to facilitate later relengthening of the sarcomere. Our results regarding the myosin cross-bridge lifetime suggest that intact and partially phosphorylated cMyBP-C, which is available in NTG and WTt/t mice, prolongs the cross-bridge lifetime, ton, by providing a particular deformation of myosin head during the post-power stroke that is more likely to prolong the ADP state or slow ATP binding, or both. Finally, haploinsufficiency due to a mutant allele for MYBPC3, as has been found in humans (5,6), may contribute to the development of cMyBP-C-related cardiomyopathies resulting in part from the loss or degradation of cMyBP-C structure and function reported here.
Acknowledgments
We thank Dr. Mark Jennings for expert technical assistance.
This study was funded by National Institutes of Health (NIH) grants R01 HL59408 (B.M.P., Y.W., J.R., D.W.M.) and T32 HL07647 (M.J.P.). S.S. was supported by an American Heart Association Scientist Development Grant (0830311N). The mass spectrometry instrumentation was made available at the Vermont Genetics Network through grant No. P20 RR16462 from the INBRE Program of the National Center for Research Resources, a component of the NIH. Use of the Advanced Photon Source was supported by the Office of Science, Basic Energy Sciences, U.S. Department of Energy, under contract No. W-31-109-ENG-38. BioCAT is an NIH-supported research center (RR-08630). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Center for Research Resources or the NIH.
Supporting Material
References
- 1.Watkins H., Conner D., Seidman C.E. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat. Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 2.Bonne G., Carrier L., Schwartz K. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat. Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 3.Richard P., Charron P., EUROGENE Heart Failure Project Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 4.Barefield D., Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 2010;48:866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marston S., Copeland O., Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 6.van Dijk S.J., Dooijes D., van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 7.Squire J.M., Roessle M., Knupp C. New X-ray diffraction observations on vertebrate muscle: organisation of C-protein (MyBP-C) and troponin and evidence for unknown structures in the vertebrate A-band. J. Mol. Biol. 2004;343:1345–1363. doi: 10.1016/j.jmb.2004.08.084. [DOI] [PubMed] [Google Scholar]
- 8.Luther P.K., Bennett P.M., Moss R.L. Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. J. Mol. Biol. 2008;384:60–72. doi: 10.1016/j.jmb.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McConnell B.K., Jones K.A., Seidman J.G. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J. Clin. Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. Erratum in J. Clin. Invest. 1999;104:1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris S.P., Bartley C.R., Moss R.L. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 11.Carrier L., Knöll R., Chien K.R. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc. Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Palmer B.M., McConnell B.K., Maughan D.W. Reduced cross-bridge dependent stiffness of skinned myocardium from mice lacking cardiac myosin binding protein-C. Mol. Cell. Biochem. 2004;263:73–80. doi: 10.1023/B:MCBI.0000041849.60591.45. [DOI] [PubMed] [Google Scholar]
- 13.Nyland L.R., Palmer B.M., Vigoreaux J.O. Cardiac myosin binding protein-C is essential for thick-filament stability and flexural rigidity. Biophys. J. 2009;96:3273–3280. doi: 10.1016/j.bpj.2008.12.3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colson B.A., Bekyarova T., Moss R.L. Radial displacement of myosin cross-bridges in mouse myocardium due to ablation of myosin binding protein-C. J. Mol. Biol. 2007;367:36–41. doi: 10.1016/j.jmb.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colson B.A., Bekyarova T., Moss R.L. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ. Res. 2008;103:244–251. doi: 10.1161/CIRCRESAHA.108.178996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freiburg A., Gautel M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur. J. Biochem. 1996;235:317–323. doi: 10.1111/j.1432-1033.1996.00317.x. [DOI] [PubMed] [Google Scholar]
- 17.Flashman E., Redwood C., Watkins H. Cardiac myosin binding protein C: its role in physiology and disease. Circ. Res. 2004;94:1279–1289. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- 18.Zoghbi M.E., Woodhead J.L., Craig R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc. Natl. Acad. Sci. USA. 2008;105:2386–2390. doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautel M., Zuffardi O., Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunst G., Kress K.R., Fink R.H. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ. Res. 2000;86:51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- 21.Weisberg A., Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc. Natl. Acad. Sci. USA. 1996;93:8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClellan G., Kulikovskaya I., Winegrad S. Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys. J. 2001;81:1083–1092. doi: 10.1016/S0006-3495(01)75765-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stelzer J.E., Patel J.R., Moss R.L. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ. Res. 2006;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 24.Stelzer J.E., Patel J.R., Moss R.L. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ. Res. 2007;101:503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- 25.Lecarpentier Y., Vignier N., Coirault C. Cardiac myosin-binding protein C modulates the tuning of the molecular motor in the heart. Biophys. J. 2008;95:720–728. doi: 10.1529/biophysj.107.127787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colson B.A., Locher M.R., Moss R.L. Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J. Physiol. 2010;588:981–993. doi: 10.1113/jphysiol.2009.183897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadayappan S., Gulick J., Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadayappan S., Osinska H., Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Previs M.J., VanBuren P., Matthews D.E. Quantification of protein phosphorylation by liquid chromatography-mass spectrometry. Anal. Chem. 2008;80:5864–5872. doi: 10.1021/ac800337v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godt R.E., Lindley B.D. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J. Gen. Physiol. 1982;80:279–297. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irving T.C., Konhilas J., de Tombe P.P. Myofilament lattice spacing as a function of sarcomere length in isolated rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H2568–H2573. doi: 10.1152/ajpheart.2000.279.5.H2568. [DOI] [PubMed] [Google Scholar]
