

Abstract
Gpbar1 (TGR5), a membrane-bound bile acid receptor, is well known for its roles in regulation of energy homeostasis and glucose metabolism. TGR5 also displays strong attenuation of macrophage reactivity in vitro, but the physiological roles of TGR5 in inflammatory response and its mechanism is unknown. Here we demonstrate that TGR5 is a negative modulator of nuclear factor κB (NF-κB)-mediated inflammation. TGR5 activation suppresses the phosphorylation of IκBα, the translocation of p65, NF-κB DNA binding activity and its transcription activity. Furthermore, TGR5 activation enhances the interaction of IκBα and β-arrestin2. Suppression of NF-κB transcription activity and its target gene expression by TGR5 agonist are specifically abolished by expression of anti-β-arrestin2 small interfering RNA. These results show that TGR5 suppresses NF-κB pathway by mediation of the interaction between IκBα and β-arrestin2. In a lipopolysaccharide (LPS)-induced inflammation model, TGR5−/− mice show more severe liver necroses and inflammation compared with wild-type (WT) mice. Activation of TGR5 by its agonist ligand inhibits the expression of inflammatory mediators in response to NF-κB activation induced by LPS in WT but not TGR5−/− mouse liver.
Conclusion
These findings identify TGR5 as a negative mediator of inflammation that may serve as an attractive therapeutic tool for immune and inflammatory liver diseases.
Keywords: bile acid receptor, G-protein coupled bile acid receptor, inflammation, NF-κB, Gpbar1
Introduction
Chronic inflammation is increasingly recognized as an important component of tumorigenesis and metabolic diseases.1, 2 For example, hepatocellular carcinoma (HCC) is a prototypical inflammation-associated cancer that often occurs secondary to chronic hepatitis. Moreover, chronic inflammation is an important mediator of insulin resistance and Type 2 diabetes in obese individuals.2 Thus the precise control of inflammation is essential for the prevention of chronic inflammatory disorders, including many types of cancers and metabolic disorders.3, 4
NF-κB has received considerable attention as a key regulator of immunity, inflammation, and carcinogenesis.1, 5 The classic NF-κB consists of a p65 (RelA) and p50 heterodimer that is sequestered in the cytoplasm of unstimulated cells by its assembly with the inhibitor IκBα. Upon stimulation with proinflammatory ligands, such as lipopolysaccharide (LPS) and tumor necrosis factor (TNF), IκBα is phosphorylated by IκB kinase (IKK), and is then subjected to ubiquitination and proteasome-mediated degradation, which results in the nuclear translocation of NF-κB, and the activation of its target genes.6 Under normal conditions, NF-κB activation is transient and tightly controlled. Conversely, chronic activation of NF-κB signaling is frequently detected in numerous human inflammatory and autoimmune diseases, cancers and diabetes.7, 8 Mounting evidence supports the notion that constitutive NF-κB activation is fundamental to the pathobiology of these human diseases.9 Therefore, defining new therapeutic targets that antagonize NF-κB signaling is crucial for further understanding the regulation of this pathway and the development of novel therapeutic strategies to inhibit prolonged activation of this pathway in these human diseases.
The bile acid receptor Gpbar1 (TGR5) is a regulator of energy homeostasis,10 bile acid homeostasis11 as well as glucose metabolism.12 TGR5 is a member of the G protein-coupled receptor family (GPCRs) which contains 7 transmembrane domains and transduces extracellular signals through heterotrimeric G proteins. Recent in vitro studies, using macrophages and Kupffer cells from wild-type (WT) animals, suggested that TGR5 may be involved in suppression of macrophage and Kupffer cell functions in response to bile acid treatment.13, 14 The physiological role of TGR5 in inflammatory response and the mechanism by which TGR5 has its immunoregulation function is still unclear.
In this study, using a specific TGR5 agonist, we identify TGR5 as a negative regulator of NF-κB-mediated inflammation in a β-arrestin2-dependent manner, and demonstrate that TGR5 ligands have utility in reducing LPS-induced inflammation in liver. These findings suggest TGR5 is a potential target for therapeutic intervention in inflammatory liver diseases.
Materials and Methods
Animals
Male mice fighting are able to cause liver inflammation and liver injury.15, 16 Therefore, in this study, we used only female mice. Eight-week-old wild-type (WT, C57BL/6J) and TGR5−/− female mice (on C57BL/6J background, Merck Research Laboratories, Kenilworth, NJ)17 were maintained in a pathogen-free animal facility under a standard 12:12-h light/dark cycle. Mice were fed a diet containing 10 mg 23(S)-mCDCA /kg diet or standard rodent chow for 3 d. After that, the mice were fasted overnight and then injected intraperitoneally (i.p.) with a single dose of LPS (20 mg/kg) or PBS, followed by feeding water ad libitum. Six hours after the injection, mice were killed, and liver was removed for further analysis. All procedures followed National Institutes of Health guidelines for the care and use of laboratory animals.
