

Abstract
Understanding the mechanism of thermodynamic stability of an RNA structure has significant implications for the function and design of RNA. We investigated the equilibrium folding of a thermophilic ribozyme and its mesophilic homologue by using hydroxyl radical protection, small-angle x-ray scattering, and circular dichroism. Both RNAs require Mg2+ to fold to their native structures that are very similar. The stability is measured as a function of Mg2+ and urea concentrations at different temperatures. The enhanced stability of the thermophilic ribozyme primarily is derived from a tremendous increase in the amount of structure formed in the ultimate folding transition. This increase in structure formation and cooperativity arises because the penultimate and the ultimate folding transitions in the mesophilic ribozyme become linked into a single transition in the folding of the thermophilic ribozyme. Therefore, the starting point, or reference state, for the transition to the native, functional thermophilic ribozyme is significantly less structured. The shift in the reference state, and the resulting increase in folding cooperativity, is likely due to the stabilization of selected native interactions that only form in the ultimate transition. This mechanism of using a less structured intermediate and increased cooperativity to achieve higher functional stability for tertiary RNAs is fundamentally different from that commonly proposed to explain the increased stability of thermophilic proteins.
A monumental increase in the knowledge of tertiary RNA structures has occurred in the last few years (1–8). These structures demonstrate that tertiary RNAs come in a wide range of shapes and folds. How a tertiary RNA achieves its stability remains elusive. The free energy of the native, functional structure of an RNA or a protein, relative to the free energy of the penultimately stable, nonfunctional structure determines the fraction of active molecules. Therefore, from the functional point of view, the stability of tertiary RNA is defined as the free energy difference between the native state and the penultimately stable state or reference state. This definition of stability is referred to as functional stability.
Proteins generally are in a two-state equilibrium between their native (N) state and a largely unfolded (U) state, in part because most isolated protein secondary structures are not independently stable. As a result, the stability of a protein is defined as the free energy difference between the N state and a reference, U state. The folding of large tertiary RNAs, however, frequently occurs with partially structured intermediate (I) states because RNA secondary structures often are independently stable (9, 10). Consequently, for tertiary RNAs, the nonfunctional I state, rather than the U state, is the defining reference state for functional stability.
The principles of tertiary RNA stability may be revealed by comparative studies of homologous RNAs from mesophilic and thermophilic organisms. Numerous studies have demonstrated that thermophilic RNAs are more stable than their mesophilic homologues. The increase in stability enables the thermophilic RNAs to function at higher temperatures. The typical explanations for the higher stability of thermophilic RNAs are increased GC content and unique posttranscriptional modifications (11–13). These changes presumably strengthen interactions in the native structure.
The existence of partially structured I state opens up a new mechanism for the increased stability of a thermophilic RNA over its mesophilic homologue. In this mechanism, the thermophilic and mesophilic RNAs have similar native states, but the reference state of the thermophilic RNA is less structured (Fig. 1). A less structured reference state results in an increase in the amount of structure formed cooperatively in the I-to-N transition of the thermophilic RNA. Conversely, more structure must be disrupted for the N state of the thermophilic RNA to unfold to the I state.
Figure 1.

Equilibrium folding models for a thermophilic (Cthermo) and a mesophilic (Cmeso) RNA. (A) The Mg2+-dependent equilibrium folding for both Cthermo and Cmeso has two identical intermediates, It and Im. Fractional population of each species is shown. (B) The free energy changes for each transition. The solid lines represent the experimentally determined ΔG as a function of Mg2+ concentration as defined by ΔG = −RT ln([N]/([It] + [Im])). (C) Free energy diagrams at Mg2+ concentrations where both ribozymes have the same functional stability, ΔGI-to-N = −2.0 kcal/mol, corresponding to 0.16 and 3.5 mM Mg2+ for the thermophilic and the mesophilic ribozyme, respectively. Near the folding transition, the It state is less stable than the Im state, so that the latter largely defines the functional stability of the native mesophilic ribozyme. The relative stability of the intermediates is switched so that the It is the defining species for the stability of the thermophilic ribozyme. For Cmeso, an increase in the stability of Im would shift it and the native species down by the same amount, and not alter the functional stability.
