

Abstract
Purpose
Abiraterone is a potent inhibitor of the steroidogenic enzyme CYP17A1 and suppresses tumor growth in patients with castration-resistant prostate cancer (CRPC). The effectiveness of abiraterone in reducing tumor androgens is not known, nor have mechanisms contributing to abiraterone resistance been established.
Experimental Design
We treated human CRPC xenografts with abiraterone and measured tumor growth, tissue androgens, androgen receptor (AR) levels, and steroidogenic gene expression vs. controls.
Results
Abiraterone suppressed serum PSA levels and improved survival in two distinct CRPC xenografts: median survival of LuCaP35CR improved from 17 to 39 days (HR 3.6, p=0.0014) and LuCaP23CR from 14 to 24 days (HR 2.5, p=0.0048). Abiraterone strongly suppressed tumor androgens, with testosterone (T) decreasing from 0.49 ± 0.22 to 0.03 ± 0.01 pg/mg (p<0.0001), and from 0.69 ± 0.36 to 0.03 ± 0.01 pg/mg (p=0.002) in abiraterone-treated 23CR and 35CR, respectively, with comparable decreases in tissue DHT. Treatment was associated with increased expression of full length AR (ARFL) and truncated AR variants (ARFL 2.3 fold, p=0.008 and ARdel567es 2.7 fold, p=0.036 in 23CR; ARFL 3.4 fold, p=0.001 and ARV7 3.1 fold, p=0.0003 in 35CR), and increased expression of the abiraterone target CYP17A1 (~2.1 fold, p=0.0001 and p=0.028 in 23CR and 35CR, respectively) and transcript changes in other enzymes modulating steroid metabolism.
Conclusions
These studies indicate that abiraterone reduces CRPC growth via suppression of intratumoral androgens and that resistance to abiraterone may occur through mechanisms that include upregulation of CYP17A1, and/or induction of AR and AR splice variants that confer ligand-independent AR transactivation.
Keywords: castration resistant prostate cancer, CYP17A1, abiraterone, steroidogenesis, androgen receptor splice variant
INTRODUCTION
Prostate cancer develops resistance to serum androgen suppression in essentially all patients with advanced disease (1). Upregulated androgen receptor (AR) expression and autonomous synthesis of androgens by neoplastic prostate epithelium (either de novo from cholesterol or through metabolism of adrenal precursors) are important contributors to castration resistant prostate cancer (CRPC) growth (2–5). Tissue androgens such as di-hydrotestosterone (DHT) may be maintained via the “classical” pathway of steroidogene-sis proceeding through dehydroepiandrosterone (DHEA), or through a “back-door” pathway using 5α-reduced steroid precursors as the primary source of DHT (6). In addition to upregulated expression of full length AR (ARFL), the generation of constitutively active AR variants by differential transcript splicing of the ligand binding domain has been described (2, 7–12). These AR splice variants have ligand independent activity, and ARv567es can also function by enhancing the response of ARFL to low ligand concentrations (10, 13).
Abiraterone is a novel agent designed to suppress growth of CRPC by inhibiting CYP17A1, a rate limiting enzyme of steroidogenesis (14). Potential sites of action include any organ capable of elaborating androgens, including testis, adrenal gland or prostate cancer tissue. Positive results in Phase I and II clinical trials, both before and after the use of docetaxel (15–17), have led to phase III studies of abiraterone, demonstrating a survival advantage over prednisone alone in men with CRPC previously treated with do-cetaxel (18).
While clinical studies of abiraterone have demonstrated responses in the majority of men with CRPC, the extent of PSA declines and measureable tumor regression are variable, and the effect of abiraterone in suppressing tumor (as opposed to serum) androgens in men with CRPC has not been determined. As shown in multiple studies, suppression of serum androgens does not correlate well with suppression of tissue androgens in men undergoing androgen deprivation (4, 19, 20). Moreover, most patients treated with abiraterone ultimately suffer tumor progression, and information delineating how resistance to abiraterone occurs is limited. Establishing the effect of abiraterone on tissue androgens and gene expression is critical for determining whether the clinical activity of abiraterone correlates with suppression of tissue androgens and for identifying potential mechanisms of resistance to abiraterone treatment.
To address these issues, we treated CRPC xenografts with abiraterone to determine the effects on tumor growth, tissue androgen concentrations, and tumor gene expression. Our results indicate that alterations in pathways of androgen metabolism and AR expression are mechanisms of molecular adaptation in response to abiraterone treatment, and consequently represent attractive targets for new therapeutic strategies.
METHODS
LuCaP Human Prostate Cancer Xenografts
The establishment and maintenance of the LuCaP23 and 35 xenografts from lymph node metastases of two individuals with CRPC as a component of the University of Washington Rapid Autopsy program has been previously described (21, 22). The AR has been sequenced and codes for a wild-type protein in both xenografts. In eugonadal hosts, these lines produce serum PSA, regress in response to castration, and subsequently re-grow as castration-resistant (CR), PSA-producing variants that were utilized in the present studies. The CR variant of LuCaP35 has previously been termed LuCaP35V, but for consistency the CR variants of both Lu-CaP23 and LuCaP35 are now designated CR. All experiments involving animals were performed in accordance with protocols approved by the University of Washington Institutional Animal Care Use Committee.
Castrate male CB-17 SCID mice (Charles River Laboratories, Wilmington MA) were implanted subcutaneously with 20mm3 pieces of LuCaP23CR or LuCaP35CR tumors. When tumors reached 250–300mm3 (length*(width^2)/2) mice (n=46 and n=28 LuCaP23CR and LuCaP35CR tumor bearing mice, respectively) were randomly assigned to vehicle control (5% benzyl alcohol, 95% safflower oil intraperitoneal (i.p.) injection) or abiraterone treatment (0.5 mmol/kg/d in vehicle) daily for 21 days following enrollment (23). (Unanticipated toxic or off-target effects were not reported in the pre-clinical studies evaluating this dose (which is ~10 fold the oral dose in clinical studies, due to differences in absorption and bioavailability)). Serum was collected by retro-orbital bleeding at interval time-points for determination of PSA. Tumors from a subset of mice in each cohort were harvested at early time points of treatment (tumor size of ~500mm3; range 7–21 days). When the remaining tumors reached approximately 1000mm3 in size, animals were euthanized according to institutional protocol and xenografts harvested and flash frozen for determination of tissue androgens and extraction of total RNA. Serum PSA was measured using the Abbott AxSYM immunoassay system (Abbot Laboratories, Chicago IL). Abiraterone was kindly provided by Cougar Biotechnology. Four mice bearing LuCaP35CR tumors survived for follow-up beyond day 40, whereas all mice bearing LuCaP23CR tumors reached the endpoint and were sacrificed by day 40 (except one at day 42).