Reagents and Plasmids
Reagents and plasmids are outlined in the Supporting Information.
Cell Culture and Transient Transfection
Cell culture and transient transfection are outlined in Supporting Information.
Isolation of Mouse Bone Marrow Derived Macrophages and Kupffer Cells
Mouse bone marrow derived macrophages were derived according to previously published methods.18 Mouse Kupffer cells were prepared as described previously.13 Cells were pretreated with 23(S)-mCDCA (10 μM). Eighteen hours after treatment, the cells were treated with LPS (1 ng/ml), and then collected for RNA isolation after a 4 h incubation.
Analysis of Alanine Aminotransferase (ALT) and Aspartate Transaminase (AST) and Histology
Analysis of ALT and AST and the staining for liver sections was described in the Supporting Information.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
Total RNA isolation from cells and tissues and quantitative real-time polymerase chain reaction were described in the Supporting Information.
Enzyme-Linked Immunosorbent Assay (ELISA)
The method for ELISA was described in the Supporting Information.
Protein Extracts Preparation and Immunoblot Analysis
Protein extracts preparation and immunoblot analysis were described in the Supporting Information.
Electrophoretic Mobility-Shift Assay (EMSA)
HepG2 cells or mouse macrophages were transfected with p65 expression plasmid or control plasmid with or without cotransfection of TGR5 plasmid. After 24-h transfection, cells were treated with 10 μM 23(S)-mCDCA or DMSO (control) for 24 h. Finally, nuclear proteins were extracted for EMSA assay. EMSA assays were performed as described.19 The following oligonucleotide was used for the EMSA assay: NF-κB-binding site; 5'- tcgagggctggggattccccat-3'.
Immunoprecipitation
The plasmids pFLAG-IκBα, pHA-β-arrestin2 and mTGR5 were co-transfected into HEK293 cells using Lipofectamine 2000. Cell treatment and immunoprecipitation for cells and liver tissue were performed as described in the Supporting Information.
β-arrestin2 siRNA
β-arrestin2 siRNA and control siRNA were purchased from Santa Cruz Biotechnology and transfected into HepG2 cells using siRNA transfection reagent (Santa Cruz Biotechnology). Cell treatment was described in the Supporting Information.
Statistics
All data represent at least three independent experiments and are expressed as the mean ± SD. The Student's t test was used to calculate P values unless stated otherwise. For multiple comparisons between groups, a two-way ANOVA followed by Bonferroni post hoc test was performed. P less than 0.05 was considered significant.
Results
TGR5−/− mouse macrophages, primary Kupffer cells and hepatic tissue display elevated expression of NF-κB-regulated proinflammatory genes
TGR5 is expressed in macrophages, primary Kupffer cells and livers.13, 14, 20 It is not expressed in hepatocytes. In this work, we found that compared with WT controls, macrophages, primary Kupffer cells and livers from TGR5−/− mice had elevated messenger RNA (mRNA) levels of some proinflammatory NF-κB target genes (Fig. 1A). These elevated genes include inducible nitric oxide synthase (iNOS), interferon-inducible protein (IP)-10 and interleukin (IL)-1α in TGR5−/− mouse macrophages; monocyte chemoattractant protein-1 (MCP-1), interferon (IFN)-γ, iNOS and IP-10 in TGR5−/− mouse primary Kupffer cells; and IL-1β and IFN-γ in TGR5−/− mouse livers, respectively The protein levels of IL-1β and IFN-γ in TGR5−/− mouse livers were also elevated compared with WT controls (Supporting Fig. 1A). These results suggest that TGR5 may be a negative modulator of hepatic inflammation.
Figure 1.





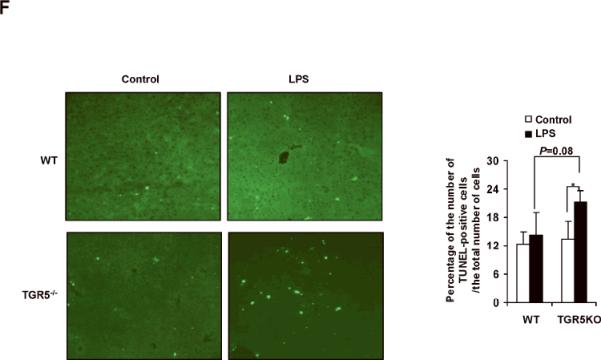
TGR5−/− mouse macrophages, Kupffer cells and liver are more sensitive to NF-κB activation. (A) TGR5−/− mouse macrophages, primary Kupffer cells and liver (n=5) display elevated expression of NF-κB-regulated proinflammatory genes. WT, wild-type; TGR5KO, TGR5−/−. *P < 0.05 versus WT group. (B) TGR5−/− mouse macrophages, primary Kupffer cells and liver (n=5) are sensitive to activation of NF-κB. * P < 0.05 and ** P < 0.005. (C) The levels of inflammatory serum markers, ALT and AST in serum from wild-type (WT) and TGR5−/− (TGR5KO) mice that were treated with either vehicle (PBS as control) or 20 mg/kg LPS (n=5). *P < 0.05 and ** P < 0.005. (D) Representative hematoxylin-eosin staining of liver sections from WT and TGR5−/− livers (magnification ×200). The arrow indicates infiltrated inflammatory cells. (E) Representative immunohistochemistry staining for F4/80 from WT and TGR5−/− livers (magnification ×200) and statistical analysis of the number of F4/80-positive cells per field. Arrows indicate F4/80 positive cells. Twenty high-power fields were counted. *P < 0.05 and **P < 0.005. (n=5). (F) Representative TUNEL staining of sections from WT and TGR5−/− livers (magnification ×200) and statistical analysis of the number of TUNEL-positive cells per total number of cells. The number of cells in at least 20 microscopic fields was counted. * P < 0.05. (n=5). Data of B, C, E and F was analyzed by two-way ANOVA for multiple comparisons as described in Materials and Methods.
