

Abstract
We describe an approach for the accurate quantitation of global protein dynamics in Caenorhabditis elegans. We adapted Stable Isotope Labeling with Amino acids in Cell culture (SILAC) for nematodes, by feeding worms a heavy lysine- and arginine-labeled E. coli strain. We also report a genetic solution to remove the arginine-to-proline conversion problem. Combining our approach with quantitative proteomics methods, we characterized the heatshock response in worms.
Results
The SILAC mass spectrometry approach facilitates the accurate and reproducible quantitation of large numbers of proteins1. In contrast, 15N-labeling which has been applied previously for proteomic analysis in C. elegans2, has drawbacks for data analysis, including the unpredictable mass shifts induced by total 15N labeling3. SILAC is typically used with tissue culture cells, and SILAC-based methods have not been reported previously for C. elegans. While SILAC has been used in a small number of multicellular organisms, problems associated with the conversion of isotope-labeled arginine to proline and other amino acids have complicated such studies4. Arginine-to-proline conversion reduces the signal from all heavy SILAC labeled peptides containing proline. The increased number of MS/MS target peptides resulting from arginine-to-proline conversion reduces the overall sensitivity of global MS/MS analysis. These additional peptide peaks also increase the likelihood of overlapping isotopic envelopes reducing peak discrimination.
We aimed to adapt SILAC methodology for C. elegans and to eliminate the arginine-to-proline conversion problem. Initially, we employed an arginine and lysine auxotrophic version of E. coli BL21 (DE3), but C. elegans did not survive well on this strain (data not shown). We therefore modified E. coli HT115, a strain commonly used for the RNAi-feeding procedure5, by generating an arginine and lysine auxotroph termed SLE1 (Supplementary Fig. 1a). Analysis of the egg laying rate and embryonic development/survival of C. elegans grown on the E. coli SLE1 strain indicated no difference (Supplementary Fig. 1b) compared to E. coli op50, which is generally used as a food source in C. elegans studies. For C. elegans labeling the SLE1 strain was grown in minimal media containing 15N4-13C6-arginine (heavy arginine) and 15N2-13C6-lysine (heavy lysine), and plated onto agarose petri dishes. C. elegans eggs were placed onto a lawn of SLE1 and the incorporation of heavy arginine and lysine into protein in the F1 generation was analysed by mass spectrometry (MS).
We observed approximately 93 % heavy isotope incorporation as illustrated for a representative peptide (Supplementary Fig. 2a). However, arginine-to-proline conversion led to ~20 % of the heavy peptide signal being diverted to a larger m/z signal. This diversion of signal intensity could be seen more clearly when equal portions of heavy and light protein were mixed (Fig. 1). Analysis of each peak shown in Supplementary Fig. 2a by tandem mass spectrometry (MS/MS) confirmed that the heavier peak contained 15N-13C5-proline (heavy proline) and no other labeled amino acids besides arginine (Supplementary Fig. 2b). The conversion of heavy arginine to heavy proline occurs in the nematode and not in SLE1 because the E. coli did not generate heavy proline containing peptides (Supplementary Table 1). Arginine-to-proline conversion occurs through the urea cycle6 (Supplementary Fig. 3, see below).
Figure 1.
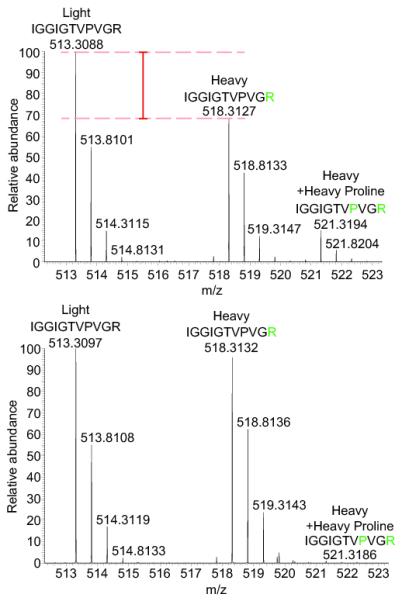
Elimination of arginine-to-proline conversion using orn-1 RNAi-feeding facilitates stable isotope labeling with amino acids. Arginine-to-proline conversion, which occurs in wild type worms (upper panel) is abolished upon orn-1 depletion (lower panel). The peak intensity of the depicted ‘light’ model peptide approximately equals the combined intensities of the corresponding heavy peptide and the peptide that contains heavy proline (n = 3). Peptides are highlighted as ‘Light’, ‘Heavy’ and ‘Heavy+ Heavy proline’. Lysates from light or heavy labeled C. elegans were mixed in equal proportions, fractionated by denaturing SEC and fraction 12 was analysed by trypsin digestion and LC-MS/MS. A representative proline containing peptide derived from EF-1α was examined.
