

Abstract
Several voltage-gated ion channels are modulated by clinically relevant doses of general anesthetics. However, the structural basis of this modulation is not well understood. Previous work suggested that n-alcohols and inhaled anesthetics stabilize the closed state of the Shaw2 voltage-gated (Kv) channel (K-Shaw2) by directly interacting with a discrete channel site. We hypothesize that the inhibition of K-Shaw2 channels by general anesthetics is governed by interactions between binding and effector sites involving components of the channel's activation gate. To investigate this hypothesis, we applied Ala/Val scanning mutagenesis to the S4-S5 linker and the post-PVP S6 segment, and conducted electrophysiological analysis to evaluate the energetic impact of the mutations on the inhibition of the K-Shaw2 channel by 1-butanol and halothane. These analyses identified residues that determine an apparent binding cooperativity and residue pairs that act in concert to modulate gating upon anesthetic binding. In some instances, due to their critical location, key residues also influence channel gating. Complementing these results, molecular dynamics simulations and in silico docking experiments helped us visualize possible anesthetic sites and interactions. We conclude that the inhibition of K-Shaw2 by general anesthetics results from allosteric interactions between distinct but contiguous binding and effector sites involving inter- and intrasubunit interfaces.
Introduction
Discoveries made over the past two decades support the notion that proteins contain relatively specific general anesthetic sites, with ion channels perhaps being the most physiologically relevant (1–5). Ligand-gated, voltage-gated, and nongated ion channels are among the best-known targets. Thus, it is hypothesized that direct modulation of diverse ion channels by general anesthetics may explain distinct physiological end points of anesthesia and common side effects of general anesthetics (2,3). Although this hypothesis has gained recognition in recent years, and crystallographic studies are confirming the presence of discrete general anesthetic sites in ion channels (6), several fundamental questions remain unanswered. What common structural features of protein cavities permit the interaction of general anesthetics with diverse and structurally distinct ion channels? What are the structural bases of the link between cavity occupancy and functional modulation by anesthetics? Answering these questions is a key step toward understanding the molecular mechanisms of general anesthesia and designing anesthetics with fewer side effects and higher therapeutic indices.
We previously demonstrated that the voltage-gated K+ (Kv) channel Shaw2 (K-Shaw2) is rapidly and reversibly inhibited by clinically relevant anesthetic doses of short- and medium-chain n-alcohols (C2-C6) and inhaled anesthetics (halothane and isoflurane) (7–11). In particular, we showed that halothane and 1-butanol (1-BuOH) share a putative binding site in the channel (7), and that n-alcohols stabilize the closed state(s) (9). Related studies have also shown that the S4-S5 linker is sufficient and necessary to confer modulation by n-alcohols and general anesthetics, and that the α-helicity of this linker is critical for the modulatory action (7,9,12–14). Moreover, the second proline in the PVP motif of the S6 segment (P410) is also unique because the mutation P410A switches the modulatory response from inhibition to potentiation (12,15). These studies led to the notion of an amphiphilic anesthetic cavity involving two key components of the activation machinery of Kv channels: the S4-S5 linker and the distal part of the S6 segment. Interactions between these regions are the basis of the electromechanical coupling underlying voltage-dependent gating of voltage-gated ion channels (16–18). Although these studies generally support the contributions of the K-Shaw2 activation gate to general anesthetic action, systematic molecular mapping is necessary to locate the sites of action. We hypothesize that the inhibition of K-Shaw2 channels by n-alcohols and inhaled anesthetics is governed by the allosteric cross talk between binding and effector sites at the channel's activation gate. These sites may overlap or exhibit distinct locations.
To test this hypothesis, we investigated the actions of 1-BuOH and halothane by combining systematic Ala/Val scanning mutagenesis (Fig. 1), electrophysiological evaluation of dose-response relations, and molecular modeling. These experiments revealed that the apparent binding cooperativity depends on critical interacting residues in the S4-S5 linker and the post-PVP S6 segment (Fig. 1), which in some instances also influence the energetics of anesthetic action and voltage-dependent gating. Additional observations demonstrated significant nonadditive energetic interactions between residue pairs possibly acting as anesthetic effector sites. Complementing these results, molecular dynamics (MD) simulations and in silico docking experiments yielded a plausible atomistic map displaying distinct but contiguous binding and effector sites formed by the moving parts of the channel's activation gate. This study sets the stage to investigate the molecular mechanisms underlying the relevant modulation of voltage-gated Na+, Ca+, and nonselective cation channels by general anesthetics (19–25).
Figure 1.
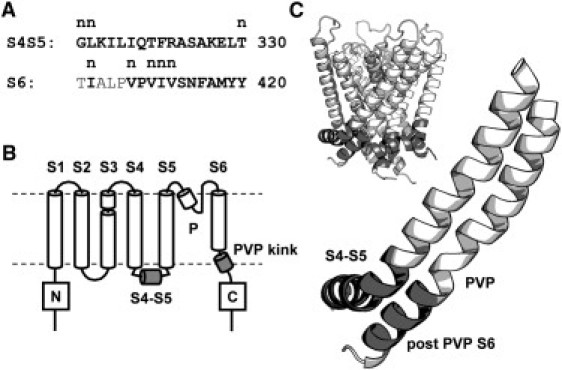
Topology and structure of the S4-S5 linker and S6 segment in the K-Shaw2 channel, with the Ala/Val scan region highlighted in gray in B and C. (A) Sequences of the S4-S5 linker and S6 segment (mutated residues shown in bold). A lowercase “n” above the sequence indicates nonexpressing mutants; numbers indicate the number of the final residue in each segment. (B) K-Shaw2 exhibits classical Kv channel membrane topology with six TM segments and intracellular termini. The first four membrane-spanning segments form the voltage sensor, and the last two segments form the pore domain. (C) Structural model of the S4-S5 linker and the S6 segment of K-Shaw2. The inset depicts a tetrameric model of the K-Shaw2 pore domain, including the S4-S5 linkers. This homology model was created by MD simulation (Supporting Material).
Materials and Methods
Experimental details about the Ala/Val scanning mutagenesis, heterologous expression, electrophysiology, MD simulations, and in silico docking are described in the Supporting Material, which includes a summary of definitions (Table S1).