TGR5−/− mouse macrophages, primary Kupffer cells and livers are sensitive to activation of NF-κB
If TGR5 is a suppressor of NF-κB-mediated inflammation, TGR5−/− mice should be more sensitive than WT mice to inflammation mediated by NF-κB. We compared the mRNA levels of proinflammatory genes in macrophages and primary Kupffer cells from WT and TGR5−/− mice after activating the NF-κB pathway with a known NF-κB pathway activator, lipopolysaccharide (LPS). LPS-treated TGR5−/− macrophages and primary Kupffer cells expressed higher mRNA levels of NF-κB target genes than did untreated TGR5−/− macrophages and primary Kupffer cells (MCP-1 and IFN-γ in macrophages; MCP-1, iNOS and IP-10 in primary Kupffer cells, see Fig. 1B). This induction was considerably reduced in WT macrophages and primary Kupffer cells. We then compared the expression of proinflammatory genes in livers from both TGR5−/− and WT mice after treatment with LPS. Induction of MCP-1, IP-10, IFN-γ and iNOS in response to LPS was significantly greater in TGR5−/− mice than WT mice (Fig. 1B) (The protein levels of some proinflammatory genes in mouse livers were measured using ELISA, see Supporting Fig. 1B). The levels of some inflammatory serum markers in TGR5−/− mice were also found significantly higher than that in WT mice after treatment with LPS (Fig. 1C). Those results suggest that certain inflammatory genes are more sensitive to LPS induction in the absence of TGR5 signaling in vivo.
The levels of ALT and AST, two markers of liver injury, were also significantly increased by treatment with LPS in TGR5−/− mice compared with WT mice (Fig. 1C). We next examined liver pathology, and found that massive inflammation was present in TGR5−/− mice, but not WT mice after administration of LPS (Fig. 1D). We then performed F4/80 immunohistochemistry staining on liver samples to determine Kupffer cell infiltration. F4/80 is a mature tissue macrophage marker. The population of F4/80 positive cells in TGR5−/− mouse liver was higher than WT even without LPS treatment (Fig. 1E). Following LPS administration, the numbers of F4/80 positive cells in WT and TGR5−/− mouse livers were increased with 47% and 57%, respectively. The results indicated that LPS increased Kupffer cell infiltration more significantly in TGR5−/− mouse liver. It has been well reported that LPS induces hepatocyte apoptosis in vitro and in vivo.21–24 The liver injury induced by LPS was further confirmed by TUNEL assays. Compared with WT controls, TUNEL-positive percentage was enhanced in the livers of TGR5−/− mice, after injection of LPS (Fig. 1F). Together, both in vitro and in vivo results suggest that macrophages, primary Kupffer cells and livers from TGR5−/− mice are more sensitive to LPS-induced inflammation.
TGR5 suppresses the expression of NF-κB-mediated proinflammatory genes induced by LPS in vitro and in vivo
We then tested whether ligand-activated TGR5 could inhibit these proinflammatory NF-κB target genes induced by LPS. 23(S)-mCDCA is a new synthetic, highly selective TGR5 agonist.25 We synthesized 23(S)-mCDCA and confirmed its TGR5-specific activity by observing the expected dose dependent increase in cAMP in WT macrophages but not TGR5−/− macrophages and by activation of a CRE-reporter construct only in TGR5-expressing cells (data not shown). Measurements of ALT, AST and alkaline phosphatase (ALP) and histological staining indicated that administering 23(S)-mCDCA to mice did not induce toxic effects (data not shown). 23(S)-mCDCA treatment repressed LPS-induced IP-10 and IL-6 expression in WT macrophages but not TGR5−/−macrophages (Fig. 2A). A similar inhibition of mRNA levels of iNOS, MCP-1, cyclooxygenase (COX)-2 and IL-6 by the TGR5 agonist was observed in response to stimulation with LPS in WT Kupffer cells but not TGR5−/− Kupffer cells (Fig. 2A). Kawamata et al.14 and Keitel et al.13 observed a similar down-regulating TNF-α, IL-6, IL-1α and IL-1β in rabbit macrophages and rat Kupffer cells upon treatment with bile acids. Furthermore, we examined the effects of the TGR5 agonist on the NF-κB pathway in vivo. Livers from WT mice that were pretreated with the TGR5 agonist 23(S)-mCDCA showed significantly less LPS-induced expression of IP-10, MCP-1, iNOS and IFN-γ mRNA than did non-pretreated livers. This inhibition was abolished, or considerably reduced in TGR5−/− mouse groups (Fig. 2B). Collectively, these results demonstrate that TGR5 plays a protective role against LPS-induced inflammation in vitro and in vivo.