To eliminate the arginine-to-proline conversion problem we employed the RNAi-feeding procedure, targeting the ornithine transaminase enzyme orn-1 (C16A3.10). This enzyme converts ornithine to L-glutamate-5-semialdehyde (Supplementary Fig. 3) and is required for arginine-to-proline conversion in S. pombe6. RNAi-feeding of C. elegans was performed according to the schemes in Fig. 2 and Supplementary Fig. 4a, and the extent of orn-1 transcript depletion was evaluated by qPCR (Supplementary Fig. 4b). Worms grown with either control or orn-1 RNAi knock-down showed similar egg laying rate and embryonic development, which indicated that orn-1 RNAi did not have a major effect on viability (Supplementary Fig. 4c). orn-1 RNAi knock-down worms were labeled with both heavy arginine and heavy lysine, or their respective light amino acids, for one generation, to determine the effect of RNAi feeding on arginine-to-proline conversion. MS analysis showed again approximately 93 % heavy arginine and lysine isotope incorporation (Fig. 1 and Supplementary Fig. 2c), and that greater than 98% of total proline was 14N-12C5-proline (light proline), indicative of a near complete elimination of arginine-to-proline conversion (Fig. 1 and Supplementary Fig. 2a). In addition, when equal portions of untreated heavy and light protein were mixed and analysed by MS, more than 95 % of the proteins had Log2 ratios that were approximately 0 (Supplementary Table 1). These data also indicate that fold-changes greater than +/− 50 % would differentiate proteins whose expression level has been altered from non-affected proteins.
Figure 2.
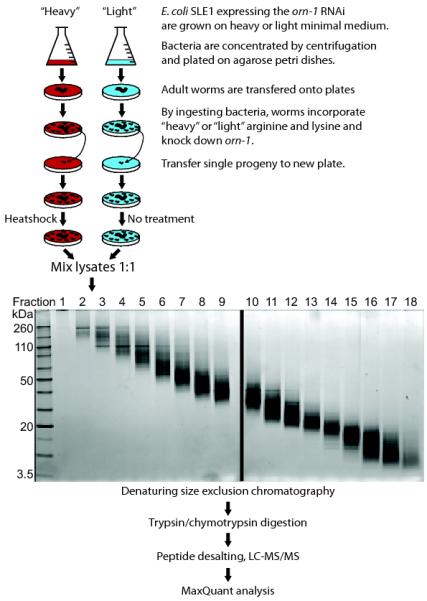
Flowchart for SILAC in nematodes, taking the analysis of the heatshock response as an example. Light medium (blue) is M9 minimal medium and arginine (R0) and lysine (K0). Heavy medium (red) is M9 minimal medium and 15N4-13C6-arginine (R10) and 15N2-13C6-lysine (K8). The RNAi procedure is described in more detail in Supplementary Fig. 4.
A large number of temperature sensitive mutants have been generated in C. elegans, allowing for a transient disruption of often essential proteins. One problem associated with this experimental procedure is background perturbations generated by the heatshock response. We therefore used the SILAC in nematodes approach to examine the extreme end of the heatshock response by shifting worms to 30 °C, according to the scheme shown in Fig. 2. The C. elegans proteome was fractionated using a detergent-free denaturing size exclusion chromatography method (Online Methods). This fractionation method proved effective as shown in Fig. 2.