Steroid measurements
Androgen levels were determined by mass spectrometry (MS) using methods we have recently described (24). This procedure resulted in a lower limit of quantitation of 1 pg per sample for testosterone and DHT respectively. Intra-assay coefficients of variation generated using human serum for high, mid and low-range samples were 3.5, 3.1 and 3.8% for testosterone and 6.3, 4.3 and 15.8% for DHT, respectively.
RNA Isolation and Quantitative RT-PCR
RNA was isolated and prepared for quantitative PCR (qRT-PCR) as previously described (5). cDNA was generated in a random-primed reverse transcription reaction, and qRT-PCR reactions were performed in triplicate using an Applied Biosystems 7700 sequence detector with 5 ng of cDNA, 1 μM of each primer pair and SYBR Green PCR master mix (Applied Biosystems, Foster City, CA). Primers (Supp Data 1) were designed using the Web-based primer design service Primer3 from the Whitehead Institute for Biomedical Research1, except for AKR1C2 and AKR1C3 (25) and RODH4, DHRS9 (NT-3alpha HSD) and 17BHSD10 (26) for which previously published primer sequences were used. Specificity of amplification was assessed based on melting point of the dissociation curve. In certain cases, 2.5ul of amplified product (total reaction volume 10ul) was also run on a 2% agarose gel to assess for product size (compared to positive control) and the presence of extraneous bands (using a Hamamatsu digital camera with acquisition software from LabWorks).
Statistical Analysis
The effect of abiraterone on tumor growth was quantified using the following linear mixed effects model:
where Volumeij is log-transformed tumor volume measurement j for mouse i, Dayij is day of measurement, Treatmentij indicates treatment group (0 = none and 1 = Abiraterone), bi is a mouse-specific independent and normally distributed random effect with mean 0 and variance σb2, and εij is an independent and normally distributed error term with mean 0 and variance σε2. Median tumor volume trajectories and 95% confidence bands were derived for mice in each treatment group based on mean predictions of empirical quantiles of 1000 bootstrap replicates generated from the original model and refit using identical model structures. Progression free survival in vehicle-control and abiraterone-treated mice (defined as tumor size <1000mm3) was determined via Kaplan Meier analysis with comparison of curves using the Mantel-Haenszel logrank test. For analysis of qRT-PCR data, the mean cycle threshold (Ct) for each gene was normalized to expression of the housekeeping gene RPL13A in the same sample (delta Ct). Unpaired two sample t-tests were used to compare mean delta Ct’s for each gene between vehicle-treated controls and abiraterone treated tumors. P values < 0.05 were considered significant. The fold change was calculated by the delta-delta CT method (fold = 2ΔΔCt ).
RESULTS
Abiraterone Inhibits the Growth of Castration Resistant Prostate Cancers
Prostate cancers progressing in the setting of castration maintain or reactivate the gene expression program regulated by the androgen receptor (AR) (27). In studies designed to identify mechanisms responsible for AR signaling in CRPC, we previously demonstrated that tissue levels of T and DHT in LuCaP23CR and LuCaP35CR xenograft tumors grown in castrate mice are similar to tumor levels measured in isogenic variants passaged in non-castrate mice (5). To determine whether CYP17A1 activity contributes to tumor growth and the maintenance of tumor androgen levels, we treated cohorts of castrate mice bearing LuCaP23CR or LuCaP35CR xenografts with the CYP17A1 inhibitor, abiraterone.
Treatment with abiraterone led to a rapid decline in serum PSA over the first 10 days of treatment in mice bearing either LuCaP23CR or LuCaP35CR tumors (Figure 1A, 1B). Abiraterone also had significant effects on tumor growth, with more rapid median growth per day in control vs. abiraterone-treated tumors (Figure 2A, 2C; LuCaP23CR: 7.4% (95% CI 6.2 – 8.0%) vs. 5% per day (95% CI 3.0% – 6.8%), p=0.0001; and Lu-Cap35CR: 4.8% (95% CI 3.9 – 5.2%) vs. 2.5% per day (95% CI 1.2% – 3.7%), p<0.0001). Accordingly, abiraterone treatment resulted in statistically significant improvements in progression free survival (PFS, defined as tumor size <1000mm3), and median survival (MS) in both xenografts (Figure 1B, 1D). The median survival of Lu-Cap23 improved from 14 to 24 days (hazard ratio (HR) for survival 2.5 (95% CI 1.6–11.2)), while the median survival of LuCaP35 improved from 17 to 39 days (HR 3.6 (95% CI 2.3–34.6)). Interestingly, serum PSA levels at tumor progression remained low in mice bearing LuCaP23CR tumors, although they began to rise in mice bearing Lu-CaP35CR tumors. These observations are consistent with a number of xenograft models demonstrating that coordinate regulation of tumor growth and PSA expression is not necessarily universal (28).
Figure 1.
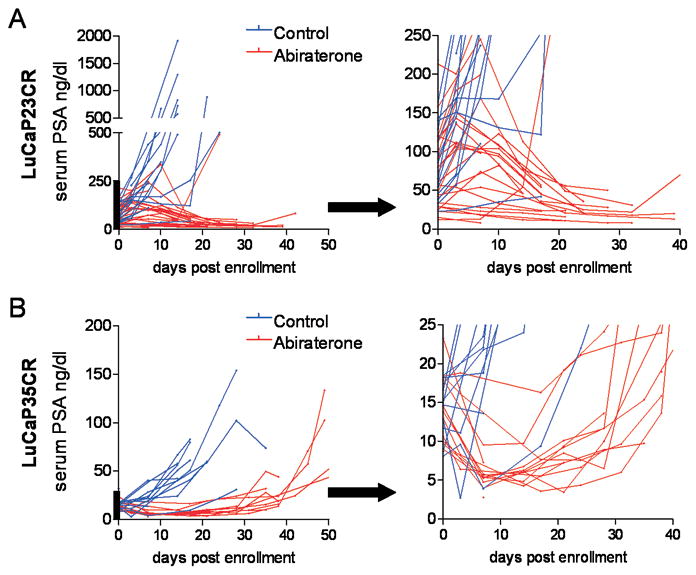
Changes in serum PSA levels in LuCaP23CR and LuCaP35CR prostate cancer xenografts in response to treatment with abiraterone. Castrate male SCID mice were implanted subcutaneously with LuCaP23CR or LuCaP35CR tumors and randomly assigned to vehicle control or abiraterone treatment for 21 days. Serum PSA curves for control (blue) or abiraterone-treated mice (red) are shown for individual mice bearing Lu-CaP23CR (A) and LuCaP35CR (B) xenografts. The segment of the y-axis denoted by the heavy bar in each graph is enlarged and presented in the adjacent panels on an expanded y-axis in order to more clearly demonstrate the decline in serum PSA over the first ~10–15 days after initiation of treatment.