Figure 2.


TGR5 suppresses the expression of NF-κB-mediated proinflammatory genes induced by LPS in vitro and in vivo. (A) TGR5 ligand treatment repressed LPS-induced proinflammatory gene expression in WT but not TGR5−/− macrophages and Kupffer cells. The macrophages or Kupffer cells were pre-treated with 23(S)-mCDCA (10 μM) for 18 h and then were treated with LPS for 6 h. (n=3). *P < 0.05 versus the only LPS-treated WT groups. (B) TGR5 ligand treatment repressed LPS-induced proinflammatory gene expression in WT but not TGR5−/− liver. (n=5–6). *P < 0.05 versus the only LPS-treated WT groups.
Activation of TGR5 antagonizes NF-κB signaling
We next tested whether TGR5 agonist inhibited NF-κB activity at the levels of gene transcription. We cotransfected HepG2 cells with a NF-κB reporter plasmid and the control plasmid phRL-TK and assessed the effects of the TGR5 ligand on the regulation of NF-κB reporter activity. Treatment with known NF-κB pathway activator 12-O-tetradecanoyl-phorbol-13-acetate (TPA) or LPS resulted in 5.1-fold and 1.8-fold higher NF-κB reporter activity, respectively (Fig. 3A). NF-κB activity induced by TPA or LPS was suppressed by 23(S)-mCDCA treatment. Transfection of these cells with TGR5 inhibited NF-κB activity in the absence of ligand, suggesting that TGR5 may suppress NF-κB activity without addition of exogenous ligand, possibly due to the fact that GPCRs have constitutive activity.26, 27 Addition of 23(S)-mCDCA further enhanced this repression (Fig. 3A). Furthermore, to eliminate the possibility that the compounds were affecting other pathways, we used p65 overexpression to activate the NF-κB reporter. Overexpression of p65 significantly activated the NF-κB reporter (Fig. 3B,C). NF-κB activity was inhibited by 23(S)-mCDCA in a ligand dose-dependent manner in the absence of the TGR5 expression vector (Fig. 3B). The expression of endogenous TGR5 in HepG2 cells was detected and endogenous TGR5 function in HepG2 cells was determined by measuring the cAMP levels (Supporting Fig.2). Therefore, TGR5 ligand suppressing the NF-κB activity in the absence of TGR5 overexpression may be through activating the endogenous TGR5 in HepG2 cells. Compared with that in the absence of the TGR5 expression vector, TGR5 overexpression enhanced the suppression of NF-κB activity by the TGR5 agonist (Fig. 3C). Moreover, the observed inhibition of NF-κB activity in response to activated TGR5 was proportional to the amounts of TGR5 vector. The inhibition of NF-κB transactivity by TGR5 activation was also confirmed in mouse macrophages (Fig. 3D). These results indicate that activation of TGR5 can antagonize NF-κB activity at the level of gene transcription.
Figure 3.