LC-MS/MS analysis of each fraction yielded a dataset of ~19,000 peptides corresponding to > 1,400 proteins, each identified with at least two peptides (Supplementary Table 1 and Supplementary Table 2). Four small heat shock proteins were amongst the 9 proteins up-regulated more than four-fold and had Maxquant Significance B values1 less than 0.05 (Fig. 3, Supplementary Table 1, and Supplementary Table 2). These data validate our technique and provide a global overview describing changes in protein abundance upon heatshock treatment. Strikingly, three cathepsin-like aspartic acid proteases of both lysozomal and non-lysozomal origin (ASP-1, ASP-2, and ASP-6), were down-regulated more than three-fold after heatshock and had Maxquant Significance B values1 less than 0.05 (Fig. 3, Supplementary Table 1, and Supplementary Table 2). ASP-1 is a lysozomal aspartic acid protease of the cathepsin-D family that is mainly expressed in intestinal cells and has been observed previously to be down-regulated in response to heatshock7. Little is known about the role of ASP-1, ASP-2, and ASP-6 in nematode biology and future studies will be needed to reveal their role in the heat stress response.
Figure 3.

Analysis the C. elegans heatshock response using SILAC in nematodes. The abundance of ~1,400 proteins is indicated on the y-axis using a log2 scale. The abundance of each protein indicated by the position of the dot on the y-axis was determined by summing up all individual light and heavy peptide intensities detected for each protein. The relative fold decrease or increase upon heat shock treatment is indicated on the x-axis. Heatshock treated worms were grown on heavy-labeled SLE1 bacteria, while untreated worms were grown on light bacteria. Proteins highlighted in solid black are up-regulated heatshock proteins, those highlighted with white fill and black outlines are down-regulated aspartic acid proteases (n = 2).
We have carried out further studies using sub-cellular fractionation of unlabelled worms. This allowed approximately three times the number of proteins to be identified by MS, as well as provided valuable information as to the sub-cellular distribution of proteins in untreated cells (Supplementary Table 1). Subcellular fractionation, in conjunction with the SILAC-based method presented here, will allow future studies to examine the dynamic re-localisation of the C. elegans proteome in response to various conditions, as recently demonstrated in human cells8.
Using the SILAC in nematodes approach we have also characterised changes in the C. elegans proteome in response to orn-1 RNAi (Supplementary Table 1). These experiments compared the proteome of worms labeled with light amino acids and treated with a control RNAi (targeting GFP), with worms labeled with heavy amino acids and orn-1 targeted RNAi (n = 1). These data showed the reduction of total ORN-1 protein by ~60 % (Supplementary Table 1). The reduced knock-down could be the result of incomplete RNAi in C. elegans neurons, which has been observed previously9. However, the residual ORN-1 activity in RNAi resistant cells appears to be minimal, because arginine-to-proline conversion was mostly abolished (see above). orn-1 RNAi also generated some changes in protein expression compared with control RNAi treated cells, as expected. However, as orn-1 RNAi should be used in both heavy and light labelled worms in the SILAC in nematodes method, these differential effects will be negated. One caveat may be that analysis of the urea cycle enzymes could be altered by orn-1 RNAi, and this should be taken into account for any experiments specifically targeting that pathway. The generation of a null mutant for the orn-1 gene by the C. elegans Knock-out Consortium has been requested by our labortatory, and will facilitate future studies without the need for RNAi-mediated orn-1 knock-down.
Our SILAC in nematodes methodology opens up many opportunities for research in C. elegans using quantitative MS-based proteomic strategies. By eliminating arginine-to-proline conversion SILAC experiments can be performed more efficiently. It will also allow the application of methods previously used in tissue culture cells, for example the relative quantitation of protein-protein interactions and the elimination of contaminants in pull-down experiments, in worms10. The generation of the E. coli SLE1 strain for simultaneous stable isotope labeling with amino acids and RNAi feeding could also be used for double RNAi experiments where possible. For example, orn-1 can be knocked-down to eliminate arginine-to-proline conversion, and another C. elegans gene could be targeted to determine the effects on the proteome. This will be especially useful for the analysis of genes where null mutations are embryonic lethal but where RNAi leaves sufficient protein product for survival. The SILAC in nematodes technique will also help to determine global proteome changes during development, aging and stress responses.