This proposed mechanism can be accomplished by linking the normally two-step folding transition in the mesophilic ribozyme into a single, more pronounced transition in the folding of the thermophilic ribozyme. In this scenario, equilibrium folding of both the thermophilic and the mesophilic RNAs occurs with two Mg2+-dependent transitions, It-to-Im and Im-to-N (Fig. 1). For the mesophilic RNA, the Mg2+ concentration required for the second transition is higher than that for the first transition, so that folding occurs in two distinct steps. For the thermophilic RNA, however, the Mg2+ concentration required for the second transition is lower than that for the first transition. Hence, once the Mg2+ concentration is high enough to drive the first transition, the second transition immediately follows and both transitions occur cooperatively. The penultimately populated species is It for the thermophilic RNA, whereas it is Im for the mesophilic RNA (Fig. 1). Therefore, the reference state for the functional stability of the native state is different for the thermophilic and mesophilic RNAs.
This study demonstrates that a thermophilic ribozyme does achieve its higher, functional stability by using a less structured intermediate and increased cooperativity. We characterized the equilibrium folding of the catalytic domain of bacterial RNase P RNAs from a thermophile, Bacillus stearothermophilus (Cthermo, 262 nucleotides), and a mesophile, Bacillus subtilis (Cmeso, 255 nucleotides). These homologous ribozymes have ≈84% identical nucleotide residues and comparable secondary structure, and both fall into the low GC content class of RNase P RNAs (14). Our results indicate that the I state of Cthermo is significantly less structured than the I state of Cmeso, even though their native structures are very similar. The adaptation of this mechanism by natural RNAs suggests that the principle of shifting the thermodynamic reference state and altering folding cooperativity can be used for the design of stable tertiary RNA structures.
Materials and Methods
RNA Preparation.
The C-domain of the B. subtilis P RNA was constructed as described (15). The C-domain of the B. stearothermophilus P RNA was derived from a circularly permuted P RNA with the 5′ end at nucleotide 250. Cthermo contains the T7 RNA polymerase promoter and nucleotides 250–425 + 1–86 of the P RNA. Transcription was carried out by using the standard in vitro transcription procedure with the T7 RNA polymerase (16) and Ear I cut plasmid DNA as the template. RNA transcripts were precipitated with ethanol, dissolved in 7 M urea and 100 mM EDTA, and purified on denaturing polyacrylamide gels containing 7 M urea and 2 mM EDTA. RNAs were eluted from the gel by the crush-and-soak method in 50 mM K-acetate/200 mM KCl, pH 7, precipitated in ethanol and stored in water at −20°C.
In all experiments, the purified RNAs were first heated in 20 mM Tris⋅HCl, pH 8.1, at 85–90°C for 2 min followed by incubation at ambient temperature for 3 min. The RNA at this stage was designated as the U state.
Folding Monitored by Hydroxyl Radical Protection.
The precise nucleotides and the fraction of the RNA protected against hydroxyl radical attack were determined by the Fe(II)-EDTA foot printing method (17). The procedure at 37°C was the same as described for the full-length P RNA at 1 mM Fe(NH4)2(SO4)2/1.2 mM EDTA (18). The reaction mixture was separated on denaturing polyacrylamide gels containing 7 M urea and the result quantitated with a Fuji phosphorimager.
Folding Monitored by Catalytic Activity.
The catalytic activity under single turnover condition ([E] ≫ [S]) of Cthermo and Cmeso was determined from 10–60°C in 50 mM Tris⋅HCl, pH 8.1, 10 mM MgCl2, and 1 mM spermine, using an in vitro selected RNA substrate (19). Cthermo was ≈1.5-fold more active than Cmeso within this temperature range. The selected substrate was no longer stable above 60°C, so that the catalytic activity could not be determined at higher temperatures.