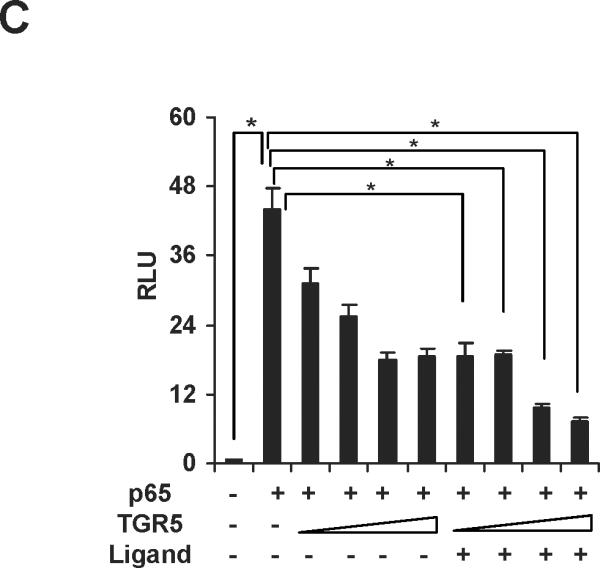



Activation of TGR5 antagonizes NF-κB transcriptional activity. (A) Activation of TGR5 inhibits NF-κB luciferase reporter activity induced by TPA and LPS. RLU, relative luciferase units. *P<0.05 (n=3). (B) TGR5 ligand dose-dependently suppresses NF-κB activity induced by p65 overexpression. HepG2 cells were cotransfected with the NF-κB reporter plasmid, the control plasmid phRL-TK, p65 expression plasmid and TGR5 expression plasmid. Cells were treated with 23(S)-mCDCA at doses of 10, 30, 60 μM. *P<0.05 (n=3). (C) TGR5 suppresses NF-κB activity induced by p65 overexpression in a TGR5 dose-dependent manner. HepG2 cells were cotransfected with the NF-κB reporter plasmid, the control plasmid phRL-TK and increasing amounts of a TGR5 expression plasmid at 0.5:1, 1:1, 2:1, or 3:1 ratios with the p65 expression plasmid. After transfection, cells were treated with 23(S)-mCDCA (10 μM) or vehicle (dimethyl sulfoxide) for 24 h. *P < 0.005. versus the group transfected with p65 only (n=3). (D) TGR5 activation suppressed NF-κB activity induced by p65 overexpression in mouse macrophages. Mouse macrophages were cotransfected with the NF-κB reporter plasmid, the control plasmid phRL-TK, p65 expression plasmid and TGR5 expression plasmid. Cells were treated with 23(S)-mCDCA at doses of 10μM for 24 h and then were harvested and the luciferase activity was determined. RLU, relative luciferase units. (n=3) *P < 0.05. (E) EMSA showed that activation of TGR5 suppresses NF-κB DNA binding activity induced by p65 overexpression in HepG2 cells. (F) EMSA showed that activation of TGR5 suppressed NF-κB DNA binding activity induced by p65 overexpression in mouse macrophages. Mouse macrophages were transfected with p65 expression plasmid or control plasmid with or without cotransfection of TGR5 plasmid. After 24-h transfection, cells were treated with 10 μM 23(S)-mCDCA or DMSO (control) for 24 h. Finally, nuclear proteins were extracted for EMSA assay.
The binding of NF-κB to its response elements was then examined via electrophoretic mobilityshift assay (EMSA) using nuclear extracts from HepG2 cells. TGR5 activation dramatically reduced the binding activity of NF-κB to DNA sequences induced by p65 overexpression (Fig. 3E). The results in HepG2 cells were also confirmed in mouse macrophages (Fig. 3F). These results suggest that TGR5 activation may suppress NF-κB transcriptional activity by decreasing the binding of NF-κB to its response elements.
TGR5 inhibits phosphorylation of IκBα and nuclear translocation of p65
The IκBα phosphorylation and nuclear p65 levels in HepG2 cells were shown in Fig. 4A. TGR5 activation by 23(S)-mCDCA dramatically suppressed the level of phosphorylated IκBα induced by TNF-α, and almost completely abolished the nuclear translocation of p65 induced by p65 overexpression. These results were further confirmed in the livers from WT and TGR5−/− mice (Fig. 4B). Increase of IκBα phosphorylation levels in response to LPS was greater in TGR5−/− mice than WT mice (Fig. 4B). In response to TGR5 ligand treatment, the increase of LPS-induced IκBα phosphorylation was completely abolished in WT mouse livers, while about 40% decrease was observed in TGR5−/− mouse livers. TGR5 agonist administration inhibited LPS-induced nuclear p65 levels in WT mice but not TGR5−/− mice (Fig. 4B). These results suggest that TGR5 activation inhibits both IκBα phosphorylation and p65 nuclear translocation, which may contribute to antagonize the transactivity of NF-κB.
Figure 4.


TGR5 inhibits phosphorylation of IκBα and translocation of p65. (A) Immunoblot analysis for phosphorylation of IκBα and translocation of p65 in HepG2 cells. TGR5 activation inhibits phosphorylation of IκBα induced by TNF-α (30 ng/ml) and nuclear translocation of p65 induced by p65 overexpression in HepG2 cells. P-IκBα, phosphorylated IκBα; T-IκBα, total IκBα. *P < 0.05. (B) Immunoblot analysis for phosphorylated IκBα (P-IκBα) and total IκBα (T-IκBα) from total protein pools, for p65 and Lamin B1 from nuclear protein pools in livers of WT and TGR5−/− mice that were treated as described in Materials and Methods. (n=5). Lamin B1 was served as a nuclear protein loading control. Ligand, 23(S)-mCDCA. *P < 0.05.