Figure 2.

Tumor growth and survival in LuCaP23CR and LuCaP35CR prostate cancer xenografts treated with abiraterone. Castrate male SCID mice were implanted subcutaneously with LuCaP23CR (A and B) or LuCaP35CR (D and E) tumors and randomly assigned to vehicle control or abiraterone treatment for 21 days. Median tumor volume trajectories with 95% confidence bands for control (blue) or abiraterone-treated mice (red) bearing the LuCaP23CR (A) and LuCaP35CR (D) xenografts. Kaplan-Meier plots of progression-free survival (defined as tumor size <1000mm3) in control or abiraterone-treated mice bearing the LuCaP23CR (B) and LuCaP35CR (E) xenografts. P-values for curve comparisons generated using the Mantel-Haenszel logrank test.
Abiraterone Treatment Reduces Tumor Androgen Levels in Castration Resistant Prostate Cancers
To determine the effect of abiraterone on tumor androgen levels, we measured levels of T and DHT in tumors resected at different time points during and after abiraterone treatment. At early time points during therapy (day 7 for LuCaP23 and days 7–21 for LuCaP35), abiraterone resulted in nearly complete suppression of T (Figure 3A, 3B) and marked suppression of DHT (Figure 3C, 3D) in both LuCaP23CR and Lu-Cap35CR tumors, respectively (p<0.0001 compared to controls for all comparisons). Levels of T (Figure 3A) and DHT (Figure 3C) remained suppressed at later time points in the majority of LuCaP23CR tumors. This included abiraterone-resistant tumors that recurred within the 21 day treatment period (Abi-R), as well as tumors which recurred after therapy had been completed (abiraterone-treated, Abi-T). In contrast, androgen levels in abiraterone-treated LuCaP35 tumors (all of which recurred after day 21) showed a trend toward reconstitution, with a small but statistically significant increase in T (p=0.032, Figure 3B) and a trend towards increase in DHT (p=0.058, Figure 3D) compared to tumors resected earlier while on therapy (d7–21). Notably, tumor androgen levels in the abiraterone treatment arms were correlated with serum PSA levels in both Lu-Ca23CR and 35CR (r=0.7168, p<0.0001; and r=0.9163, p<0.0001; for Pearson correlations with tumor DHT, respectively), consistent with biologic activity associated with androgen levels in the recurrent tumors.
Figure 3.

Tumor testosterone (upper panels) and DHT levels (lower panels) in control and abiraterone-treated LuCaP23CR (A,C) and LuCaP35CR (B,D) xenografts. Androgen levels in abiraterone-treated xenografts were evaluated by mass spectrometry in tumors resected at early (days 7–21) or late time points after therapy. Abiraterone-resistant tumors (Abi-R) recurred (defined as progression to >1000mm3) and were resected during the 21-day period of abiraterone treatment. Abiraterone-treated tumors (Abi-T) recurred and were resected after the completion of abiraterone treatment. This occurred between days 24–42 for LuCap23CR tumors, and between days 29–67 for LuCaP35CR (none of which recurred during the abiraterone-treatment period). P values represent unpaired two-sided t-tests between the indicated groups. P values <0.05 were considered significant. One p value in panel D (in italics) is included as trending toward statistical significance (p<0.10).
Abiraterone Treatment Alters the Expression of Full-Length and Splice-Variant Forms of the AR
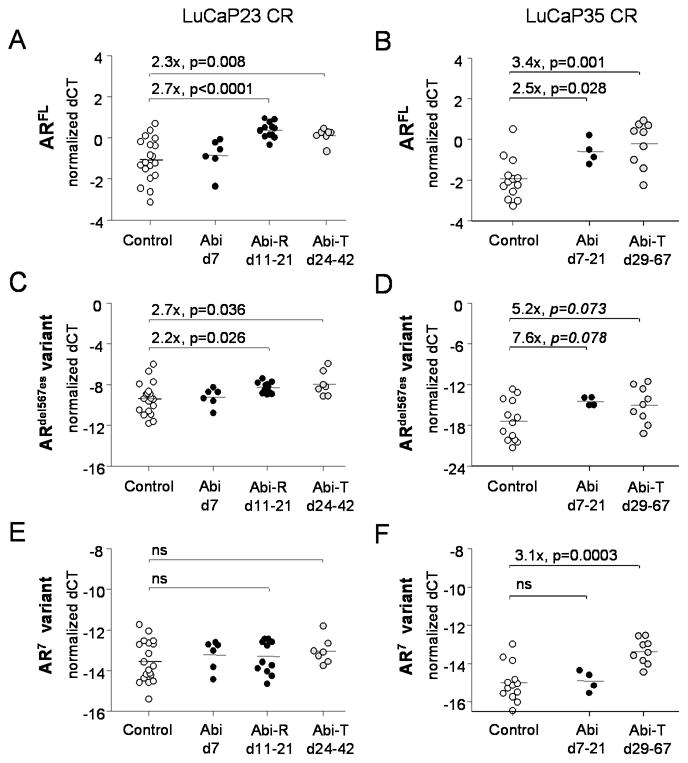
Castration resistant prostate tumors frequently express elevated levels of full-length AR (ARFL) and, as recently reported, increases in expression of AR splice variants that confer constitutive ligand-independent activity (2, 7–11)(12). To determine whether castration resistant tumors recurring after abiraterone treatment demonstrate further alterations in AR expression, we quantitated mRNAs encoding full length ARFL, and the ARdel567es and ARV7 variants recently identified in CRPC metastases (the latter also separately reported as ARV3) (2, 7–12). Compared to controls, LuCaP23CR demonstrated no changes in AR expression at early time point of therapy (Abi d7, Figure 4A, 4C, 4E), but demonstrated significant changes at later time points. Specifically, both abiraterone-resistant (Abi-R) and abiraterone-treated (Abi-T) LuCaP23CR tumors demonstrated increased expression of ARFL (Abi-R 2.7 fold, p<0.0001; Abi-T 2.3 fold p=0.008; Figure 4A) and of ARdel567es (Abi-R 2.2 fold, p<0.026; Abi-T 2.7 fold p=0.036, Figure 4C) with no change in ARV7 (Figure 4E).
Figure 4.