The fraction of the catalytically competent C-domain as a function of Mg2+ concentration at 10°C was determined by the amount of cleavage of an in vitro selected substrate (19, 20). Varying concentrations of Mg2+ were added to the U state and the mixture incubated at 10°C for 10 min. The folding half-life of Cthermo at this temperature was less than 20 s (data not shown). The ribozyme was then mixed with an equal volume of renatured substrate in 50 mM Tris⋅HCl, pH 8.1, 200 mM MgCl2, and 2 mM spermine. The cleavage reaction proceeded for 7–15 s and the reaction was stopped on adding twice the volume of 9 M urea and 90 mM EDTA. The reaction product was separated from the unreacted substrate on 15% denaturing polyacrylamide gels and the fraction of the product quantitated by phosphorimaging. Compared with a control reaction with Cthermo preincubated at 37°C, >80% of Cthermo incubated at 10°C was catalytically active.
Mg2+ and Urea Titration Monitored by Circular Dichroism.
The U state was obtained as described above. When needed, Mg2+ was added to appropriate concentrations and the RNA incubated at 25°C for 5 min to obtain the I state and the N state. The circular dichroism (CD) and absorbance measurements were conducted with a Jasco J715 (Easton, MD) spectropolarimeter interfaced to a Hamilton electronic titrator.
The two Mg2+ dependent transitions were described according to a semiemperical cooperative binding model
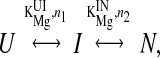 |
where n and KMg are the Hill constant and Mg2+-midpoint of each transition, respectively (21). Because the two transitions were well separated in this system, each transition was fit independently according to
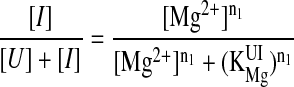 |
1 |
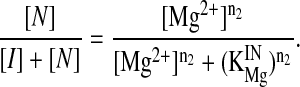 |
2 |
Folding Monitored by Small-Angle X-Ray Scattering (SAXS).
SAXS experiments were carried out at the SAXS instrument on the BESSRC ID-12 beam-line of Argonne National Laboratory's Advanced Photon Source as described (22, 23). Two parameters, P(r) function and Rg, were determined in this work. The P(r) function has a maximum at the most probable distance in the object and goes to zero at the maximum dimension, dmax, of the object. P(r) functions were calculated by using the indirect Fourier inversion algorithms developed by Moore (24)
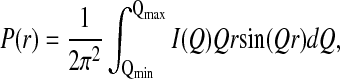 |
3 |
where I(Q), is the scattering intensity, Q = 4πsinθ/λ, λ is the x-ray wavelength, and θ is the half scattering angle. The Rg was determined from the second moment of P(r).
Results and Discussion
The catalytic domains of bacterial RNase P RNAs from B. stearothermophilus and B. subtilis are homologous ribozymes that have >80% identical nucleotide residues. They have comparable secondary structure as derived from phylogeny, and perform the same function in the cell (14). We have carried out a detailed equilibrium folding analysis of Cmeso in an earlier study (25). The present study compares the equilibrium folding of Cthermo and Cmeso to reveal the underlying principles of how Cthermo achieves its higher stability.
The Native Structure of Cthermo and Cmeso.
In the presence of 10 mM Mg2+, both RNAs adopt very similar native structure as determined by hydroxyl radical protection (Fig. 2 A and B), a method that detects the burial of ribose moieties in the native structure (17, 26), and by SAXS (Fig. 2C), a method that identifies the overall size and shape of a molecule in solution (27). Hydroxyl radical protection indicates that except for the P19 region, the protected residues in both ribozymes are located at the same positions, including those that form the active site in J18/2, J19/4, P4, and J3/4. The P19 region is protected only in the mesophilic ribozyme. This protection in P19, unlike protection in other regions that strictly requires the presence of Mg2+, is observed on addition of K+ alone (data not shown). Therefore, the extra protection in P19 is due to a difference in the local structure within this region. Furthermore, the size and shape of the native structures for both ribozymes are very similar as determined by SAXS. The radius of gyration, Rg (Table 1), as well as the P(r) function (Fig. 2C) are identical for both ribozymes within experimental error.
Figure 2.