Results
Anesthetic dose-inhibition relations of wild-type and mutant K-Shaw2
To map the contributions of individual residue side chains to putative anesthetic binding and effector sites in K-Shaw2 and the resulting inhibition, we applied Ala/Val scanning mutagenesis to the S4-S5 linker and the post-PVP S6 segment, and functional characterization in Xenopus oocytes under two-electrode voltage-clamp conditions (Supporting Material). These regions were chosen because 1), they play central roles in Kv channel gating, and general anesthetics influence K-Shaw2 gating; and 2), previous work has demonstrated the importance of these regions in the modulation of K-Shaw2 by general anesthetics. As shown previously, both n-alcohols (e.g., 1-BuOH) and halothane inhibit wild-type K-Shaw2 currents in a reversible and dose-dependent manner (Fig. 2, A and B). Assuming that the Hill equation describes the average dose-response curves (Supporting Material), we obtained best-fit parameters of K0.5 = 11.2 mM and nH = 1.6 for 1-BuOH, and K0.5 = 0.26 mM and nH = 1.2 for halothane (Fig. 2 B, Table S2, and Supporting Material). These values are comparable to those previously reported and indicate that K-Shaw2 is sensitive to clinically relevant doses of halothane (minimum alveolar concentration = 0.25 mM). Because 1-BuOH is significantly less volatile than halothane, and these drugs share a putative binding site in K-Shaw2 (7), we first used 1-BuOH as a tool to probe the inhibition of all mutants (Table S2). This initial screening allowed us to select mutants for subsequent investigation with halothane. Several single S4-S5 linker mutations had modest but significant effects on the K0.5; Q320A, T321V, F322A, and L329A increased it, whereas S325A and A326V decreased it. By contrast, most mutations in the post-PVP S6 segment (A417V, M418A, Y419A, and Y420A) increased the K0.5 to a greater extent (e.g., Fig. 2 C). Several mutations had a relatively modest impact on the nH, which typically remained between 1.4 and 2. However, a few yielded larger changes. The nH-values of T321V, E328A, and L329A in the S4-S5 linker, and Y420A in S6 are reduced (1.2, 1.2, 1.2, and 1.1, respectively), whereas the nH-values of K316A, S414A, and M418A are increased (2.1, 2.1, and 2.2, respectively). Relative to single mutations, the impact of double mutations on K0.5 and nH is generally larger. Although one double mutation within the S4-S5 linker (Q320A/A326V) affected the 1-BuOH response significantly, even larger changes were caused by paired mutations across the putative interface between the S4-S5 linker and the post-PVP S6 segment (e.g., Fig. 2 C and Table S2). The K0.5-values for Q320A/A326V, Q320A/A417V, Q320/M418A, Q320A/Y420A, A326V/A417V, and A326V/Y419A are 32, 47, 22, 48, 94, and 58 mM, respectively. The nH-values for A326V/A417V and A326V/Y419A are 4.5 and 3.4, respectively, and thus are the largest of the entire scan. Nevertheless, across the entire scan, there is no significant correlation between K0.5 and nH (Fig. S1 B).
Figure 2.
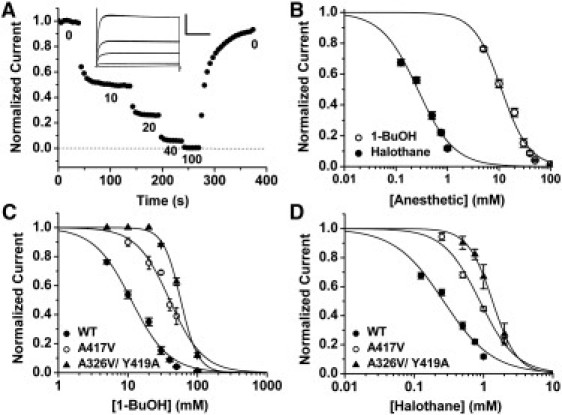
Dose-inhibition analysis of the wild-type K-Shaw2 channel and representative Ala/Val mutants. (A) Normalized current versus time plot of wild-type K-Shaw2. Upon application of increasingly higher doses of 1-BuOH (mM), there is an incremental inhibition of the current. This inhibition reaches equilibrium and is reversible upon washout. The inset shows the whole-oocyte K-Shaw2 currents corresponding to the experiment shown in the main panel evoked by a step from −100 to +60 mV; scale for inset indicates 4 μA (y axis) and 100 ms (x axis). All mutants were subjected to this experimental strategy. (B) Semilogarithmic plots of normalized current versus anesthetic concentration for wild-type K-Shaw2. The solid lines depict the best Hill equation fits, which yielded the following parameters for halothane: K0.5 = 0.26 mM, nH = 1.2; and the following best-fit parameters for 1-BuOH: K0.5 = 11.2 mM, nH = 1.6. (C) The effects of Ala/Val mutations on the 1-BuOH dose-inhibition relation. A417V shifted the relation to the right (K0.5 = 39.4 mM, nH = 1.7) and the double mutation A326V/Y419A caused an even larger shift and increased the apparent cooperativity (K0.5 = 57.7 mM, nH = 3.4). (D) The effects of Ala/Val mutations on the halothane dose-inhibition relation. A417V showed a rightward shift (K0.5 = 0.88, nH = 1.7), and the double mutation A326V/Y419A exhibited an even larger shift (K0.5= 1.34, nH = 2.5). In all cases (B–D), the points represent the average of 4–45 independent determinations.
We next examined the properties of the halothane dose-response curve for a group of selected single and double mutants with significantly altered responses to 1-BuOH (Fig. 2 D and Table S3). The K0.5-values of Q320A, A326V, A417V, and Y420A are clearly increased (0.5, 1.2, 0.9, and and 1 mM, respectively), and the nH-values are modestly changed for Q320A, S325A, A326V, A417V, and Y419A (1.6, 1, 1.6, 1.6, and 1, respectively). Three double mutations have a more profound impact on the K0.5 of halothane: Q320A/A326V, Q320A/Y420A, and A326V/A417V yielded 1.8, 1.4, and >>4 mM, respectively. Essentially, A326V/A417V is insensitive to halothane, and whereas the nH for Q320A/A326V is 2.8, that of A326V/A417V is 1.8. As for 1-BuOH, there was no significant correlation between the K0.5- and nH-values of halothane (Fig. S1 B). Nevertheless, consistent with the presence of overlapping sites for 1-BuOH and halothane in K-Shaw2, the overall impact of the mutations on the K0.5 for these anesthetics is similar (e.g., compare Fig. 2, C and D), hence there is a significant correlation between the K0.5-values of 1-BuOH and halothane (Fig. S1). These results add new support to the notion of a general anesthetic action involving the interface between the S4-S5 linker and the post-PVP S6 segment of K-Shaw2.