TGR5 antagonizes NF-κB pathway in a β-arrestin2-dependent manner
The ubiquitously expressed protein β-arrestin2 is a multifunctional signaling molecule. It is originally identified as a negative regulator of GPCR signaling.28 It has been demonstrated that β-arrestin2 is able to bind to NF-κB inhibitor IκBα in the cytoplasm to inhibit NF-κB activity.29, 30 In this work, we further investigated the function of β-arrestin2 in NF-κB signaling. HEK293 cells cotransfected with TGR5, β-arrestin2, and IκBα were challenged with TGR5 agonist 23(S)-mCDCA, and then the cell extracts were subjected to immunoprecipitantion. TGR5 activation enhanced β-arrestin2 interaction with IκBα (Fig. 5A). These results were also confirmed in mouse livers. TGR5 ligand administration increased β-arrestin2 interaction with IκBα in WT but not in TGR5−/−mouse livers (Fig. 5B). Knockdown of β-arrestin2 abolished the inhibition of TGR5 activation on NF-κB transactivity and its target gene expression induced by p65 overexpression (Fig. 5C–E). These results indicate that TGR5 inhibits NF-κB in a β-arrestin2-dependent manner.
Figure 5.





TGR5 antagonizes NF-κB pathway in a β-arrestin2-dependent manner. (A) HEK293 cells were cotransfected with FLAG-IκBα, HA-β-arrestin2, and TGR5 plasmids. After serum starvation for 12 h, cells were stimulated with 23(S)-mCDCA (10 μM) for the indicated times. Proteins immunoprecipitated with FLAG-specific antibody or HA-specific antibody were subject to immunoblotting with antibodies against FLAG or HA as indicated. *P < 0.05. (n=3) (B) TGR5 ligand administration increased interaction between β-arrestin2 and IκBα in wild-type (WT) mouse livers but not TGR5−/− mouse livers. Mice were fed a diet containing 10 mg 23(S)-mCDCA /kg diet or standard rodent chow for 3 d. After that, the mouse livers were collected for co-immunoprecipitation. (n=5–6). *P < 0.05. (C) Immunoblot analysis shows that β-arrestin2 protein level was decreased by anti-β-arrestin2 small interfering RNA (β-arrestin2 siRNA). (D) Suppression of NF-κB transcription activity by TGR5 agonist is specifically abolished by expression of anti-β-arrestin 2 small interfering RNA. *P < 0.05. (n=3) (E) Suppression of NF-κB-mediated target gene expression by TGR5 agonist is specifically abolished by expression of anti-β-arrestin 2 small interfering RNA. *P < 0.05. (n=3)
Discussion
GPCRs comprise the largest protein family of transmembrane receptors that sense molecules outside the cell and activate inside signal transduction pathways through agonist binding to an orthosteric binding site. GPCRs regulate cell migration, proliferation, differentiation and survival and play a major role in development and progression of many diseases such as inflammatory diseases and cancer.31, 32 Many GPCRs induce NF-κB activation,33, 34 while only a few of GPCRs inhibit NF-κB-mediated inflammation.35 Two GPCRs, the A2A and A2B adenosine receptors, suppress NF-κB pathway in a specific gene and cell type-dependent manner.35–37 Activation of β2-adrenergic receptor, a subtype of GPCRs, inhibits NF-κB activity by means of β-arrestin interaction with IκBα.29 Our data show that TGR5 is a potential suppressor of NF-κB-dependent inflammatory response. TGR5 activation is able to enhance β-arrestin2 interaction with IκBα. TGR5 antagonizing NF-κB signaling was abolished by expression of anti-β-arrestin 2 small interfering RNA (Fig. 5). These results suggest that TGR5 inhibits NF-κB in a β-arrestin2-dependent manner, and the inhibition of NF-κB-mediated inflammation by some GPCRs could share the same mechanism. It is interesting to study the mechanism of the TGR5-dependent β-arrestin2-IκBα interaction. We ruled out the possibility that TGR5 may interact with β-arrestin2. We also ruled out another possibility that activation of TGR5 may reduce the interaction of casein kinase II and β-arrestin2 and thus enhance the β-arrestin2-IκBα interaction.29 It is interesting to continue the study in the future work.
We noted that TGR5 activation repressed specific sets of NF-κB target genes, but not all the target genes in response to LPS in vitro and in vivo. This phenomenon has also been observed for bile acid nuclear receptor FXR.38 It will be interesting to define the mechanism by which TGR5 activation inhibits NF-κB in a gene-specific manner.
NF-κB is the central transcriptional regulator of inflammatory and immune responses.39 Constitutive NF-κB activation has been implicated in the malignant progression of numerous human inflammatory diseases, metabolic diseases, cancers and diabetes.40 Inhibiting the aberrant activation of NF-κB signaling can slow down or stop these disease processes.41, 42 In this study, our analysis results of inflammatory gene expression reveal that TGR5 has anti-inflammatory properties in mouse liver. Our data show that TGR5 activation prevents the phosphorylation of IκBα, nuclear translocation of p65 and NF-κB DNA binding activity.