METHODS
Methods are available in the online version of the paper.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Wellcome Trust (Grant#: 083524/Z/07/Z, 073980/Z/03/Z, 08136/Z/03/Z, and 0909444/Z/09/Z) and by the EU FP7 Prospects network (Grant#: HEALTH-F4-2008-201648). A.I.L. is a Wellcome Trust Principal Research Fellow and A.G. a Senior Wellcome Trust Senior Research Fellow. S.C. is supported by a Royal Society of Edinburgh fellowship. D.P.X. is an Association for International Cancer Research fellow. We thank Tracy Palmer (Department of Molecular Microbiology, College of Life Sciences, University of Dundee, Dundee, United Kingdom) for providing the Keio library and Frank Sargent for helpful discussions as well as Fiona Wheatley for her help.
Online Methods
Materials
Coomassie Plus (Bradford) reagent and Triscarboxyethylphosphine (TCEP) (Bond-breaker neutral pH solution) was from Pierce. Trypsin was from Promega. Oasis HLB 96-well μ-elution plates were from Waters. The Pepmap C18 columns and trapping cartridges were from Dionex. Complete protease inhibitor cocktail tablets and PhosStop phosphatase inhibitor tablets were from Roche. CBQCA assay kit was from Invitrogen. All other materials were obtained from Sigma.
C. elegans strains and maintenance
C. elegans N2 Bristol strain11 was used and maintained at 20 °C unless otherwise indicated. For isotopic labeling C. elegans were grown on NGM-N plates (see below). L1 larval stage worms were separated from an unsynchronized population by filtering through a nylon filter (11 μm, NY11, Millipore) and 5 L1 stage C. elegans were incubated on a fresh NGM-N plate (plate 1). C. elegans were monitored every day and L4 larvae stage C. elegans of the following generation (F1) were picked onto a fresh plate (plate 2). This procedure was followed unless indicated otherwise.
SDS-PAGE
Size fractionation was confirmed by SDS-PAGE analysis on 4-12 % (wt/vol) Bis-Tris NuPage gels using MES running buffer (Invitrogen) according to manufacturer's instructions but with the addition of 25 mM TCEP, in the LDS sample buffer (Invitrogen). Equal amounts of protein were loaded with a maximum of 10 μg per lane. SYPRO Ruby staining was performed as per manufacturer's instructions (Invitrogen).
Generation of E. coli mutant strains
The SLE1 auxotrophic derivative of E. coli HT115 was generated using bacteriophage P1 transduction13 to introduce in-frame deletions in argA and lysA employing the the E. coli Keio Collection single gene mutant library14. Briefly, the argA and lysA kanamycin restistance cassette replacement mutants were retrieved from the library and used to prepare P1 lysates. Generalised P1 transduction was then performed according to standard methods13 to sequentially introduce each mutation into the recipient HT115 strain. Positive transductants were selected using kanamycin resistance and confirmed by testing auxotrophy for the corresponding amino acid (arginine or lysine). Following the successful introduction of each mutation, the kanamycin resistance cassette was excised to leave an unmarked deletion, via the transient expression of the FLP recombinase as described15. Auxotrophy was confirmed by assessing growth in the presence or absence of the relevant amino acid(s) at 40 μg ml-1 in M9 minimal media (Na2HPO4 5.8 g l-1, KH2PO4 3 g l-1, NaCl 0.5 g l-1, NH4Cl2 1 g l-1, Glucose 0.2 % (wt/vol), MgSO4 1 mM, Thiamine 0.01 % (wt/vol). The full genotype of SLE1 is argA, lysA, F-, mcrA, mcrB, IN(rrnD-rrnE)1, lambda -, rnc14::Tn10 (DE3 lysogen: lavUV5 promoter -T7 polymerase). The SLE1 strain will be available to the community via the Caenorhabditis Genetics Center at the University of Minnesota.