Energetic impact of mutations on the modulation of K-Shaw2 by general anesthetics
To evaluate the energetic impact of all mutations on the modulation of K-Shaw2 by the anesthetics, we used the parameters derived from the analysis of dose-response curves to calculate the apparent free-energy change (ΔG = nHRTlnK0.5; Supporting Material). The Hill coefficient (nH), which is >1 in the wild-type and changes significantly in several mutants (Table S2 and Table S3), scales the free-energy change. Then, we defined ΔΔG = ΔGMUT – ΔGWT. For 1-BuOH, the absolute impact of 12/15 single S4-S5 linker mutations is relatively small or modest (≤1 kcal/mol; Fig. 3 A and Table S2). However, the ΔΔG of the remaining three mutants (E328A, L329A, and T321V) is significantly larger and unfavorable (+1.4, +1.5, and +1.5 kcal/mol, respectively). The energetic impact of single S6 mutations is also modest, but in two instances it is >1 kcal/mol (+1.2 and +2.1 kcal/mol for A417V and Y420A, respectively). In addition, single and double mutations that change the nH have the most significant energetic impact. To further examine this interaction, we plotted the ΔΔG of all single and double mutants against the nH and found a significant correlation (Fig. 3 C). In contrast, ΔΔG and K0.5 are not correlated (Fig. S1 C). We then examined selected double mutants, including the S4-S5 linker and S6 mutations, that significantly affected nH and/or K0.5 (e.g., Q320A, T321V, S325A, A326V, T330V, M418A, A417V, Y419A and Y420A; Table S2), and applied double-mutant cycle analysis (DMCA) to investigate the additivity of the energetic impacts of the mutations (Fig. 3 B; Table S2). This approach can help identify putative anesthetic effector sites. In this context, effector sites are residues that directly or allosterically interact with each other to control how the anesthetic perturbs gating upon interaction with a binding site. The coupling energies of 7/13 double mutants (Q320A/T321V, Q320A/M418A, S325A/A326V, S325A/A417V, A326V/A417V, A326V/Y419A, and A326V/Y420A) are substantial (−3.5, 1, −2.1, −1.5, −2, −2.9, and −4 kcal/mol, respectively).
Figure 3.
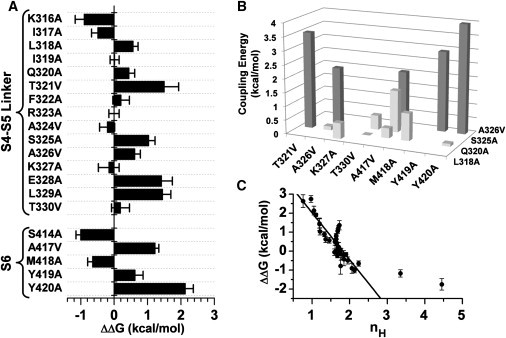
Energetic impact of Ala/Val mutations on the modulation of K-Shaw2 by 1-BuOH. (A) Apparent free-energy changes induced by single S4-S5 and S6 mutations. These changes are calculated relative to the wild-type apparent free-energy change as indicated in the main text. (B) Absolute coupling energies for double K-Shaw2 mutants; note that A326V appears on both axes due to intra-S4-S5 mutations. DMCA was used to calculate these energies as described in Table S1. Dark gray bars depict coupling energies > 2 kcal/mol. (C) Scatter plot of ΔΔG versus nH. The line is the best-fit linear regression (R2 = 0.75). Two outliers with high nH-values were excluded from the regression analysis. These mutants exhibit very low 1-BuOH sensitivities, and very high doses were not investigated, which limited the estimation of the Hill equation parameters (Table S1).
Overall, the thermodynamic analysis indicates that specific residues in the S4-S5 linker and the post-PVP S6 segment may play significant structural roles in determining the action of general anesthetics on K-Shaw2. Furthermore, the DMCA results suggest that direct or indirect interactions between residue pairs (Q320A/T321V, S325A/A326V, A326V/A417V, A326V/Y419A, and A326V/Y420A) are especially critical, and the energetic impact of the mutations on anesthetic action is closely tied to changes in the apparent cooperativity (nH) of anesthetic action. To visualize the key residues on the channel structure, we created an atomic membrane equilibrated MD model of K-Shaw2 (Supporting Material) and mapped the energetic effects (ΔΔG) of the mutations (Fig. S2). This map reveals distinct interfacial patches involving relatively high-impact residues in the S4-S5 linker and the post-PVP S6 segment.
Based on the effects of mutations on the Hill equation parameters for 1-BuOH, we also investigated the energetic impact of selected single and double mutations on the interaction between halothane and K-Shaw2 (Fig. 4 A; Table S3). Despite some significant changes in the Hill equation parameters, the ΔΔG values of individual S4-S5 linker and S6 mutants (e.g., Q320A, S325A, A326V, A417V, M418A, Y419A, and Y420A) are relatively small (−1.2, +1.1, −0.5, −0.9, −1.1, +1.2, and −0.1 kcal/mol). Nevertheless, the coupling energies of 5/6 double mutants tested with halothane and subjected to DMCA are substantial (Fig. 4 B). Q320A/A326V, S325A/A326V, S325A/A417V, and A326V/Y419A yield 2.8, 2.6, 2.8, and 4.3 kcal/mol, respectively, and A326V/A417V, which virtually eliminates the inhibition by halothane, has an even larger coupling energy (≫4 kcal/mol). Moreover, as found for 1-BuOH, ΔΔG and nH are highly correlated (Fig. 4 C). In general, the results obtained with halothane corroborate the significance of residues in the S4-S5 linker and the post-PVP S6 segment as important players in the interaction of K-Shaw2 with general anesthetics.
Figure 4.
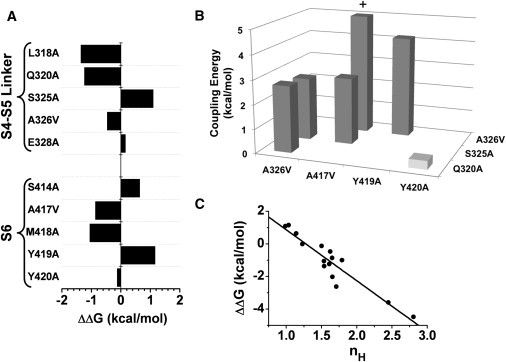
Energetic impact of Ala/Val mutations on the modulation of K-Shaw2 by halothane. (A) Apparent free-energy changes induced by single S4-S5 and S6 mutations (see legend to Fig. 3 for additional information). (B) Absolute coupling energies for double K-Shaw2 mutants (see legend to Fig. 3). The + sign above the tallest bar indicates that the coupling energy is estimated to be ≫5 kcal/mol. (C) Scatter plot of ΔΔG versus nH. The line is the best-fit linear regression (R2 = 0.87).