Expression of full length and truncated AR splice variants in LuCaP23CR and LuCaP35CR tumors treated with abiraterone compared to vehicle control. Transcript levels for full length AR (ARFL; A, B), the AR7 splice variant (C, D), and theARdel567es splice variant (E, F) were measured by qRT-PCR in frozen LuCaP23CR (A, C, E) and LuCaP35CR (B, D, E) tumors. White circle denote vehicle treated controls. Black circles denote tumors resected while on abiraterone treatment, either at early time points (d7–21 or at abiraterone-resistant re-growth (Abi-R). Gray circles denote abiraterone-treated tumors (Abi-T) which recurred and were resected after completion of abiraterone treatment. Fold changes are calculated from the difference in mean delta Ct’s between abiraterone treated and vehicle treated controls (delta-delta CT method; fold = 2ddCt). P values from two sample t-tests. P values < 0.05 were considered significant. The p values in panel D (in italics) are included as trending toward statistical significance (p<0.10). n.s. not significant.
LuCaP35CR tumors showed increases in ARFL expression at both early and late time points after treatment (Abi d7–21 2.5 fold, p=0.028; and Abi-T 3.4 fold, p=0.001; Figure 4B). AR variant expression was also altered, with changes in ARdel567es trending toward significance (Abi d7–21, 7.6 fold, p=0.078; and Abi-T 5.2 fold p=0.073; Figure 4D), and ARV7 significantly increased in Abi-T tumors (3.1 fold, p=0.0003; Figure 4F). Notably, the magnitude of full length and AR variant expression is higher at baseline in LuCaP23CR vs. LuCaP35CR tumors (ARFL 1.8 fold higher, p=0.0265; ARdel567es 255 fold p<0.0001; ARV7 2.8 fold, p=0.0001). It is possible a difference in baseline AR levels may facilitate the more rapid growth rate observed in LuCaP23CR and the ability of this tumor to recur in a setting of ongoing ligand suppression.
Abiraterone Treatment Alters the Expression of AR Regulated Genes and Transcripts Encoding Steroidogenic Enzymes
The ultimate objective of CYP17A1 inhibition by abiraterone is suppression of intratumoral androgens with concomitant inhibition of AR mediated signaling. We therefore determined the impact of abiraterone on tumoral expression of androgen regulated genes. We also evaluated expression of steroidogenic genes throughout the androgen biosynthetic pathway, as alteration in steroidogenic activity might mitigate the impact of abiraterone on intratumoral androgen levels.
Expression of Androgen Regulated Genes
Consistent with decreases in tumor androgen levels associated with abiraterone treatment, both LuCap23CR and LuCap35CR demonstrate significant decreases in expression of androgen regulated genes (e.g, NKX3.1, FKBP5, TMPRSS2; Table 1). Interestingly, tissue PSA expression was not altered by abiraterone treatment in LuCaP23CR (Figure 5A), remaining highly expressed despite the rapid and sustained suppression of serum PSA levels associated with abiraterone treatment (Figure 1A). In contrast, treatment with abiraterone was clearly associated with inhibition of tissue PSA expression in LuCaP35CR (Figure 5B), particularly at the early timepoints (d7–21) when tumors were under active treatment. Moreover, tissue and serum PSA levels were directly correlated in abiraterone-treated LuCaP35CR tumors (Pearson correlation r=0.8226; p=0.0006) but not in LuCaP23CR. These observations suggest that androgen-mediated effects on serum PSA levels may reflect alterations in release/secretion of PSA with or without associated changes in tumoral PSA transcription.
Table 1.
Changes in AR and Steroidogenic Gene Expression in Abiraterone-Treated CRPC Xenografts
| LuCaP 23 CR | LuCaP 35 CR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Time point of resection | Early (d7) | Abiraterone t Resistant (d11–21) | Abiraterone tt Treated (d24–42) | Early (d7–21) | Abiraterone tt Treated (d29–67) | ||||||
| Gene and Function | fold* | p value** | fold | p value | fold | p value | fold | p value | fold | p value | |
| AR | ARFL | 2.7 | p<0.0001 | 2.3 | 0.008 | 2.5 | 0.028 | 3.4 | 0.001 | ||
| ARdel567es | 2.2 | 0.026 | 2.7 | 0.036 | 7.6 | 0.078 | 5.2 | 0.073 | |||
| ARV7 | 3.1 | 0.0003 | |||||||||
|
| |||||||||||
| AR regulated genes | PSA | −3.4 | 0.003 | −2.0 | 0.023 | ||||||
| NKX3.1 | −1.2 | 0.026 | −1.9 | 0.038 | −1.6 | 0.036 | |||||
| FKBP5 | −1.6 | 0.004 | −3.0 | p<0.0001 | −2.7 | p<0.0001 | −3.8 | 0.002 | |||
| TMPRSS2 | −1.3 | 0.005 | −1.6 | p<0.0001 | |||||||
|
| |||||||||||
| Cholesterol import and processing | SCARB1 | −1.5 | 0.042 | ||||||||
| LDLR | −3.0 | 0.001 | −1.9 | 0.003 | |||||||