Both ribozymes have similar native structures. (A) Hydroxyl radical protection in 20 mM Tris⋅HCl, pH 7.5, 10 mM MgCl2, 37°C (18, 26). (B) Protection mapped onto the phylogenetically derived secondary structure (35). Residues protected in the presence of Mg2+ are shaded. Differences in the sequence are shown in lowercase. Residues that cannot be analyzed are shown as smaller, plain letters. (C) Pair-distribution function of the native state determined by SAXS.
Table 1.
Equilibrium folding parameters of Cthermo and Cmeso
| Parameter | Cthermo | Cmeso |
|---|---|---|
| I-to-N transition | ||
| Mg2+ Hill coefficient (n) | 7.8 ± 0.3 | 2.9 ± 0.4* |
| Surface burial (m, kcal mol−1M−1) | 3.8 ± 0.2 | 1.0 ± 0.1* |
| Heat capacity (ΔCp, kcal mol−1K−1) | 2.5 ± 0.3 | 0.46 ± 0.12 |
| Transition midpoint (KMg, mM, 37°C) | 0.11 ± 0.01 | 1.2 ± 0.2* |
| Hypochromicity at 260 nm | ΔAbsthermo > ΔAbsmeso | |
| I state structure | ||
| Radius of gyration (Rg, Å) | 60 ± 1 | 46 ± 1† |
| N state structure | ||
| Radius of gyration (Rg, Å) | 37.6 ± 0.5 | 38.0 ± 0.5† |
| Hydroxyl radical footprinting | Cthermo ≈ Cmeso | |
Lastly, both RNAs also have comparable catalytic activity for an in vitro selected RNA substrate (data not shown). Hence, the native states of Cthermo and Cmeso are structurally and functionally similar.
Starting from an Mg2+-free U state, both ribozymes fold as a function of Mg2+ concentration to the N state through at least one thermodynamic intermediate (Fig. 3; ref. 25). Therefore, the minimal thermodynamic folding scheme is U ←—→ I ←—→ N for both ribozymes. Because the thermodynamic reference state is the catalytically inactive I state, its structure and free energy directly affect the population of the N state and, hence, its functional stability.
Figure 3.

Structural transitions of Cthermo. (A) Mg2+-titration monitored by CD at 260 and 287 nm at 37°C. The KMg and n values are the same at both wavelengths. (B) Urea titrations at Mg2+ concentrations of 0.2 (black), 0.4 (red), 0.6 (green), and 0.8 mM (blue). All experiments were performed in 20 mM Tris⋅HCl, pH 8.1, 0.3 μM RNA, 37°C. (Inset) Stability extrapolated to 0 M urea vs. RTln[Mg2+]. The slope of this plot is the Hill constant, n = 7.8 ± 0.3. (C) Temperature melting at the indicated, constant Mg2+ concentration (in mM) monitored by CD at 287 nm. (D) Temperature dependence of the Mg2+ midpoint for the observed I-to-N transition. KMg was obtained either from Mg2+-titrations at fixed temperature (circles) or from temperature melting at fixed Mg2+ concentrations (triangles). The heat capacity change, ΔCp, was obtained from the temperature-dependent stability, ΔG(T) = ΔH* − TΔS* + ΔCp[(T − 300) − T ln(T/300)], where ΔH* and ΔS* are the enthalpy and entropy changes at 300 K.
The I-to-N Transition of Cthermo and Cmeso.
The Mg2+-induced structural transitions are monitored by CD at 10–65°C (Fig. 3A) and by catalytic activity at 10°C for both Cmeso (19, 20) and Cthermo (above 10°C, the folding rate of Cthermo is too fast for activity measurement by manual methods). The I-to-N structural transition is characterized by a midpoint, KMg, and a Hill constant, n, which can be interpreted as the number of Mg2+ ions bound in this transition. Within experimental error, the Hill constant and the KMg value determined by catalytic activity are identical to those determined from CD measurement (data not shown). At 37°C, the KMg for Cthermo is near 0.1 mM, or 10-fold lower than KMg for Cmeso, indicating that the thermophilic ribozyme can form its native structure at much lower Mg2+ concentration (Table 1). The n value for Cthermo is eight, nearly 3-fold larger than that for Cmeso, indicating that significantly more Mg2+ ions are cooperatively bound in the I-to-N transition for Cthermo.