Interplay between voltage-dependent gating and general anesthetic action
The S4-S5 linker and the post-PVP S6 segment play critical roles in the electrochemical coupling that underlies voltage-dependent gating (16–18). To assess whether the effects of mutations on the inhibition by 1-BuOH and halothane are byproducts of possible gating effects, we measured the shift in activation voltage (ΔV), time to 50% activation (T0.5), and degree of inactivation at the end of a 400-ms pulse to +70 mV (I400/Ipeak; Supporting Material and Table S4). If anesthetics preferentially interact with resting/closed K-Shaw2 channels, the energetics of anesthetic action would be proportional to the relative stability of the closed conformation(s). Accordingly, some mutants (E328A, L329A, A417V, Q320A/A326V, and Q320A/A417V) exhibit relatively large leftward shifts (>5 mV) in voltage-dependent activation (i.e., a relative destabilization of closed states) and corresponding positive ΔΔG values (>1 kcal/mol) calculated from experiments at a fixed voltage that most likely does not saturate the open probability (Supporting Material and Table S4).
These results suggest that any other mutation(s) whose main effect would be to shift voltage-dependent gating might also secondarily affect anesthetic action in other Kv channels, and the inhibition by the anesthetics might exhibit state dependence. To test these premises, we exploited the triple ILT mutation (V369I, I372L, and S376T) in the voltage sensor of ShakerB channels, which confers a depolarized K-Shaw2-like activation phenotype (26). Relative to the wild-type counterpart, the activation curve of the ShakerB-ILT mutant exhibits a dramatic rightward shift of +123 mV, demonstrating a strong relative stabilization of the channel's closed state(s) (Fig. S3 A). Despite this large change, the ShakerB-ILT mutant is resistant to 1-BuOH (K0.5 = 47 mM, nH = 1.4; Fig. S3 C) when tested at a strongly depolarized voltage (+100 mV) corresponding to ∼50% activation. Testing it at less depolarized voltages (+70 to +80) modestly increases the inhibition to a maximum of ∼70% at 50 mM 1-BuOH (Fig. S3 B). Interestingly, ShakerB wild-type yields similar anesthetic resistance (K0.5 = 57 mM, nH = 1.5; Fig. S3 C) and state-dependent inhibition despite its normal voltage dependence (Fig. S4 A). Thus, the inhibition is maximal at voltages where channels may populate closed-activated states, and becomes weaker as the open state becomes progressively more represented. Nevertheless, the maximal inhibition of mutant and wild-type ShakerB channels does not match that of K-Shaw2, which is also state-dependent but nearly complete (96%) at 50 mM 1-BuOH and over a voltage range where the inhibition increases to a maximum and levels off (Fig. 2 and Fig. S4 C).
In the light of these results, we made two additional predictions: First, the converse triple VIS mutation (I302V, L304I, and T308S) in K-Shaw2 (K-Shaw2-VIS) may exhibit left-shifted voltage-dependent activation and a concomitant decrease in the apparent anesthetic affinity. Accordingly, voltage-dependent activation of the K-Shaw2-VIS mutant is somewhat leftward shifted (ΔV = −20 mV) and is slightly less sensitive to 1-BuOH than the wild-type counterpart (K0.5 = 19 mM, nH = 1.5; Fig. S3 C). Second, the nongated K-Shaw2 P410D mutant may become insensitive to general anesthetics. This mutation targets the second P in the PVP motif, rendering K-Shaw2 constitutively open (27). Thus, the current-voltage relation of K-Shaw2 P410D is approximately linear and displays a reversal potential consistent with a conductance highly selective for K+ (Fig. 5, A and B). Moreover, as an extreme example of state-dependent inhibition, the P410D mutant is highly resistant to general anesthetics (Fig. 5 C). Relative to wild-type K-Shaw2, P410D is 22 times less sensitive to 1-BuOH (K0.5 = 250 mM) and 23 times less sensitive to halothane (K0.5 = 6.1 mM). This result also confirms previous conclusions that general anesthetics do not significantly affect the properties of the open state at the unitary level (9).
Figure 5.
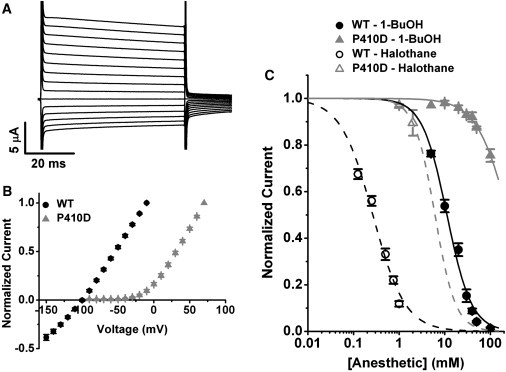
General anesthetic action on the constitutively open K-Shaw2 P410D mutant channel. (A) P410D currents evoked from a holding voltage of −100 mV by voltage steps ranging from −150 to 0mV at 10-mV intervals. (B) Comparison of normalized current-voltage relations from wild-type and the P410D mutant. The current of the constitutively open P410D mutant reverses polarity at −98 mV, as expected for a K+-selective Kv channel. (C) Comparison of the 1-BuOH and halothane dose-inhibition plots from wild-type and the P410D mutant. For the P410D mutant, the dashed and solid gray lines are the best Hill equation fits with the following parameters for 1-BuOH: K0.5 = 250 mM, nH = 1.2; and the following best-fit parameters for halothane: K0.5 = 6.1 mM, nH = 1.9. Wild-type data are replotted from Fig. 2.
Although the examples described above clearly link voltage-dependent gating to the modulation of K-Shaw2 by general anesthetics, there is no systematic general correlation between the energetic impact of all Ala/Val mutations and the gating properties of the K-Shaw2 channel (Fig. S1 and Table S4). Only in a few of the cases mentioned above (∼15% of mutants) did this correlation become apparent. In a larger number of cases (45%), activation gating changes are not associated with significant changes in the energetic impact of the mutations, and in some others the effects are inversely related (T321V, Q320A/Y420A, A326V/A417V, and A326V/Y419A). Thus, mutations that affect gating may also affect anesthetic action because the inhibition is gating-state-dependent; however, the lack of a general relationship suggests that many mutations in the investigated regions primarily influence binding and/or effector sites of general anesthetics in K-Shaw2.