| LIPE | 4.5 | p<0.0001 | 7.1 | p<0.0001 | |||||||
| SCAP | 1.8 | 0.003 | 2.6 | p<0.0001 | 1.3 | 0.024 | |||||
| CYP11A1 | 1.3 | 0.022 | 1.2 | 0.031 | 1.6 | 0.0003 | |||||
| STAR | 1.5 | 0.002 | 1.5 | 0.018 | |||||||
| STARD3 | 1.8 | 0.003 | 2.4 | p<0.0001 | |||||||
| STARD4 | −1.6 | p<0.0001 | −1.4 | 0.007 | −2.6 | 0.004 | 1.6 | 0.057 | |||
| STARD5 | 1.5 | 0.005 | 1.7 | 0.006 | 1.8 | 0.003 | |||||
|
| |||||||||||
| CYP17A1 and co-factors | CYP17A1 | 1.6 | 0.022 | 1.7 | 0.0002 | 2.1 | 0.0001 | 2.2 | 0.028 | ||
| HNF1A | 1.7 | 0.004 | 1.8 | 0.003 | 2.1 | 0.003 | |||||
| HNF4A | −1.7 | 0.01 | 22.7 | 0.015 | |||||||
| NR5A1 (SF-1) | 1.3 | 0.02 | 1.3 | 0.04 | 1.5 | 0.010 | |||||
| CYB5A | 1.4 | 0.0001 | |||||||||
| POR | 1.6 | p<0.0001 | 2.1 | p<0.0001 | −1.5 | 0.0002 | 1.2 | 0.043 | |||
| DAX1 | 2.8 | 0.03 | 2.0 | 0.043 | |||||||
| DUSP | 1.7 | 0.026 | −3.1 | p<0.0001 | |||||||
|
| |||||||||||
| Conversion of C-19 steroids to T and DHT | HSD3B1 | ||||||||||
| HSD3B2 | 1.5 | 0.050 | 1.7 | 0.063 | |||||||
| HSD17B3 | 1.3 | 0.006 | 1.4 | 0.003 | 2.2 | 0.001 | |||||
| AKR1C3 | 1.6 | 0.086 | 1.9 | 0.001 | 1.6 | 0.053 | 5.2 | p<0.0001 | |||
| SDR5A1 | −1.4 | 0.004 | |||||||||
| SDR5A2 | 1.9 | 0.028 | 3.5 | p<0.0001 | 4.5 | p<0.0001 | |||||
| SDR5A3 | 1.2 | 0.038 | 1.4 | p<0.0001 | 1.7 | p<0.0001 | |||||
| CYP19A | 1.7 | 0.009 | 2.5 | 0.001 | 2.5 | 0.037 | |||||
|
| |||||||||||
| Pre-receptor regulation of DHT | AKR1C2 | 4.2 | p<0.0001 | 3.9 | 0.001 | 3.3 | 0.0001 | ||||
| HSD17B10 | |||||||||||
| RODH 4 | −2.9 | 0.000 | −4.8 | p<0.0001 | −5.9 | p<0.0001 | −1.8 | 0.024 | |||
| RDH5 | 2.8 | 0.0001 | |||||||||
| RLHSD (HSD17B6) | −4.3 | 0.003 | −26.6 | p<0.0001 | −26.2 | p<0.0001 | −27.3 | p<0.0001 | −4.8 | 0.001 | |
| DHRS9 (HSD17B13) | 3.7 | 0.0001 | 7.5 | p<0.0001 | −6.3 | 0.0003 | |||||
|
| |||||||||||
| Gluc-uronide formation | UGDH | 1.7 | 0.006 | 2.3 | p<0.0001 | −1.6 | 0.016 | ||||
| UGT2B15 | 4.6 | p<0.0001 | 9.3 | p<0.0001 | 5.1 | 0.002 | |||||
|
|
|||||||||||
| UGT2B17 | 2.6 | 0.0001 | 3.6 | p<0.0001 | UGT2B17 is deleted in LuCaP35CR | ||||||
Abiraterone Resistant tumors recurred (size 1000mm3) and were resected while on abiraterone.
Abiraterone Treated tumors recurred (size 1000mm3) after the 21 day treatment period.
Fold change from difference in mean delta Ct’s (delta-delta CT method; fold = 2ddCt) between controls and abiraterone-treated tumors in the indicated groups (Early, Resistant or Treated). Fold changes for genes which were not statistically significant are omitted
P values from two sample t-tests. P values ≤ 0.05 were considered significant. Genes with p values <0.10 were considered trending toward significance if fold change was also ≥ 1.5 (shown in italics).
Figure 5.
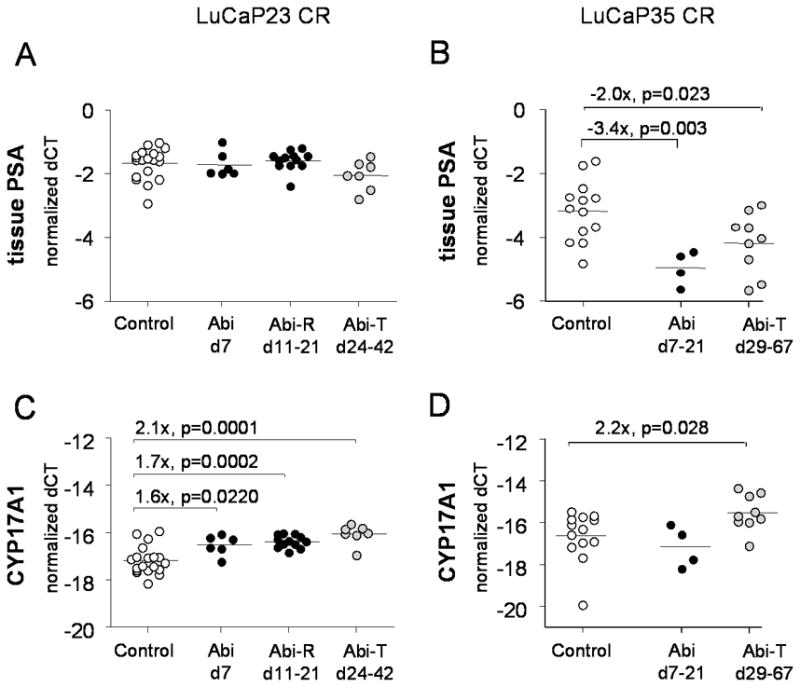
Expression of PSA and CYP17A1 in LuCaP23CR and LuCaP35CR tumors treated with abiraterone compared to vehicle control. Transcript levels for PSA (A, B) and CYP17A1 were measured by qRT-PCR in frozen LuCaP23CR (A, C) and Lu-CaP35CR (B, D) tumors. White circle denote vehicle treated controls. Black circles denote tumors resected while on abiraterone treatment, either at early time points (d7–21 or at abiraterone-resistant re-growth (Abi-R). Gray circles denote abiraterone-treated tumors (Abi-T) which recurred and were resected after completion of abiraterone treatment. Fold changes are calculated from the difference in mean delta Ct’s between abiraterone treated and vehicle treated controls (delta-delta CT method; fold = 2ddCt). P values from two sample t-tests. P values < 0.05 were considered significant.
Expression of Genes Required for Androgen Synthesis
Abiraterone-treated tumors demonstrated increased expression of numerous genes throughout the androgen biosynthetic pathway (Figure 6). Both LuCaP23CR and LuCaP35CR tumors responded to abiraterone treatment with increased expression of the target gene CYP17A1 (Figure 5C, Lu-CaP23CR Abi-R 1.7 fold, p=0.0002; Abi-T 2.1 fold, p=0.0001; Figure 5D, LuCaP35CR Abi-T 2.1 fold, p=0.0278). The expression of AKR1C3 and HSD17B3, two key enzymes mediating conversion of adrenal androgen intermediates to T, was also increased in both LuCaP23CR and LuCap35CR tumors compared to controls (Table 1). Of note, expression changes in abiraterone-resistant LuCaP23CR tumors recurring while on therapy (Abi-R) were generally similar in direction and magnitude to changes observed in abira-terone-treated tumors recurring after completion of therapy (Abi-T), suggesting the effects of abiraterone were sustained beyond the immediate treatment interval.
Figure 6.