The KMg and n values define the stability of an Mg2+-dependent RNA structure according to ΔG = −nRT ln([Mg2+]/KMg) (25, 28). Just as observed previously for Cmeso, the ΔG and n value for Cthermo obtained by an alternative method, urea titration at constant Mg2+ concentration, are identical to those obtained by Mg2+ titration (Fig. 3B).
The increased stability of Cthermo is also evident in thermal melting measurements performed at constant Mg2+ concentrations (Fig. 3C). For example, at 0.2 mM Mg2+, the melting temperature for the native state of Cthermo is nearly 50°C, whereas the melting temperature for the native state of Cmeso is too low (<5°C) for convenient measurement.
Importantly, much more structure is formed in the I-to-N structural transition for Cthermo as compared with that for Cmeso. This increased amount of structure formation is reflected by an increase in the urea-induced denaturant response parameter, the m-value, as well as in the heat capacity change, ΔCp (Fig. 3 B and D and Table 1). Both the m-value (28) and ΔCp (29, 30) can be considered to correlate with the amount of surface area buried in the I-to-N transition. The nearly 4-fold larger m-value and the 5-fold larger ΔCp for Cthermo indicate that significantly more surface area is buried in the I-to-N transition of Cthermo.
The I State Structure of Cthermo and Cmeso.
SAXS indicates that the I state of Cthermo is significantly less compact than the I state of Cmeso (Fig. 4). For each ribozyme, the change in the radius of gyration (Rg) with increasing Mg2+ concentration identifies the same structural transitions observed by other methods (Fig. 4A). The dimension of the It, the I state of the thermophilic ribozyme is much larger than the Im state of its mesophilic homologue (Rg ≈ 60 Å vs. ≈ 46 Å). The It state of the thermophilic ribozyme also has a much more extended pair–distance distribution function, P(r) (Fig. 4B). These data further establish that Cthermo has a significantly more expanded intermediate state than Cmeso, even though their native structures are very similar.
Figure 4.
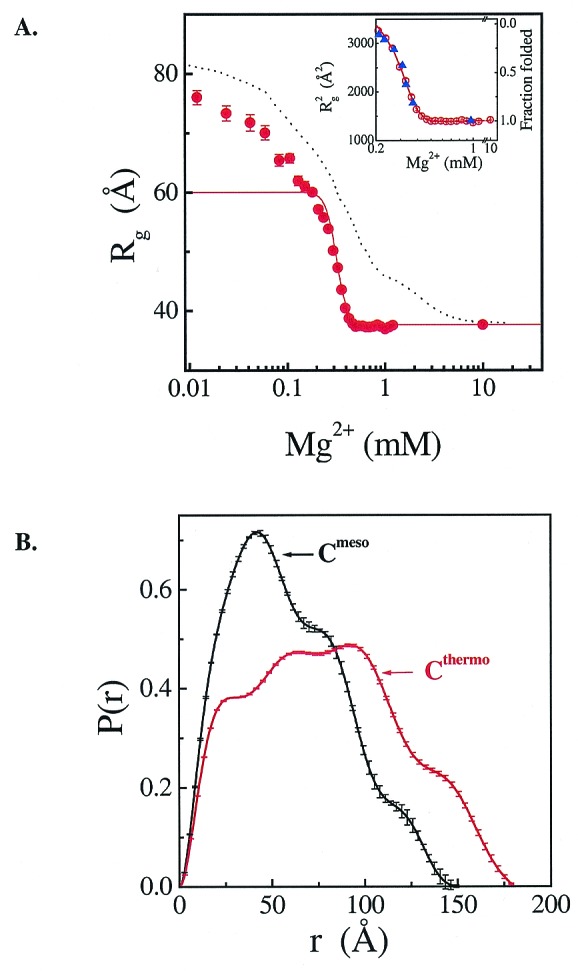
I state dimensions determined by SAXS. (A) Mg2+-titration of 0.3 mg/ml (3.8 μM) Cthermo (circles) and Cmeso (dash curve; from ref. 23). Hill-type analysis of the I-to-N transition for Cthermo (solid line and Inset) yields n = 7.9 ± 0.4, identical to that obtained from urea melting, and KMg = 0.31 ± 0.01 mM, identical to that obtained from hydroxyl radical protection at the same, high RNA concentration (triangles in the Inset). (B) The pair-distribution function of the I state of Cthermo and Cmeso.