Activation of NF-κB in Kupffer cells promotes liver cancer development through IL-6 and liver inflammatory responses.43 Blockage of NF-κB by deletion of IKKβ in Kupffer cells in addition to hepatocytes strongly inhibited diethylnitrosamine (DEN)-induced HCC development.43 Thus, the suppression of NF-κB might be a therapeutical strategy for treating liver cancer because the loss of NF-κB in Kupffer cells might suppress cancer. TGR5 is highly expressed in Kupffer cells of liver.13, 14 In this study, we demonstrated that TGR5 activation is able to strongly suppress NF-κB-induced inflammation in vitro and in vivo, which suggests that TGR5 may be a desirable therapeutic target for liver cancer treatment.
It has been reported that TGR5 could be a potential target for the treatment of diabesity and associated metabolic disorders.10, 12, 44, 45 For example, Watanabe et al. reported that TGR5 activation by bile acids induces energy expenditure in muscle and brown adipose tissue.10 Thomas et al. found that TGR5 activation improves glucose tolerance and insulin sensitivity in fat-fed mice.12 These diseases, such as obesity, insulin resistance and type 2 diabetes, are also closely associated with chronic inflammation characterized by abnormal cytokine production, increased acute-phase reactants, and activation of a network of inflammatory signaling pathways.4, 46, 47 Inhibition of NF-κB related inflammation is able to improve glucose metabolism in vivo.48, 49 Here, our data show that TGR5 is a negative modulator of NF-κB-mediated inflammation. Therefore, there is a potential link between anti-inflammation and treatment of obesity and diabetes through TGR5. TGR5 may be an attractive therapeutic target for metabolic disorders through not only regulation of energy and glucose homeostasis but also suppression of NF-κB signaling.
In conclusion, our results reveal that TGR5 is a negative regulator of NF-κB-mediated hepatic inflammation, and indicate that TGR5 ligands have utility in anti-inflammation. These findings suggest TGR5 is a potential target for anti-inflammatory drug design and its agonist ligands offer possible therapies to prevent and treat inflammatory liver diseases.
Supplementary Material
Acknowledgments
We thank Dr. Galya Vassileva in Merck Research Laboratories and Merck Research Laboratories for TGR5−/− mice, Dr. Peter Tontonoz, Dr. Bruce Blumberg, Xufeng Chen, Akio Kruoda and Dr. Gang Pei for plasmids and Sofia Loera for conducting the immunohistochemical staining in the Anatomic Pathology Core Facility of City of Hope.
Grant support: This work is supported by City of Hope GI Cancer Program GI Cancer Research Pilot Fund to Y.-D.W. and NCI 1R01CA139158-01A2 to W.H..
Abbreviation used
- COX-2
cyclooxygenase-2
- EMSA
electrophoretic mobilityshift assay
- GPCR
G protein-coupled receptor
- IFN-γ
interferon–γ
- iNOS
nitric oxide synthase
- IP-10
interferon-inducible protein-10
- LPS
lipopolysaccharide
- MCP-1
monocyte chemoattractant protein-1
- TNF
tumor necrosis factor
- TPA
12-O-tetradecanoyl-phorbol-13-acetate
Footnotes
Author Contributions: Y.-D.W. and W.-D.C. conceived the project and designed all the experiments. Y.-D.W. and W.-D.C. performed all the experiments and analyzed data. Y.-D.W., W.-D.C., B.M.F. and W.H. discussed the results. D.Y. and B.M.F. synthesized TGR5 agonist 23(S)-mCDCA. Y.-D. W. and W.-D.C. prepared and wrote the paper. B.M.F. and W.H. reviewed and edited the paper.
Disclosures: The authors have declared that no conflict of interest exists.