Bacterial growth conditions
E. coli HT115 cells knocked-out for lysA and argA (SLE1) were grown in M9 minimal media. M9 minimal media was prepared by mixing 100 ml of 10 × M9 salts (420 mM Disodium phosphate, 240 mM Monopotassium phosphate, 90 mM Sodium chloride, 190 mM Ammonium chloride) and 893 ml of deionised water, and autoclaved (120 °C for 20 min). After cooling to 55 °C, 5 ml of 40 % (wt/vol) glucose, 1 ml of 1 M MgSO4 and 1 ml of 1 % (wt/vol) thiamine were added. Lysine and Arginine were added to 40 μg ml-1 final concentrations from a 73 mg ml-1 or 42 mg ml-1 stock (in PBS) respectively. A single bacterial colony freshly streaked on an LB plate from a frozen stock was used to start a 200 ml culture. Bacteria were incubated at 37 °C under agitation (220 rpm) until OD600 nm = 1 was reached. Bacteria were concentrated by centrifugation (8000 g, 20 min) to OD600 nm = 50 and 5 ml plated onto NGM-N plates. Plates were then stored at 20 °C and used within 7 days.
orn-1 RNAi feeding construct
For the ornithine transaminase orn-1 (gene ID: C16A3.10), plasmid pAG608 was generated using the following primers and Not-1 restriction. Forward TATATATAGCGGCCGCACTTCTCGCACACTACCACG, reverse TATATATAGCGGCCGCTTAGTTTTGCTTTGCAAAATCG. E. coli SLE1 argA, lysA were made chemically competent by incubating 5 ml of freshly grown bacteria in LB (OD600 nm = 0.4) with ice cold CaCl2 (100 μM) for 2 hours.
RNAi-feeding
RNA interference experiments were performed according to the feeding method5 with the following modifications: Cells carrying the corresponding feeding vector were grown in 2 ml of M9 minimum media supplemented with either heavy or light arginine and lysine (40 μg ml-1), and carbenicillin (1 μg ml-1) at 37 °C until OD600 nm = 1. Double-stranded RNA expression was induced by the addition of IPTG (1 mM final) for 3 hours. Bacteria were pelleted and resuspended in 1 ml of worm M9 buffer supplemented with IPTG 1 mM, carbenicillin 1 μg ml-1 to which ~500 L1 larvae stage worms were added. This mixture was transferred into a 50 ml falcon tube to allow oxygenation, and incubated at 25 °C overnight under gentle agitation (150 rpm). Worms and bacteria were spun down the following day, plated on 9 cm NGM-N plates and incubated at 20 °C. The following worm generation (F1) was analyzed for the corresponding phenotype.
Quantitative PCR
RNA was extracted by resuspending worms in Qiazol (Qiagen) disruption with 0.7 mm zirconia beads (Biospec Products). 500 ng of RNA was reverse-transcribed using a Quantitect kit (Qiagen) in a final volume of 20 μl of which 0.5 μl was used for the quantitative PCR. MesaGreen mix (Eurogentec) was used following manufacturer instructions on an iCycler iQ5 (Biorad). Cycling conditions were : 1 × [5 min 95 °C] and 50 × [15 sec 95 °C, 20 sec 60 °C, 40 sec 72 °C] fluorescence was measured after each 72 °C step. Relative expression levels were determined using tbg-1 (γ-tubulin) transcript as standard. Experiments were done in triplicate.
Labeling C. elegans plates
C. elegans are grown on NGM plates that do not contain any nitrogen source (called NGM-N plates). For one liter of NGM-N medium, 3 g of NaCl and 12 g of agarose (Invitrogen, molecular grade) were mixed with 970 ml of deionised water and autoclaved (120 °C for 20 min). After cooling down to 55 °C the following compounds were added: 1 ml of 1 M CaCl2, 1 ml of 1M MgSO4, 25 ml of 1 M KPO4, 1 ml of cholesterol 5 mg ml-1 (in ethanol), 1 ml of Nystatin (10,000 units ml-1). 9 cm plates were used.