Shifting the Thermodynamic Reference State.
These results demonstrate that the structure of the I state for each ribozymes is quite different, despite the similarity of their native states. How is the change in the reference state achieved for the thermophilic RNA? Close inspection of the UV and CD spectra (Fig. 5), as well as SAXS data, indicate that at the Mg2+ concentration where It is formed, the mesophilic ribozyme has a similar, partially folded structure. This result suggests that at low Mg2+ concentrations, an intermediate akin to It in Fig. 1 is populated in equilibrium folding of the mesophilic ribozyme. With increasing Mg2+ concentration, this It species of the mesophilic ribozyme folds sequentially through the Im species, and finally to the native state. Hence, Im is the reference state for the functional stability of the native structure for the mesophilic ribozyme (Fig. 1).
Figure 5.
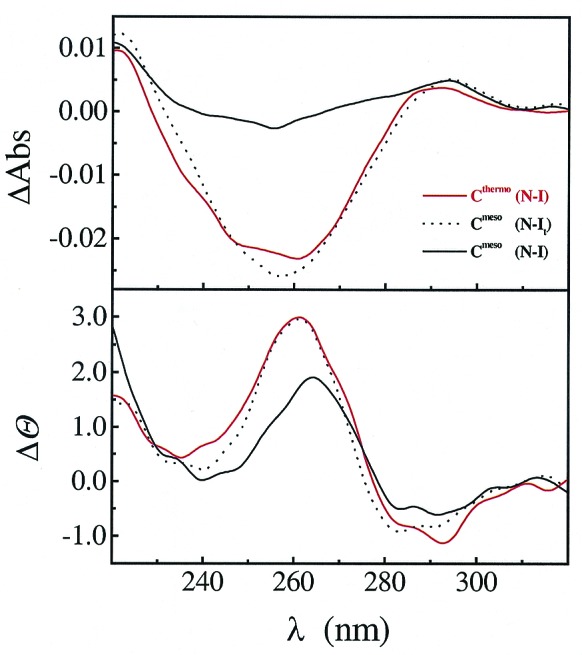
Experimental support for the presence of It state in the folding of Cmeso. At the same low concentration of Mg2+ (0.075 mM), the UV and CD spectra of the mesophilic and thermophilic ribozymes are similar. This similarity, shown as difference spectra at 37°C, indicates that an It state is populated in the equilibrium folding of both ribozymes.
We believe, however, that the Im species of the thermophilic ribozyme exists but does not accumulate in equilibrium folding. We posit that for Cthermo, some interactions that exclusively form in the Im-to-N transition are selectively stabilized, so that the Mg2+ concentration required for this transition is lower than that required for the preceding It-to-Im transition. This inversion in the Mg2+ requirement in Cthermo results in the coincident formation of Im and N. Effectively, the two-distinct It-to-Im and Im-to-N transitions become a single It-to-N transition. Thus, It becomes the reference state for the functional stability of the native structure of the thermophilic RNA (Fig. 1C).
In this model, the total amount of structure formed in the It-to-N transition for Cthermo should be the sum of that formed in the It-to-Im and Im-to-N transitions. Consistently, larger n, m, and ΔCp as well as increased changes in hypochromicity accompany the It-to-N transition of Cthermo compared with the Im-to-N transition of Cmeso. The increase in structure formation between the I and N state of Cthermo is also reflected by the larger differences in Rg and P(r) function for these two species compared with that for Cmeso.