Modeling the K-Shaw2 structures in the open and closed states
To investigate putative general anesthetic binding sites at the atomic level, we first created full-atomistic structural models for the transmembrane domain (TM) of K-Shaw2 by means of comparative protein modeling (28). Models of the resting-closed and activated-open states of K-Shaw2 were based on previously reported modeling of the homologous ShakerB-like Kv1.2 channel (Supporting Material). The K-Shaw2 models were equilibrated in a fully hydrated phospholipid membrane by an ∼18 ns MD simulation. In each of the channel subunits in the tetrameric channel, the TM remained stable in its starting resting-closed or activated-open conformations throughout the simulations (Fig. S5). In the MD run, the root mean-square deviation values for the whole TM domain, as well as for segments S1–S6 and the S4-S5 linker, range from 1.0 to 3.0 Å (Fig. S6), which agrees with the structural drift quantified in previous simulation studies of KcsA (29) and other K+ channels (30,31). The good stereochemical quality of the relaxed K-Shaw2 structures was confirmed by further Procheck analyses (32).
In silico docking of 1-BuOH and halothane onto K-Shaw2
Whereas the resting closed state of K-Shaw2 is preferentially sensitive to general anesthetics (see above), Kv1.2 is resistant. High anesthetic doses (50 mM 1-BuOH or 2 mM halothane) inhibit this channel by ≤20% (data not shown). We exploited these differences to identify likely general anesthetic cavities in membrane-equilibrated atomistic models of K-Shaw2 (closed and open) and Kv1.2 (closed). We subjected each isoform and conformation to in silico docking by probing the binding of both 1-BuOH and halothane to 10 equilibrium structures to account for the influence of molecular flexibility on ligand binding (Supporting Material). Then, we used a subtraction method to resolve unique 1-BuOH and halothane docking sites on the K-Shaw2 closed structure (Supporting Material). To carry out this strategy, we first estimated the effective molecular binding constant (Bi) of the ligand for a given binding site i on the receptor structures. We computed Bi as the ligand energy-weighted binding probability by considering the entire space of docking solutions. We then compared the affinity of the ligand for a given site on the K-Shaw2 closed conformation with that of the K-Shaw2 open conformation and the Kv1.2 closed conformation by quantifying the difference between the effective molecular binding constants:
The in silico docking analyses revealed that 1-BuOH binds to K-Shaw2 (closed and open) and Kv1.2 (closed) at multiple sites with a diverse range of binding affinities (Fig. S7). Specifically, 1-BuOH binds to eight distinct locations on the K-Shaw2 closed structure (hereafter called sites 1–8). The calculation also shows that 1-BuOH binds to the K-Shaw2 open conformation through all of these sites except site 3. Despite a similar pattern for the interaction with the channel in both conformations, 1-BuOH interacts with the closed K-Shaw2 conformation with enhanced affinities in sites 3, 4, 6, and 8. Compared with the Kv1.2 closed structure, sites 3, 6, and 8 in the closed K-Shaw2 channel also exhibit higher affinity. Further docking calculations demonstrate that halothane binding sites on the K-Shaw2 closed structure overlap with all sites determined for 1-BuOH but site 6 (Fig. 6). The subtraction analysis identified sites 2, 3, and 5 as possible halothane interaction spots in the closed K-Shaw2 conformation. Compared with K-Shaw2 (open) and Kv1.2 (closed), these sites display enhanced affinities.
Figure 6.
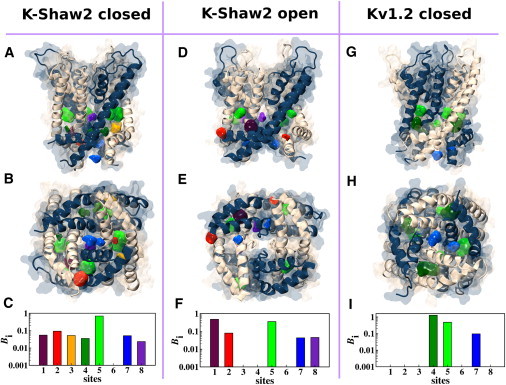
In silico docking of halothane to K-Shaw2 (closed and open conformations) and Kv1.2 (closed). Shown are lateral (A) and bottom (B) views of K-Shaw2 in the closed conformation (S4-S5 linker through the S6 segment). In silico docking solutions returned eight possible sites (sites 1–8; Supporting Material). Colored blobs represent the computed density isosurface of the ligand on a given receptor site. (C) Calculated ligand-binding constants (Bi) for sites 1–8 (Supporting Material). (D–F) Idem for the open conformation of K-Shaw2. (G–I) Idem for the closed conformation of Kv1.2. Bi = 0 indicates that the ligand has no affinity for the site.
Although they are structurally heterogeneous, the identified sites are cavities located at the interfacial region between helical segments of the channel, i.e., the S4-S5 linker, S5, and S6. For a cutoff distance of ≤4.6 Å, Table 1 highlights the amino acid side chains of K-Shaw2 that may interact with the anesthetics at these sites. Sites 3, 5, and 8 are delineated by hydrophobic amino acids, whereas sites 2 and 6 are predominantly hydrophilic. Overall, whereas K-Shaw2 inhibition by 1-BuOH implicates sites 3, 6, and 8, the inhibition of the channel by halothane implicates sites 2, 3, and 5. Thus, the intersubunit site 3 emerges as a putative unique binding site for the interaction of 1-BuOH and halothane with K-Shaw2 (Fig. 7). In this site, the residue side chains in close proximity to the anesthetic molecule (Table S5) do not correspond to those of high-impact Ala/Val mutations (Figs. 3 and 4; Table 1). The residue side chains lining site 3 are L315, L318, I319, and F322 in the S4-S5 linker from the same subunit, whereas L331, L332, F335 (in S5), and L407 (in S6) are from a neighboring subunit. These results suggest that Ala/Val mutations in the regions of interest may not primarily perturb physical contacts, and highlight the importance of allosteric interactions involving distinct binding and effector sites in the mechanism that governs the inhibition of K-Shaw2 by general anesthetics.
Table 1.
Residues ≤ 4.7 Å from the ligand at selected sites
| Ligand | Site | S4-S5 linker | S5 | S6 |
|---|---|---|---|---|
| 3 | L315 L318 I319 F322 | L331 L332 F335 | L407 | |
| 1-Butanol | 6 | K316 Q320 | Y419 T423 | |
| 8 | V409 | |||
| 2 | I317 | K327 E328 | V411 S414 N415 F416 Y420 | |
| Halothane | 3 | L315 L318 I319 F322 | L332 F335 | L407 |
| 5 | L332 V333 L336 | V402 L403 T404 L407 P408 | ||
Figure 7.