References
- 1.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 2.Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 298:E419–428. doi: 10.1152/ajpendo.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 4.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 5.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 6.O'Connell MA, Bennett BL, Mercurio F, Manning AM, Mackman N. Role of IKK1 and IKK2 in lipopolysaccharide signaling in human monocytic cells. J Biol Chem. 1998;273:30410–30414. doi: 10.1074/jbc.273.46.30410. [DOI] [PubMed] [Google Scholar]
- 7.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 9.D'Acquisto F, Ianaro A. From willow bark to peptides: the ever widening spectrum of NF-kappaB inhibitors. Curr Opin Pharmacol. 2006;6:387–392. doi: 10.1016/j.coph.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 11.Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, Nakamura T, et al. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol. 2006;191:197–205. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- 12.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- 14.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez O, Viladrich M, Ramirez I, Soley M. Liver injury after an aggressive encounter in male mice. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1908–1916. doi: 10.1152/ajpregu.00113.2007. [DOI] [PubMed] [Google Scholar]
- 16.Hirschfield GM, Gallimore JR, Kahan MC, Hutchinson WL, Sabin CA, Benson GM, et al. Transgenic human C-reactive protein is not proatherogenic in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 2005;102:8309–8314. doi: 10.1073/pnas.0503202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–430. doi: 10.1042/BJ20060537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;112:1176–1185. doi: 10.1172/JCI18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen WD, Wang YD, Zhang L, Shiah S, Wang M, Yang F, et al. Farnesoid x receptor alleviates age-related proliferation defects in regenerating mouse livers by activating forkhead box m1b transcription. Hepatology. 2010 doi: 10.1002/hep.23390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, Selbach O, et al. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. doi: 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]
- 21.Kudo H, Takahara T, Yata Y, Kawai K, Zhang W, Sugiyama T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 2009;51:168–175. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 22.Hamada E, Nishida T, Uchiyama Y, Nakamura J, Isahara K, Kazuo H, et al. Activation of Kupffer cells and caspase-3 involved in rat hepatocyte apoptosis induced by endotoxin. J Hepatol. 1999;30:807–818. doi: 10.1016/s0168-8278(99)80133-0. [DOI] [PubMed] [Google Scholar]
- 23.Eipel C, Bordel R, Nickels RM, Menger MD, Vollmar B. Impact of leukocytes and platelets in mediating hepatocyte apoptosis in a rat model of systemic endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2004;286:G769–776. doi: 10.1152/ajpgi.00275.2003. [DOI] [PubMed] [Google Scholar]
- 24.Xiang L, Klintman D, Thorlacius H. Allopurinol inhibits CXC chemokine expression and leukocyte adhesion in endotoxemic liver injury. Inflamm Res. 2003;52:353–358. doi: 10.1007/s00011-003-1184-6. [DOI] [PubMed] [Google Scholar]
- 25.Pellicciari R, Sato H, Gioiello A, Costantino G, Macchiarulo A, Sadeghpour BM, et al. Nongenomic actions of bile acids. Synthesis and preliminary characterization of 23-and 6,23-alkyl-substituted bile acid derivatives as selective modulators for the G-protein coupled receptor TGR5. J Med Chem. 2007;50:4265–4268. doi: 10.1021/jm070633p. [DOI] [PubMed] [Google Scholar]
- 26.Tao YX. Constitutive activation of G protein-coupled receptors and diseases: insights into mechanisms of activation and therapeutics. Pharmacol Ther. 2008;120:129–148. doi: 10.1016/j.pharmthera.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- 28.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 29.Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, et al. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell. 2004;14:303–317. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- 30.Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci U S A. 2004;101:8603–8607. doi: 10.1073/pnas.0402851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fraser CC. G protein-coupled receptor connectivity to NF-kappaB in inflammation and cancer. Int Rev Immunol. 2008;27:320–350. doi: 10.1080/08830180802262765. [DOI] [PubMed] [Google Scholar]
- 32.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, You Y, Lin PC, Xue L, Morris SW, Zeng H, et al. Bcl10 plays a critical role in NF-kappaB activation induced by G protein-coupled receptors. Proc Natl Acad Sci U S A. 2007;104:145–150. doi: 10.1073/pnas.0601894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye RD. Regulation of nuclear factor kappaB activation by G-protein-coupled receptors. J Leukoc Biol. 2001;70:839–848. [PubMed] [Google Scholar]
- 35.Linden J. New insights into the regulation of inflammation by adenosine. J Clin Invest. 2006;116:1835–1837. doi: 10.1172/JCI29125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, et al. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. 2006;116:2173–2182. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lappas CM, Sullivan GW, Linden J. Adenosine A2A agonists in development for the treatment of inflammation. Expert Opin Investig Drugs. 2005;14:797–806. doi: 10.1517/13543784.14.7.797. [DOI] [PubMed] [Google Scholar]
- 38.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48:1632–1643. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 40.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bharti AC, Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Biochem Pharmacol. 2002;64:883–888. doi: 10.1016/s0006-2952(02)01154-1. [DOI] [PubMed] [Google Scholar]
- 42.Sarkar FH, Li Y. NF-kappaB: a potential target for cancer chemoprevention and therapy. Front Biosci. 2008;13:2950–2959. doi: 10.2741/2900. [DOI] [PubMed] [Google Scholar]
- 43.He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas C, Auwerx J, Schoonjans K. Bile acids and the membrane bile acid receptor TGR5--connecting nutrition and metabolism. Thyroid. 2008;18:167–174. doi: 10.1089/thy.2007.0255. [DOI] [PubMed] [Google Scholar]
- 45.Fiorucci S, Mencarelli A, Palladino G, Cipriani S. Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol Sci. 2009;30:570–580. doi: 10.1016/j.tips.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond) 2008;32(Suppl 7):S52–54. doi: 10.1038/ijo.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8:923–934. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yin J, Zuberi A, Gao Z, Liu D, Liu Z, Ye J. Shilianhua extract inhibits GSK-3beta and promotes glucose metabolism. Am J Physiol Endocrinol Metab. 2009;296:E1275–1280. doi: 10.1152/ajpendo.00092.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.