It should be noted that enhanced stabilization of interactions that are already formed in the Im intermediate of Cmeso will not accomplish the desired change in the reference state nor result in an increase in cooperativity and functional stability of Cmeso. For example, both ribozymes in this study are considered to have a relatively low GC content, although the GC content for Cthermo is slightly larger than that for Cmeso (56% vs. 48%). Increasing the GC content in a helix should not increase the stability of the native state if the helix is already formed in Im, because both the intermediate and the native states would be stabilized equivalently. This view is supported by the observation that the stability of Cmeso does not change on substitution of the P3 and P18 hairpins by their very GC-rich Cthermo counterparts that increase the total GC content from 48% to 54%, essentially the level in Cthermo (data not shown). Likewise, increased cooperativity and functional stability would not be achieved by destabilization of native interactions in Im because this destabilization would be realized in the native ribozyme as well, and would lead to no net change in the free energy gap.
The ability of RNA structures to achieve a higher degree of cooperativity through selective stabilization is useful for the design of tertiary RNA structures. Interactions that form in the ultimate structural transition (e.g., the Im-to-N transition) are the most crucial interactions for functional stability. Altering the stability of these “hot spots” can also alter the reference state, resulting in enhanced cooperativity in equilibrium folding and increased stabilization of the native structure. The present results indicate that the Cthermo ribozyme very efficiently utilizes this design principle.
The significant stabilization of a thermophilic RNA through an increase in folding cooperativity is a fundamentally different strategy than that proposed for thermophilic proteins (31, 32). In principle, stabilization through increased cooperativity is available for any system that folds via stable intermediates (33, 34). Because many proteins fold without populated intermediates near their melting transitions, the reference state is generally considered to be a denatured state largely devoid of structure. The lack of stable intermediates implies that, unlike tertiary RNAs, the folding of many proteins already is maximally cooperative, and that enhancing stability by altering the reference state is not a viable option.
Acknowledgments
We thank Drs. C. Correll, K. Dill, D. Herschlag, N. Kallenbach, S. W. Englander, and O. Uhlenbeck and the reviewers for insightful comments. This work was supported by grants from the National Institutes of Health (R01GM57880), U.S. Department of Energy Basic Energy Sciences-Materials Science (under contract W-31-109-ENG-38), University of Chicago–Argonne National Laboratory Collaborative Seed Grant Program, and Packard Foundation Interdisciplinary Science Program.
Abbreviations
- N
native
- U
unfolded
- I
intermediate
- CD
circular dichroism
- Cmeso
the catalytic domain of the Bacillus subtilis P RNA including nucleotides 240–409 + 1–85
- Cthermo
the catalytic domain of the Bacillus stearothermophilus P RNA including nucleotides 250–425 + 1–86
- P RNA
ribozyme component of the RNase P RNA
- SAXS
small-angle x-ray scattering
References
- 1.Cate J H, Gooding A R, Podell E, Zhou K, Golden B L, Kundrot C E, Cech T R, Doudna J A. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 2.Ferre-D'Amare A R, Zhou K, Doudna J A. Nature (London) 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 3.Golden B L, Gooding A R, Podell E R, Cech T R. Science. 1998;282:259–264. doi: 10.1126/science.282.5387.259. [DOI] [PubMed] [Google Scholar]