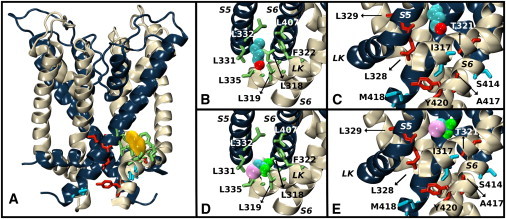
Average equilibrium conformation of the K-Shaw2 closed-state structure bound to 1-BuOH and halothane. (A) Side view of K-Shaw2 with backbones colored in navy blue and off-white. The density blob (yellow) depicts 1-BuOH bound to site 3. Residue side chains < 4.7 Å from 1-BuOH are represented as green sticks (Table 1). (B) Close-up view of 1-BuOH in site 3. (C) Close-up view of 1-BuOH effector sites. Red-colored side chains indicate that Ala/Val mutations had an unfavorable energetic impact on the inhibition of K-Shaw2 by 1-BuOH, and blue-colored side chains indicate a favorable energetic impact. (D) As in panel B, but displaying halothane in site 3. (E) As in panel C, but displaying the effects of mutations on the inhibition by halothane.
Discussion
To gain insights into the structural basis of general anesthetic action on voltage-gated ion channels, we investigated K-Shaw2, a unique Kv channel that displays inhibition by n-alcohols and inhaled anesthetics at clinically relevant concentrations. Our results support the notion of distinct but contiguous binding and effector sites implicating components of the channel's activation gate. The following discussion highlights the mechanistic implications of the our results.
Cooperative modulation of K-Shaw2 by general anesthetics
Although the inhibition of wild-type K-Shaw2 by n-alcohols and halothane is weakly cooperative (nH = 1.2–1.6), the investigated mutations not only change the K0.5, they systematically change nH (Figs. 2–4; Table S2 and Table S3). Compared with the wild-type, some mutations lower the nH (0.8) and others increase it (nH = 2–4). In some extreme cases, such as A326V/A417V and A326V/Y419A, an apparent association between large nH and large K0.5 suggests that higher multiple-site occupancy is needed to achieve inhibition when the apparent affinity of the anesthetic sites is very low. Cooperativity is expected because Kv channels are tetrameric and therefore have four possible binding interfaces that may interact allosterically. The general systematic correlations between ΔΔG and nH for both 1-BuOH and halothane (Figs. 3 C and 4 C) suggest that the mutations may mainly influence effector sites allosterically coupled to binding sites. Indicating a reciprocal communication between anesthetic binding and effector sites, the roles of these effector sites are then twofold: 1), regulate cooperative multisite occupancies; and 2), engage in intra- and intersubunit interactions, which are discouraged or enhanced upon anesthetic binding to modulate gating. The latter emerges from the significant nonadditive energetic impact of several double mutations on the inhibitions of K-Shaw2 by 1-BuOH and halothane. Independently of the channel's oligomeric structure, whether this cooperativity may instead or additionally result from local rearrangements within the binding cavity remains to be investigated.
Colocalization of gating and general anesthetic sites in K-Shaw2
The canonical mechanism of voltage-dependent activation gating in cation channels involves electromechanical coupling between the movement of voltage sensors and opening of the S6 bundle crossing that constitutes the main internal activation gate (16–18,33–36). This coupling is made possible by the S4-S5 linkers, which interact with the distal intracellular portion of the S6 segments. Accordingly, it is not surprising that gating alterations may result from mutations in these regions (Fig. S1 and Table S4). Also, because general anesthetic action on K-Shaw2 is gating-state-dependent, perturbations (mutations or voltage) that destabilize the resting/closed states or strongly favor the open state would be predicted to have a negative impact on anesthetic action. We found several clear examples of these predictions (see Results). Interestingly, however, we also found several instances in which the mutations had a significant energetic impact on the inhibition by 1-BuOH and halothane, but had no major effect on voltage-dependent gating, such as S325A, S414A, Y420A, and A326V/Y420A (Table S4). The lack of a systematic general correlation between gating effects and the energetic impact on anesthetic action indicates that a subset of mutations mainly disrupt anesthetic effector sites. Our results are thus consistent with the coexistence of gating and anesthetic sites in the S4-S5 linker and the post-PVP S6 segment of K-Shaw2.
The constitutively open K-Shaw2 P410D mutant yielded one of the most striking results because it is essentially insensitive to the tested general anesthetics. We propose that the conformational change that underlies the opening of the activation gate dramatically rearranges the structure of the anesthetic binding sites. This idea is supported by in silico docking experiments demonstrating that the preferred site (site 3) has very low affinity in the open state of K-Shaw2 and is therefore virtually absent (Fig. 6 and Fig. S7). These interpretations are in line with the notion of a relatively large conformational change underlying the opening of the internal activation gate upon voltage-dependent activation in Kv channels (37).
Structural correlates of general anesthetic action in K-Shaw2
The K-Shaw2 S4-S5 linker presents major primary-sequence variations compared with the homologous region in related Kv channels. For instance, residues I319, F322, and A326 are not found in general anesthetic-resistant Kv channels. Interestingly, the side chains of hydrophobic residues I319 and F322 are modeled in close proximity (≤4.6 Å) to halothane in site 3 (Fig. 7), the preferred consensus site of anesthetic action in K-Shaw2. L315, L318 (S4-S5 linker), L332, F335 (S5), and L407 (S6) are also in close proximity to halothane but are better conserved across Kv channel subfamilies. Given the predicted architecture of site 3, how can we reconcile the thermodynamic map of individual mutations with the predicted locations of the implicated side chains? Generally, the ΔΔG of individual Ala/Val mutations in S4-S5 and S6 is small or modest (Figs. 3 and 4). The recently published crystal structures of a ligand-gated ion channel bound to propofol or desflurane provides possible answers (6). The authors suggested that the mobility of propofol in its pocket may explain discrepancies between side-chain positions and the effects of the respective mutations on the inhibitory action of the anesthetics. A previous study also showed this fluidity in the hydrophobic general anesthetic binding site of apoferritin (5). This explanation is plausible because typically, binding of general anesthetics to proteins is weakly constrained by specific structural features (1,4). In response to modest mutational perturbations, a general anesthetic may adopt an alternative low-energy state in the protein pocket. Therefore, a single Ala/Val mutation may not be disruptive enough to affect the binding of halothane and 1-BuOH to K-Shaw2, as the ligand may move to establish new nonspecific contacts with the protein backbone and/or membrane phospholipids to facilitate stable binding (38). In future work, investigators should validate this model by investigating the disruption of anesthetic effects by systematic perturbations of residues in binding site 3.