- 32.Fischetti R., Stepanov S., Bunker G.B. The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J. Synchrotron Radiat. 2004;11:399–405. doi: 10.1107/S0909049504016760. [DOI] [PubMed] [Google Scholar]
- 33.Palmer B.M., Georgakopoulos D., Kass D.A. Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circ. Res. 2004;94:1249–1255. doi: 10.1161/01.RES.0000126898.95550.31. [DOI] [PubMed] [Google Scholar]
- 34.Palmer B.M., Noguchi T., LeWinter M.M. Effect of cardiac myosin binding protein-C on mechanoenergetics in mouse myocardium. Circ. Res. 2004;94:1615–1622. doi: 10.1161/01.RES.0000132744.08754.f2. [DOI] [PubMed] [Google Scholar]
- 35.Palmer B.M., Suzuki T., Maughan D.W. Two-state model of acto-myosin attachment-detachment predicts C-process of sinusoidal analysis. Biophys. J. 2007;93:760–769. doi: 10.1529/biophysj.106.101626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pohlmann L., Kröger I., Carrier L. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ. Res. 2007;101:928–938. doi: 10.1161/CIRCRESAHA.107.158774. [DOI] [PubMed] [Google Scholar]
- 37.Yagi N., Okuyama H., Kajiya F. Sarcomere-length dependence of lattice volume and radial mass transfer of myosin cross-bridges in rat papillary muscle. Pflugers Arch. 2004;448:153–160. doi: 10.1007/s00424-004-1243-z. [DOI] [PubMed] [Google Scholar]
- 38.Farman G.P., Allen E.J., de Tombe P.P. Interfilament spacing is preserved during sarcomere length isometric contractions in rat cardiac trabeculae. Biophys. J. 2007;92:L73–L75. doi: 10.1529/biophysj.107.104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsubara I., Goldman Y.E., Simmons R.M. Changes in the lateral filament spacing of skinned muscle fibres when cross-bridges attach. J. Mol. Biol. 1984;173:15–33. doi: 10.1016/0022-2836(84)90401-7. [DOI] [PubMed] [Google Scholar]
- 40.Matsubara I., Umazume Y., Yagi N. Lateral filamentary spacing in chemically skinned murine muscles during contraction. J. Physiol. 1985;360:135–148. doi: 10.1113/jphysiol.1985.sp015608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsubara I., Maughan D.W., Yagi N. Cross-bridge movement in rat cardiac muscle as a function of calcium concentration. J. Physiol. 1989;417:555–565. doi: 10.1113/jphysiol.1989.sp017818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alpert N.R., Brosseau C., Warshaw D.M. Molecular mechanics of mouse cardiac myosin isoforms. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H1446–H1454. doi: 10.1152/ajpheart.00274.2002. [DOI] [PubMed] [Google Scholar]
- 43.Tyska M.J., Warshaw D.M. The myosin power stroke. Cell Motil. Cytoskeleton. 2002;51:1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- 44.Sugiura S., Kobayakawa N., Sugi H. Comparison of unitary displacements and forces between 2 cardiac myosin isoforms by the optical trap technique: molecular basis for cardiac adaptation. Circ. Res. 1998;82:1029–1034. doi: 10.1161/01.res.82.10.1029. [DOI] [PubMed] [Google Scholar]
- 45.Palmiter K.A., Tyska M.J., Warshaw D.M. Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. J. Physiol. 1999;519:669–678. doi: 10.1111/j.1469-7793.1999.0669n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herron T.J., Korte F.S., McDonald K.S. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H1217–H1222. doi: 10.1152/ajpheart.2001.281.3.H1217. [DOI] [PubMed] [Google Scholar]
- 47.Korte F.S., Herron T.J., McDonald K.S. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H801–H812. doi: 10.1152/ajpheart.01227.2004. [DOI] [PubMed] [Google Scholar]
- 48.Maughan D.W., Godt R.E. Radial forces within muscle fibers in rigor. J. Gen. Physiol. 1981;77:49–64. doi: 10.1085/jgp.77.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawai M., Wray J.S., Zhao Y. The effect of lattice spacing change on cross-bridge kinetics in chemically skinned rabbit psoas muscle fibers. I. Proportionality between the lattice spacing and the fiber width. Biophys. J. 1993;64:187–196. doi: 10.1016/S0006-3495(93)81356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yagi N., Saeki Y., Kurihara S. Cross-bridge and calcium behavior in ferret papillary muscle in different thyroid states. Jpn. J. Physiol. 2001;51:319–326. doi: 10.2170/jjphysiol.51.319. [DOI] [PubMed] [Google Scholar]
- 51.Nagayama T., Takimoto E., Kass D.A. Control of in vivo left ventricular [correction] contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation. 2007;116:2399–2408. doi: 10.1161/CIRCULATIONAHA.107.706523. [DOI] [PubMed] [Google Scholar]
- 52.Stelzer J.E., Dunning S.B., Moss R.L. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ. Res. 2006;98:1212–1218. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- 53.Stelzer J.E., Fitzsimons D.P., Moss R.L. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys. J. 2006;90:4119–4127. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korte F.S., McDonald K.S., Moss R.L. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 2003;93:752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 55.Veigel C., Molloy J.E., Kendrick-Jones J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat. Cell Biol. 2003;5:980–986. doi: 10.1038/ncb1060. [DOI] [PubMed] [Google Scholar]
- 56.Kad N.M., Patlak J.B., Warshaw D.M. Mutation of a conserved glycine in the SH1-SH2 helix affects the load-dependent kinetics of myosin. Biophys. J. 2007;92:1623–1631. doi: 10.1529/biophysj.106.097618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.