- 4.Correll C C, Wool I G, Munishkin A. J Mol Biol. 1999;292:275–287. doi: 10.1006/jmbi.1999.3072. [DOI] [PubMed] [Google Scholar]
- 5.Cate J H, Yusupov M M, Yusupova G Z, Earnest T N, Noller H F. Science. 1999;285:2095–2104. doi: 10.1126/science.285.5436.2095. [DOI] [PubMed] [Google Scholar]
- 6.Batey R T, Rambo R P, Lucast L, Rha B, Doudna J A. Science. 2000;287:1232–1239. doi: 10.1126/science.287.5456.1232. [DOI] [PubMed] [Google Scholar]
- 7.Ban N, Nissen P, Hansen J, Moore P B, Steitz T A. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 8.Nissen P, Hansen J, Ban N, Moore P B, Steitz T A. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 9.Turner D H. Curr Opin Struct Biol. 1996;6:299–304. doi: 10.1016/s0959-440x(96)80047-9. [DOI] [PubMed] [Google Scholar]
- 10.Tinoco I, Jr, Bustamante C. J Mol Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 11.Brown J W, Haas E S, Pace N R. Nucleic Acids Res. 1993;21:671–679. doi: 10.1093/nar/21.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowalak J A, Dalluge J J, McCloskey J A, Stetter K O. Biochemistry. 1994;33:7869–7876. doi: 10.1021/bi00191a014. [DOI] [PubMed] [Google Scholar]
- 13.Galtier N, Lobry J R. J Mol Evol. 1997;44:632–636. doi: 10.1007/pl00006186. [DOI] [PubMed] [Google Scholar]
- 14.Pace N R, Brown J W. J Bacteriol. 1995;177:1919–1928. doi: 10.1128/jb.177.8.1919-1928.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan T, Zhong K. Biochemistry. 1994;33:14207–14212. doi: 10.1021/bi00251a032. [DOI] [PubMed] [Google Scholar]
- 16.Milligan J F, Uhlenbeck O C. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 17.Latham J A, Cech T R. Science. 1989;245:276–282. doi: 10.1126/science.2501870. [DOI] [PubMed] [Google Scholar]
- 18.Pan T. Biochemistry. 1995;34:902–909. doi: 10.1021/bi00003a024. [DOI] [PubMed] [Google Scholar]
- 19.Pan T, Jakacka M. EMBO J. 1996;15:2249–2255. [PMC free article] [PubMed] [Google Scholar]
- 20.Loria A, Pan T. RNA. 1996;2:551–563. [PMC free article] [PubMed] [Google Scholar]
- 21.Cantor C, Schimmel P. Biophysical Chemistry: Part III. New York: Freeman; 1980. [Google Scholar]
- 22.Seifert S, Winans R E, Tiede D M, Thiyagarajan P. J Appl Crystallogr. 2000;33:782–784. [Google Scholar]
- 23.Fang X, Littrell K, Yang X, Henderson S J, Siefert S, Thiyagarajan P, Pan T, Sosnick T R. Biochemistry. 2000;39:11107–11113. doi: 10.1021/bi000724n. [DOI] [PubMed] [Google Scholar]
- 24.Moore P B. J Appl Crystallogr. 1980;13:168–175. [Google Scholar]
- 25.Fang X, Pan T, Sosnick T R. Biochemistry. 1999;38:16840–16846. doi: 10.1021/bi991700n. [DOI] [PubMed] [Google Scholar]
- 26.Celander D W, Cech T R. Science. 1991;251:401–407. doi: 10.1126/science.1989074. [DOI] [PubMed] [Google Scholar]
- 27.Kratky O, Pilz I. Q Rev Biophys. 1978;11:39–70. doi: 10.1017/s0033583500001918. [DOI] [PubMed] [Google Scholar]
- 28.Shelton V M, Sosnick T R, Pan T. Biochemistry. 1999;38:16831–16839. doi: 10.1021/bi991699s. [DOI] [PubMed] [Google Scholar]
- 29.Privalov P L, Filimonov V V. J Mol Biol. 1978;122:447–464. doi: 10.1016/0022-2836(78)90421-7. [DOI] [PubMed] [Google Scholar]
- 30.Holbrook J A, Capp M W, Saecker R M, Record M T., Jr Biochemistry. 1999;38:8409–8422. doi: 10.1021/bi990043w. [DOI] [PubMed] [Google Scholar]
- 31.Jaenicke R, Bohm G. Curr Opin Struct Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 32.Szilagyi A, Zavodszky P. Struct Fold Des. 2000;8:493–504. doi: 10.1016/s0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 33.Tilton R F, Jr, Dewan J C, Petsko G A. Biochemistry. 1992;31:2469–2481. doi: 10.1021/bi00124a006. [DOI] [PubMed] [Google Scholar]
- 34.Tang K E, Dill K A. J Biomol Struct Dyn. 1998;16:397–411. doi: 10.1080/07391102.1998.10508256. [DOI] [PubMed] [Google Scholar]
- 35.Haas E S, Brown J W, Pitulle C, Pace N R. Proc Natl Acad Sci USA. 1994;91:2527–2531. doi: 10.1073/pnas.91.7.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]