Residues in the effector sites that determine the stability of the closed state through more-specific interactions are, however, more sensitive to mutations. Residues in this category are Q320, T321, S325, A326, E328, L329, A417, M418, Y419, and Y420. The correlation between the nH and the energetic impact of all mutations and the relatively large coupling energies between a subset of residue pairs indicate that K-Shaw2 effector residues influence binding cooperativity and dictate anesthetic action more strongly than those predicted to be in closer proximity to the anesthetic molecule in the hydrophobic cavity. The double mutant A326V/A417V is an interesting example of stringent interactions controlling gating at effector sites, because although the substitutions are mild, the sensitivity of the double mutant to the anesthetics is nearly eliminated.
Relation to other studies
Several earlier studies focused on the interactions between general anesthetics and archetypical eukaryotic Kv channels expressed heterologously, and demonstrated interesting modulations occurring at relatively high doses (38–41). We found that ShakerB and Kv1.2 channels are inhibited ≤10% by relatively high doses of halothane (1–2 mM), and their 1-BuOH K0.5-values are 50 and 120 mM, respectively (data not shown). Thus, these Kv channels are considered general-anesthetic-resistant. In a recent study, Finol-Urdaneta et al. (42) reconstituted the prokaryotic Kv channel (KvAP) in planar lipid bilayers and investigated the modulation of this channel by surface-active substances (including general anesthetics). The authors observed interesting inhibitory modulations and proposed that, depending on the conformational susceptibility of ion channels, a mechanism of general anesthetic action may involve nonspecific membrane effects (e.g., changes in surface tension and phospholipid packing). We did not test these possibilities in our experiments; however, our results do not rule them out. Given the S4-S5 linker's interfacial location at the membrane, it may help transduce lipid bilayer-mediated effects of general anesthetics. Further work is necessary to test this hypothesis.
Generally, the K-Shaw2 mechanism of anesthetic action agrees well with recently reported high-resolution crystallographic observations directly demonstrating the binding of propofol and desflurane to a discrete site in a prokaryotic H+-gated ion channel (GLIC) homologous to eukaryotic Cys-loop receptors (6). Here, the anesthetic cavity involves an interface between helical segments of the channel protein in a region that plays a critical role in gating. This cavity can accommodate either propofol or desflurane, but the binding determinants are distinct and the anesthetics exhibit significant mobility when occupying the site. As proposed for K-Shaw2, general anesthetics also stabilize the closed state of GLIC and a subset of related ligand-gated ion channels.
Conclusions
We conclude that helical regions of the S4-S5 linker and the S5 and S6 segments of K-Shaw2 form inter- and intrasubunit interfaces that delineate a putative general anesthetic binding pocket/cavity. In this cavity, the side chains of hydrophobic residues surround the anesthetic molecule, and neighboring side chains in the activation gate are allosterically coupled to general anesthetic partitioning into the hydrophobic cavity to ultimately modulate the stability of the closed state. The general structural features of the modeled cavity (inter- and intrasubunit helical regions and a hydrophobic interior) are consistent with those found by crystallographic methods in other proteins exhibiting relevant general anesthetic-binding sites (5,6). We hypothesize that the regions identified in the K-Shaw2 channels may also contribute to general anesthetic action in voltage-gated Na+, Ca2+, and nonselective cation channels (19–25). Gating of these channels is also essentially dependent on the interaction between the S4-S5 linkers and the S6 segments (16–18,33–36).
Acknowledgments
We thank Mr. Brian Urbani for technical support, and Dr. Qing Meng (from the Eckenhoff laboratory) for HPLC work. We also thank Dr. Rod Eckenhoff for critically reading the preliminary manuscript.
This study was supported by the National Institutes of Health (research grant R01 AA010615 to M.C., and training grant T32 AA007463 to A.F.B.), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (141009/2009-8 to W.T.).
Supporting Material
References
- 1.Eckenhoff R.G., Johansson J.S. Molecular interactions between inhaled anesthetics and proteins. Pharmacol. Rev. 1997;49:343–367. [PubMed] [Google Scholar]
- 2.Franks N.P. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- 3.Hemmings H.C., Jr., Akabas M.H., Goldstein P.A., Trudell J.R., Orser B.A., Harrison N.L. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol. Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Urban B.W. The site of anesthetic action. Handb. Exp. Pharmacol. 2008:3–29. doi: 10.1007/978-3-540-74806-9_1. [DOI] [PubMed] [Google Scholar]
- 5.Vedula L.S., Brannigan G., Economou N.J., Xi J., Hall M.A., Liu R., Rossi M.J., Dailey W.P., Grasty K.C., Klein M.L., Eckenhoff R.G., Loll P.J. A unitary anesthetic-binding site at high resolution. J. Biol. Chem. 2009;36:24176–24184. doi: 10.1074/jbc.M109.017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nury H., Van R.C., Weng Y., Tran A., Baaden M., Dufresne V., Changeux J.P., Sonner J.M., Delarue M., Corringer P.J. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharji A., Klett N., Go R.C., Covarrubias M. Inhalational anaesthetics and n-alcohols share a site of action in the neuronal Shaw2 Kv channel. Br. J. Pharmacol. 2010;159:1475–1485. doi: 10.1111/j.1476-5381.2010.00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Covarrubias M., Rubin E. Ethanol selectively blocks a noninactivating K+ current expressed in Xenopus oocytes. Proc. Natl. Acad. Sci. USA. 1993;90:6957–6960. doi: 10.1073/pnas.90.15.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Covarrubias M., Vyas T.B., Wei A. Alcohols inhibit a cloned potassium channel at a discrete saturable site. Insights into the molecular basis of general anesthesia. J. Biol. Chem. 1995;270:19408–19416. doi: 10.1074/jbc.270.33.19408. [DOI] [PubMed] [Google Scholar]
- 10.Eckenhoff R.G., Xi J., Shimaoka M., Bhattacharji A., Covarrubias M., Dailey W.P. Azi-isoflurane, a photolabel analog of the commonly used inhaled general anesthetic isoflurane. ACS Chem. Neurosci. 2010;1:139–145. doi: 10.1021/cn900014m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shahidullah M., Harris T., Germann M.W., Covarrubias M. Molecular features of an alcohol binding site in a neuronal potassium channel. Biochemistry. 2003;42:11243–11252. doi: 10.1021/bi034738f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharji A., Kaplan B., Harris T., Qu X., Germann M.W., Covarrubias M. The concerted contribution of the S4–S5 linker and the S6 segment to the modulation of a Kv channel by 1-alkanols. Mol. Pharmacol. 2006;70:1542–1554. doi: 10.1124/mol.106.026187. [DOI] [PubMed] [Google Scholar]