The pathways of androgen biosynthesis. In the classical pathway (solid gray arrow), C21 precursors (pregnenolone and progesterone) are converted to the C19 adrenal androgens DHEA and androstenedione (AED) by the sequential hydroxylase and lyase activity of CYP17A1. DHEA and AED are converted to testosterone by a series of reactions involving the activity of HSD3B1 and 2, HSD17B3 and AKR1C3 enzymes. Testosterone is converted to the potent androgen DHT by the activity of SRD5A1 and 2. Oxida-tive 3 α-HSD enzymes (including HSD17B6 (RL-HSD), HSD17B10, HSD17B13 (DHRS9), RODH4 and RDH5) can act to inhibit the pre-receptor catabolism of DHT. An alternative pathway (short gray arrows) has also been proposed in which C21 precursors are first acted upon by SRD5A and the reductive activity of AKR1C2, followed by CYP17A1, HSD17B3 and subsequent oxidation to DHT Adapted from Mostaghel EA, Nelson PS. Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Best Pract Res Clin Endocrinol Me-tab. 2008;22:243, with permission (pending).
Overall, genes required for androgen biosynthesis were expressed in both Lu-CaP23CR and LuCaP35CR tumors, and observed changes in expression are consistent with an up-regulated capacity for androgen biosynthesis. As might be anticipated, the relative magnitude of steroidogenic gene induction in CRPC xenografts, before and after abiraterone treatment, is lower than that observed in clinical studies of CRPC tissues, in which CRPC metastases were compared to primary, untreated prostate tumors (5).
Expression of Genes Mediating Pre-receptor Regulation of DHT Levels
Tumor androgen levels reflect the sum total of activity in both androgen biosynthetic and androgen catabolic pathways. The back-conversion of 3α-diol to DHT, which can be mediated by enzymes with 3 alpha-hydroxysteroid reductase (3α-HSD) activity, has primarily been attributed to RL-HSD, although RODH4, RDH5, HSD17B10, and DHRS9 can also mediate this reaction and are expressed to varying degrees in prostate tissue (29, 30). We therefore examined whether genes involved in pre-receptor regulation of DHT levels via back-conversion of 3-α-androstanediol (3-α-diol) to DHT were also altered by abirate-rone treatment. Interestingly, changes in expression of oxidative genes mediating back-conversion of 3α-diol to DHT in abiraterone treated tumors were mixed, with increases in genes such as DHRS9 in LuCaP23CR, and RDH5 in LuCaP35, but significant decreases in expression of RL-HSD in both LuCaP23CR and LuCaP35CR (Table 1).
The marked suppression of RL-HSD in response to ligand inhibition with abirate-rone is consistent with a recent report demonstrating suppression of RL-HSD levels in CWR22 PCa xenografts in response to castration (30), and may explain the relatively low levels of androgens measured in abiraterone-treated tumors despite general induction of genes mediating androgen biosynthesis. Notably, tumor DHT levels were strongly correlated with RODH4 (r=0.7348, p<0.0001) and RL-HSD (r=0.6431, p=0.0005) in Lu-CaP23CR, and with RODH4 (r=0.6211, p=0.0235), 17BHSD10 (r=0.6332, p=0.0202), DHRS9 (r=0.7815, p=0.0016) and RL-HSD (r=0.6329, p=0.0202) in LuCaP35CR (Pear-son product correlations). These data suggest that despite an overall decrease in expression of genes such as RL-HSD, androgen levels in abiraterone treated tumors are, nevertheless, specifically associated with expression of genes inhibiting catabolism of DHT.
As tissue DHT levels in abiraterone-treated (Abi-T) LuCaP35CR tumors ranged from ~0.1 to >4pg/mg (Figure 4D), we also determined whether these differences in androgen reconstitution were related to changes in the steroidogenic transcriptome. Notably, Abi-T LuCaP35CR tumors that recurred with high vs. low tissue androgens (defined as tissue DHT above or below the median of ~0.5pg/mg) demonstrated 2.5 fold higher PSA (p=0.007), 2.9 fold higher CYP17A1 (p=0.033), markedly higher HNF4A (97 fold, p=0.020, a CYP17A1 cofactor), and 8.2 and 5.4 higher levels of genes involved in preventing DHT catabolism (p=0.011 and p=0.005 for RL-HSD and DHRS9, respectively), consistent with a critical role for genes involved in both androgen synthesis and pre-receptor regulation of DHT catabolism in determining tissue androgen levels. (Statistically significant differences in expression of AR or AR variants were not observed, data not shown).
DISCUSSION
Clinical studies with abiraterone have demonstrated striking responses in men with CRPC, though not all patients respond and most patients eventually develop resistance. While abiraterone is known to inhibit CYP17A1 in vitro, the effectiveness of abiraterone in suppressing intratumoral androgens has not been established, and it remains unclear which CRPC phenotypes and genotypes are susceptible to CYP17A1 inhibition and how tumors develop resistance. Using two distinct CRPC models, we demonstrate that treatment with abiraterone significantly inhibited tumor growth, serum PSA, and intratumoral androgen levels, supporting the hypothesis that abiraterone’s primary mechanism of action is through effects on tissue androgens. Furthermore, both CRPC models responded to CYP17A1 inhibition with mechanisms that maintain AR signaling. This included upregulated expression of full length AR and ligand independent AR variants, as well as induction of steroidogenic genes (including the target gene, CYP17A1), several of which showed strong correlations with DHT levels in the recurrent tumors. Thus, in the setting of tumor progression on abiraterone, the rationale for focusing further therapeutic efforts on more potent AR antagonists and agents suppressing AR ligands remains strong.
Although specific mechanisms driving induction of alternative AR splicing have not been established, generation of AR splice variants following suppression of tumor androgens by abiraterone is consistent with the castration-mediated induction of AR splice variants observed in castration sensitive prostate cancer models (10, 12, 13). Interestingly, studies of testosterone replacement in this setting (either in vitro, or when given within days of castration in vivo) have been shown to inhibit castration-associated increases in AR variant expression (10, 12). However, we did not observe lower levels of full length or variant AR expression in those tumors with higher levels of androgens at recurrence (data not shown). These observations suggest that factors regulating the initial induction of AR splice variant expression could differ from those maintaining variant expression at later time points of recurrent growth. Moreover, these observations demonstrate that certain tumors may simultaneously engage or accrue multiple resistance pathways directed at preserving the AR axis.