Generation of C. elegans lysates
C. elegans were collected from plates by flushing with PBS and washed three times with PBS. C. elegans were pelleted and resuspended in ice cold 100 μl PBS containing Complete protease inhibitors, PhosStop and 5 mM N-ethyl maleimide prior to snap-freezing in liquid nitrogen. For lysis, C. elegans were thawed on ice and powdered guanidine-HCl (76 mg) was added to generate a ~6 M guanidine-HCl solution. TCEP (reducing agent) was added to a concentration of 25 mM, the solution was vortexed and then heated to 65 °C for 10 min. Zirconia beads (0.7 mm, Biospec Products) were added to make a ~50 % (vol/vol) slurry and the sample was bead-beated (Mini Beadbeater 8, Biospec Products) for 1 min at room temperature. The lysate was then centrifuged for 10 min at 17,000 g at room temperature. A Bradford assay was performed on the supernatant and for SILAC mixing, equal proportions of protein were combined.
Sub-cellular fractionation of C. elegans
The QProteome Cell Compartment fractionation kit (Qiagen) was used to fractionate worms according to the manufacturer's tissue fractionation protocol. Briefly, C. elegans were collected by flushing with PBS and washed three times with PBS. Freshly harvested C. elegans were pelleted and resuspended in ice cold buffer 1 containing Complete protease inhibitors. Zirconia beads (0.7 mm, Biospec Products) were added to make a ~50 % (vol/vol) slurry and the sample was bead-beated (Mini Beadbeater 8, Biospec Products) for 5 s at 4 °C and lysis was checked by microscopy. The lysate was then processed for the remaining steps as per the manufacturer's instructions. A BCA assay was performed on each fraction prior to denaturing gel filtration chromatography of each.
Denaturing Gel Filtration Chromatography, Trypsin Digestion and Peptide Clean-up
In order to identify and quantify as many proteins as possible we employed two strategies to maximize protein fractionation (Fig. 2). First, we separated proteins by molecular weight using denaturing size exclusion chromatography with a combination of urea and thiourea as denaturants. This enabled fractionation of small amounts of protein with minimal sample loss. It also allowed us to perform protein digestion in solution, which overcomes the problem of incomplete peptide extraction associated with the in-gel digestion procedure. Furthermore, the reduction of handling steps reduces protein contamination problems, for instance generated by keratin. Second, we digested the size exclusion chromatography fractions with either trypsin or chymotrypsin to generate complementary digests of the fractionated proteins. Trypsin (which cleaves after arginine and lysine) and chymotrypsin (which cleaves after bulky hydrophobic residues) yield quite different peptide fragments. Combining MS/MS datasets obtained upon trypsin and chymotrypsin cleavage increases sequence coverage and the number of protein identifications.
Size fractionation and protein digestion were performed as described below. Using a Dionex Ultimate 3000 HPLC system, lysates in 6 M guanidine-HCl were injected (20 μl per injection – 80 μg protein) onto a mAbPacSEC column (Dionex) equilibrated with 6 M Urea, 2 M Thiourea, 0.1 M Tris-HCl pH 7.0. The flow rate was 0.2 ml min-1 and 16 × 100 μl fractions were collected using a low protein binding 96-deep well plate (Eppendorf). Trypsin/chymotrypsin digestions and peptide desalting was performed as per the scheme shown in Supplementary Fig. 5. Briefly, three volumes of 0.1 M Tris-HCl pH 8.0, 1 mM CaCl2 were added to each fraction to dilute the urea. 500 ng of trypsin or chymotrypsin were subsequently added to each well. The plate was sealed with a rubber mat, vortexed and incubated overnight at 37 °C. Trifluoroacetic acid was added to 1% (vol/vol) final concentration and peptides were purified using an Oasis HLB 96-well μ-elution plate. Peptides were eluted in 100 μl of 50 % (vol/vol) acetonitrile and speedivaced to dryness prior to resuspension in 5 % (vol/vol) formic acid. Peptide concentrations were determined using the CBQCA assay (Invitrogen) after 100-fold dilution of peptide samples in water.