- 13.Covarrubias M., Bhattacharji A., Germann M.W. Alcohol and anesthetic action at the gate of a voltage-dependent K+ channel. In: Mashimo T., Ogli K., Uchida I., editors. Basic and Systemic Mechanisms of Anesthesia. Elsevier B.V.; Amsterdam: 2005. pp. 55–60. [Google Scholar]
- 14.Harris T., Shahidullah M., Ellingson J.S., Covarrubias M. General anesthetic action at an internal protein site involving the S4–S5 cytoplasmic loop of a neuronal K+ channel. J Biol. Chem. 2000;275:4928–4936. doi: 10.1074/jbc.275.7.4928. [DOI] [PubMed] [Google Scholar]
- 15.Harris T., Graber A.R., Covarrubias M. Allosteric modulation of a neuronal K+ channel by 1-alkanols is linked to a key residue in the activation gate. Am. J. Physiol Cell Physiol. 2003;285:C788–C796. doi: 10.1152/ajpcell.00113.2003. [DOI] [PubMed] [Google Scholar]
- 16.Lee S.Y., Banerjee A., MacKinnon R. Two separate interfaces between the voltage sensor and pore are required for the function of voltage-dependent K(+) channels. PLoS. Biol. 2009;7:e47. doi: 10.1371/journal.pbio.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long S.B., Campbell E.B., MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 18.Lu Z., Klem A.M., Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cacheaux L.P., Topf N., Tibbs G.R., Schaefer U.R., Levi R., Harrison N.L., Abbott G.W., Goldstein P.A. Impairment of hyperpolarization-activated, cyclic nucleotide-gated channel function by the intravenous general anesthetic propofol. J. Pharmacol. Exp. Ther. 2005;315:517–525. doi: 10.1124/jpet.105.091801. [DOI] [PubMed] [Google Scholar]
- 20.Chen X., Sirois J.E., Lei Q., Talley E.M., Lynch C., III, Bayliss D.A. HCN subunit-specific and cAMP-modulated effects of anesthetics on neuronal pacemaker currents. J. Neurosci. 2005;25:5803–5814. doi: 10.1523/JNEUROSCI.1153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrington J., Stern R.C., Evers A.S., Lingle C.J. Halothane inhibits two components of calcium current in clonal (GH3) pituitary cells. J. Neurosci. 1991;11:2226–2240. doi: 10.1523/JNEUROSCI.11-07-02226.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horishita T., Harris R.A. n-Alcohols inhibit voltage-gated Na+ channels expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 2008;326:270–277. doi: 10.1124/jpet.108.138370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouyang W., Hemmings H.C., Jr. Isoform-selective effects of isoflurane on voltage-gated Na+ channels. Anesthesiology. 2007;107:91–98. doi: 10.1097/01.anes.0000268390.28362.4a. [DOI] [PubMed] [Google Scholar]
- 24.Ouyang W., Jih T.Y., Zhang T.T., Correa A.M., Hemmings H.C., Jr. Isoflurane inhibits NaChBac, a prokaryotic voltage-gated sodium channel. J. Pharmacol. Exp. Ther. 2007;322:1076–1083. doi: 10.1124/jpet.107.122929. [DOI] [PubMed] [Google Scholar]
- 25.Ratnakumari L., Hemmings H.C., Jr. Inhibition of presynaptic sodium channels by halothane. Anesthesiology. 1998;88:1043–1054. doi: 10.1097/00000542-199804000-00025. [DOI] [PubMed] [Google Scholar]
- 26.Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J. Gen. Physiol. 1998;111:421–439. doi: 10.1085/jgp.111.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sukhareva M., Hackos D.H., Swartz K.J. Constitutive activation of the shaker Kv channel. J. Gen. Physiol. 2003;122:541–556. doi: 10.1085/jgp.200308905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y., Skolnick J. The protein structure prediction problem could be solved using the current PDB library. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1029–1034. doi: 10.1073/pnas.0407152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shrivastava I.H., Sansom M.S. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J. 2000;78:557–570. doi: 10.1016/S0006-3495(00)76616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jogini V., Roux B. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys. J. 2007;93:3070–3082. doi: 10.1529/biophysj.107.112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treptow W., Tarek M. Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys. J. 2006;90:L64–L66. doi: 10.1529/biophysj.106.080754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. PM:9008363. [DOI] [PubMed] [Google Scholar]
- 33.Catterall W.A. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J., Mitcheson J.S., Tristani-Firouzi M., Lin M., Sanguinetti M.C. The S4–S5 linker couples voltage sensing and activation of pacemaker channels. Proc. Natl. Acad. Sci. USA. 2001;98:11277–11282. doi: 10.1073/pnas.201250598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labro A.J., Raes A.L., Grottesi A., Van Hoorick D., Sansom M.S., Snyders D.J. Kv channel gating requires a compatible S4–S5 linker and bottom part of S6, constrained by non-interacting residues. J. Gen. Physiol. 2008;132:667–680. doi: 10.1085/jgp.200810048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prole D.L., Yellen G. Reversal of HCN channel voltage dependence via bridging of the S4–S5 linker and Post-S6. J. Gen. Physiol. 2006;128:273–282. doi: 10.1085/jgp.200609590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y., Holmgren M., Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 38.Li J., Correa A.M. Single-channel basis for conductance increase induced by isoflurane in Shaker H4 IR K(+) channels. Am. J. Physiol. Cell Physiol. 2001;280:C1130–C1139. doi: 10.1152/ajpcell.2001.280.5.C1130. [DOI] [PubMed] [Google Scholar]
- 39.Correa A.M. Gating kinetics of Shaker K+ channels are differentially modified by general anesthetics. Am. J. Physiol. 1998;275:C1009–C1021. doi: 10.1152/ajpcell.1998.275.4.C1009. [DOI] [PubMed] [Google Scholar]
- 40.Friederich P., Benzenberg D., Urban B.W. Interaction of volatile anesthetics with human Kv channels in relation to clinical concentrations. Anesthesiology. 2001;95:954–958. doi: 10.1097/00000542-200110000-00026. [DOI] [PubMed] [Google Scholar]
- 41.Li J., Correa A.M. Kinetic modulation of HERG potassium channels by the volatile anesthetic halothane. Anesthesiology. 2002;97:921–930. doi: 10.1097/00000542-200210000-00026. [DOI] [PubMed] [Google Scholar]
- 42.Finol-Urdaneta R.K., McArthur J.R., Juranka P.F., French R.J., Morris C.E. Modulation of KvAP unitary conductance and gating by 1-alkanols and other surface active agents. Biophys. J. 2010;98:762–772. doi: 10.1016/j.bpj.2009.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.