The molecular alterations occurring in CRPC tumors following abiraterone treatment suggest tumor-specific methods of addressing resistance, either through optimizing steroidogenic blockade or by inhibiting AR signaling. Although substantially suppressed, androgen levels remained detectable in abiraterone-treated tumors. Importantly, a 2 to 3 fold increase in expression of full length AR is known to render low androgen levels physiologically relevant in promoting AR driven growth (2), suggesting clinical treatment of abiraterone-resistant patients with more stringent ligand inhibition may be of benefit. More complete reduction of steroidogenesis might be achieved through enhancing local concentrations of CYP17A1 inhibitors, or targeting transcriptional activation of the enzyme. CYP17A1 is regulated by SF-1 and other co-factors (31), and its regulatory domains contain multiple cAMP responsive elements, providing several targets for modulation of enzyme expression, such as using phosphodiesterase inhibitors (32). Combining CYP17A1 blockade with inhibitors of other critical components of the pathway such as HSD3B1 or SRD5A2 could also offset adaptive upregulation of CYP17A1 (33).
Data regarding expression of C′ terminal truncated AR splice variants in CRPC continues to emerge, and will be a critical area of investigation as more potent ligand synthesis and AR inhibitors become utilized in the treatment of CRPC. AR splice variants may act by potentiating activity of full length AR as well as mediating constitutive AR transactivation (10, 13). Thus, increased expression of AR splice variants in abiraterone-treated tumors may be an important biomarker of resistance and target for therapy. Incorporation of potent AR inhibitors, such as MDV3100, or agents targeting the AR N-terminal domain, such as EPI-001, could be utilized for tumors adapting to CYP17A1 inhibition via induction of full-length AR and/or AR splice variants lacking the ligand binding C-terminal domain (13, 34–36). Studies to delineate whether sequential or concurrent use of these agents with abiraterone can improve tumor growth inhibition and/or suppress adaptation in xenograft models will be important to provide rationale for human studies evaluating these agents in the treatment of clinical disease.
The response to abiraterone in our study is most likely due to suppression of de novo intratumoral steroidogenesis (due to the reported lack of CYP17A1 in rodent adrenal glands) (37, 38), whereas in human studies the response to abiraterone may reflect inhibition of both adrenal and/or intratumoral CYP17A1 activity. Importantly, the proposed mechanism driving clinical activity of abiraterone in both scenarios is a decrease in intra-tumoral androgens (whether from suppression of adrenal steroidogenesis, intratumoral steroidogenesis, or both). Our observations confirm this expected mechanism of activity. Furthermore, increases in expression of full length AR and ligand-independent AR splice variants following abiraterone treatment are likely to be clinically relevant mechanisms of resistance regardless of whether tumoral CYP17A1 activity is present.
Another consideration for translation of our results to the clinical setting is that men with CRPC are likely to be treated with abiraterone plus prednisone (or dexametha-sone) rather than abiraterone alone. In castrate men treated with abiraterone, glucocorti-coids are primarily used to inhibit pituitary-mediated secretion of ACTH (induced by adrenal CYP17A1 inhibition) which can exacerbate side effects of mineralocorticoid excess, but may potentially inhibit tumor growth directly (39). ACTH may also drive clinically significant increases in production of androgenic precursors by the adrenal gland, as evidenced by PSA declines following the addition of dexamethasone in men who had progressed on abiraterone (15). Thus, the inclusion of prednisone in men with CRPC is likely to accentuate any decrease in tumor androgens caused by abiraterone, and may accentuate the types of tumoral responses observed in the abiraterone-treated xeno-grafts.
Our hypothesis that tumoral androgens in these xenograft studies are derived from de-novo steroidogenesis is consistent with previous observation that a subset of CRPC metastases have increased levels of transcripts for CYP17A1 and HSD3B1 (necessary for de novo synthesis) and the demonstration by Locke et al that CRPC xenografts are capable of synthesizing DHT from acetate (5, 6, 40). However, alternative androgenic precursors (of adrenal or other origin) may also be present in the circulation as potential substrates. Unfortunately, serum samples adequate to assess circulating androgen levels were not available for analysis in this study. In addition, the duration of abiraterone treatment in our study was 21 days. It is possible that more prolonged treatment would have resulted in a more robust induction of the adaptive changes already observed. Finally, me-tastatic CRPC is characterized by significant heterogeneity, while our conclusions reflect an analysis of only two CRPC phenotypes, and we had small numbers in some of the treatment groups. These limitations can be addressed by evaluating diverse panels of CRPC xenografts over a more prolonged time-course of therapy, and through studies of human tumor biopsies obtained at abiraterone resistance.
In conclusion, our finding that abiraterone suppresses intratumoral androgens and inhibits CRPC growth supports the hypothesis that tissue androgen levels are major contributors to AR signaling and mediators of CRPC progression. Though hypothesized, this is the first demonstration that the efficacy of abiraterone is related to its ability to suppress tumor androgen levels and complements previously published data from phase I studies regarding suppression of serum androgens (15, 41). Our results also identify potential mechanisms of adaptation to CYP17A1 blockade, including increased expression of AR, AR splice variants, and the steroidogenic transcriptome. Importantly, these adaptive mechanisms can potentially be targeted by using higher dose levels of abiraterone or combinations with potent AR antagonists currently in development. These data provide optimism that more effective suppression of AR signaling will continue to be an important means of effectively treating advanced prostate cancer.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Abiraterone is a novel CYP17A1 inhibitor recently shown to extend the survival of men with castration resistant prostate cancer (CRPC). However, many men ultimately progress and mechanisms of resistance to abiraterone treatment in vivo have not been elucidated. We demonstrate in two xenograft models of CRPC that the clinical response to abiraterone is accompanied by marked suppression of tumor androgen levels, and identify increased expression of AR, ligand independent AR splice variants, and the steroido-genic transcriptome (including the CYP17A1 target gene) as potential mechanisms of adaptation to CYP17A1 blockade. These data suggest resistance can potentially be targeted by using higher dose levels of abiraterone or combinations with potent AR antagonists currently in development, and demonstrate that more effective suppression of AR signaling remains an important means of effectively treating advanced prostate cancer.
Acknowledgments
We thank Holly Nguyen, Andrew Morgan, Jared Lucas and Roger Coleman for expert technical assistance, Roman Gulati for statistical support, Cougar Biotechnology for providing abiraterone, and the Lucas Family for their generous support.
Support: Prostate Cancer Foundation (Career Development Award to EAM and Synergy Award to SB, RBM and PSN); Damon Runyon Cancer Research Foundation (Damon Runyon-Genentech Clinical Investigator Award CI-40-08 to EAM); National Institutes of Health (Career Development Award K23 CA122820 to EAM; Pacific Northwest Prostate Cancer SPORE P50 CA97186 (RV, PSN); the Cancer Center Support Grant to Fred Hutchinson Cancer Research Center (P30 CA015704 pilot to RBM)); and the Veterans Affairs Research Service (SRP).