LC-MS/MS and Maxquant Analysis
Using a Dionex Ultimate 3000 nanoHPLC system, 1 μg of peptides in 5 % (vol/vol) formic acid were injected onto an Acclaim PepMap C18 nano-trap column (Dionex). After washing with 2 % (vol/vol) acetonitrile 0.1 % (vol/vol) formic acid peptides were resolved on a 150 mm × 75 μm Acclaim PepMap C18 reverse phase analytical column over a 100 min organic gradient with a flow rate of 300 nl min-1. Peptides were ionised by nano-electrospray ionisation at 1.2 kV using a fused silica emitter with an internal diameter of 5 μm (New Objective). Tandem mass spectrometry analysis was carried out on a LTQ-Velos Orbitrap mass spectrometer (Thermo Scientific). The data dependent acquisition method used was the FT10 protocol as described previously12. Data were processed, searched and quantified using the Maxquant software package version 1.1.1.36 as described previously1, using the default settings and employing a combined C. elegans and E. coli Uniprot databases. The settings used for the Maxquant analysis were: 2 failed cleavages were allowed; enzymes were Trypsin or Chymotrypsin; Variable modifications included in the analysis were methionine oxidation, deamidation of glutamine or asparagine, N-terminal pyro-glutamic acid formation, protein N-terminal acetylation. To identify heavy proline containing peptides 15N-13C5-proline was added as a variable modification. A mass tolerance of 7 ppm was used for precursor ions and a tolerance of 0.5 Da was used for fragment ions. Using the default Maxquant settings a maximum false positive rate of 1 % was allowed for both peptide and protein identification. This cutoff was used for accepting individual spectra as well as whole proteins in the Maxquant output. This threshold has previously been shown to be a rigorous method for identifying true positive matches 1. Protein quantitation data was always derived from a minimum of two or more peptides per protein, and Maxquant Significance B values are provided as described previously to identify significant fold changes1. All replicates indicated are biological replicates.
Footnotes
AUTHOR CONTRIBUTIONS: A.P.B and E.P. cloned, passaged and treated all C. elegans samples. M.L. performed all protein analysis. M.L., A.P.B, A.G., and A.I.L. wrote the paper. A.P.B., R.T.H., G.B., and S.C. generated the E .coli auxotrophic strains. D.X., A.G. and A.I.L. mentored and financed the project.
COMPETING FINANCIAL INTERESTS: The authors declare no competing financial interests.
References
- 1.Cox J, Mann M. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 2.Krijgsveld J, et al. Nat. Biotechnol. 2003;21:927–931. doi: 10.1038/nbt848. [DOI] [PubMed] [Google Scholar]
- 3.Ong SE, et al. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 4.Sury MD, Chen JX, Selbach M. Mol. Cell. Proteomics. 2010;9:2173–2183. doi: 10.1074/mcp.M110.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Genome Biol. 2001;2 doi: 10.1186/gb-2000-2-1-research0002. RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rappsilber J, Bicho CC, Alves FD, Chen ZA, Sawin KE. Mol. Cell. Proteomics. 2010;9:1567–1577. doi: 10.1074/mcp.M110.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madi A, et al. Proteomics. 2003;3:1526–1534. doi: 10.1002/pmic.200300490. [DOI] [PubMed] [Google Scholar]
- 8.Lamond AI, Boisvert FM, Lam YW, Lamont D. Mol. Cell. Proteomics. 2010;9:457–470. doi: 10.1074/mcp.M900429-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calixto A, Chelur D, Topalidou I, Chen XY, Chalfie M. Nat. Methods. 2010;7:554–U102. doi: 10.1038/nmeth.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulon S, et al. Mol Cell. 2010;39:912–924. doi: 10.1016/j.molcel.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haas W, et al. Mol. Cell. Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Miller J. A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory Press; New York: 1992. [Google Scholar]
- 14.Baba T, et al. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100049. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datsenko KA, Wanner BL. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.