Footnotes
References
- 1.Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 2.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 3.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 4.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, et al. Intra-prostatic Androgens and Androgen-Regulated Gene Expression Persist after Testosterone Suppression: Therapeutic Implications for Castration-Resistant Prostate Cancer. Cancer Res. 2007;67:5033–41. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 5.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 7.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcias G, Erdmann E, Lapouge G, Siebert C, Barthelemy P, Duclos B, et al. Identification of novel truncated androgen receptor (AR) mutants including unreported pre-mRNA splicing variants in the 22Rv1 hormone-refractory prostate cancer (PCa) cell line. Hum Mutat. 2010;31:74–80. doi: 10.1002/humu.21138. [DOI] [PubMed] [Google Scholar]
- 12.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases is Associated with Castration-Resistance and Short Survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarman M, Barrie SE, Llera JM. The 16,17-Double Bond Is Needed for Irreversi-ble Inhibition of Human Cytochrome P45017alpha; by Abiraterone (17-(3-Pyridyl)androsta-5,16-dien-3beta-ol) and Related Steroidal Inhibitors. J Med Chem. 1998;41:5375–81. doi: 10.1021/jm981017j. [DOI] [PubMed] [Google Scholar]
- 15.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–8. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–95. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, Smith MR, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol. 2010;28:1496–501. doi: 10.1200/JCO.2009.25.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abirate-rone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohler JL, Gregory CW, Ford OH, 3rd, Kim D, Weaver CM, Petrusz P, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–8. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 20.Page ST, Lin DW, Mostaghel EA, Hess DL, True LD, Amory JK, et al. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J Clin En-docrinol Metab. 2006;91:3850–6. doi: 10.1210/jc.2006-0968. [DOI] [PubMed] [Google Scholar]
- 21.Corey E, Quinn JE, Buhler KR, Nelson PS, Macoska JA, True LD, et al. LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate. 2003;55:239–46. doi: 10.1002/pros.10198. [DOI] [PubMed] [Google Scholar]
- 22.Ellis W, Vessella R, Buhler K, Bladou F, True L, Bigler S, et al. Characterization of a novel androgen-sensitive, prostate-specific antigen-producing prostatic carcinoma xenograft: LuCaP 23. Clin Cancer Res. 1996;2:1039–48. [PubMed] [Google Scholar]
- 23.Barrie SE, Potter GA, Goddard PM, Haynes BP, Dowsett M, Jarman M. Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17–20 lyase) J Steroid Biochem Mol Biol. 1994;50:267–73. doi: 10.1016/0960-0760(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 24.Page ST, Kalhorn TF, Bremner WJ, Anawalt BD, Matsumoto AM, Amory JK. Intratesticular androgens and spermatogenesis during severe gonadotropin suppression induced by male hormonal contraceptive treatment. J Androl. 2007;28:734–41. doi: 10.2164/jandrol.107.002790. [DOI] [PubMed] [Google Scholar]
- 25.Ji Q, Chang L, VanDenBerg D, Stanczyk FZ, Stolz A. Selective reduction of AKR1C2 in prostate cancer and its role in DHT metabolism. Prostate. 2003;54:275–89. doi: 10.1002/pros.10192. [DOI] [PubMed] [Google Scholar]
- 26.Steckelbroeck S, Watzka M, Reissinger A, Wegener-Toper P, Bidlingmaier F, Bliesener N, et al. Characterisation of estrogenic 17[beta]-hydroxysteroid dehydrogenase (17[beta]-HSD) activity in the human brain. The Journal of Steroid Biochemistry and Molecular Biology. 2003;86:79–92. doi: 10.1016/s0960-0760(03)00251-6. [DOI] [PubMed] [Google Scholar]
- 27.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–27. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denmeade SR, Sokoll LJ, Dalrymple S, Rosen DM, Gady AM, Bruzek D, et al. Dissociation between androgen responsiveness for malignant growth vs. expression of prostate specific differentiation markers PSA, hK2, and PSMA in human prostate cancer models. Prostate. 2003;54:249–57. doi: 10.1002/pros.10199. [DOI] [PubMed] [Google Scholar]
- 29.Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the Major Oxidative 3{alpha}-Hydroxysteroid Dehydrogenase in Human Prostate That Converts 5{alpha}-Androstane-3{alpha},17{beta}-diol to 5{alpha}-Dihydrotestosterone: A Potential Therapeutic Target for Androgen-Dependent Disease. Mol Endocrinol. 2006;20:444–58. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- 30.Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotes-tosterone in prostate cancer. Cancer Res. 2011;71:1486–96. doi: 10.1158/0008-5472.CAN-10-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu B, Yang WH, Gerin I, Hu CD, Hammer GD, Koenig RJ. Dax-1 and steroid receptor RNA activator (SRA) function as transcriptional coactivators for steroidogenic factor 1 in steroidogenesis. Mol Cell Biol. 2009;29:1719–34. doi: 10.1128/MCB.01010-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Urs AN, Allegood J, Leon A, Merrill AH, Jr, Sewer MB. Cyclic AMP-Stimulated Interaction between Steroidogenic Factor 1 and Diacylglycerol Kinase {theta} Facilitates Induction of CYP17. Mol Cell Biol. 2007;27:6669–85. doi: 10.1128/MCB.00355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evaul K, Li R, Papari-Zareei M, Auchus RJ, Sharifi N. 3beta-hydroxysteroid de-hydrogenase is a possible pharmacological target in the treatment of castration-resistant prostate cancer. Endocrinology. 2010;151:3514–20. doi: 10.1210/en.2010-0138. [DOI] [PubMed] [Google Scholar]
- 34.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al. Anti-tumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375:1437–46. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 37.van Weerden WM, Bierings HG, van Steenbrugge GJ, de Jong FH, Schroder FH. Adrenal glands of mouse and rat do not synthesize androgens. Life Sci. 1992;50:857–61. doi: 10.1016/0024-3205(92)90204-3. [DOI] [PubMed] [Google Scholar]
- 38.Luu-The V, Pelletier G, Labrie F. Quantitative appreciation of steroidogenic gene expression in mouse tissues: new roles for type 2 5alpha-reductase, 20alpha-hydroxysteroid dehydrogenase and estrogen sulfotransferase. J Steroid Biochem Mol Biol. 2005;93:269–76. doi: 10.1016/j.jsbmb.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Kassi E, Moutsatsou P. Glucocorticoid receptor signaling and prostate cancer. Cancer Lett. doi: 10.1016/j.canlet.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 40.Mostaghel EA, Wright JL, Kwon EM, Montgomery RB, Ostrander EA, Vessella R, et al. Genetic variation in SLCO2B1, SLCO1B3, and prostate cancer risk and mortality. 2010 Genitourinary Cancers Symposium; 2010; 2010. [Google Scholar]
- 41.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.