

Abstract
Binding of erythropoietin (Epo) to the Epo receptor (EpoR) is crucial for production of mature red cells. Although it is well established that the Epo-bound EpoR is a dimer, it is not clear whether, in the absence of ligand, the intact EpoR is a monomer or oligomer. Using antibody-mediated immunofluorescence copatching (oligomerizing) of epitope-tagged receptors at the surface of live cells, we show herein that a major fraction of the full-length murine EpoR exists as preformed dimers/oligomers in BOSC cells, which are human embryo kidney 293T-derived cells. This observed oligomerization is specific because, under the same conditions, epitope-tagged EpoR did not oligomerize with several other tagged receptors (thrombopoietin receptor, transforming growth factor β receptor type II, or prolactin receptor). Strikingly, the EpoR transmembrane (TM) domain but not the extracellular or intracellular domains enabled the prolactin receptor to copatch with EpoR. Preformed EpoR oligomers are not constitutively active and Epo binding was required to induce signaling. In contrast to tyrosine kinase receptors (e.g., insulin receptor), which cannot signal when their TM domain is replaced by the strongly dimerizing TM domain of glycophorin A, the EpoR could tolerate the replacement of its TM domain with that of glycophorin A and retained signaling. We propose a model in which TM domain-induced dimerization maintains unliganded EpoR in an inactive state that can readily be switched to an active state by physiologic levels of Epo.
Binding of erythropoietin (Epo) to the Epo receptor (EpoR) is crucial for production of mature red cells. Homodimerizing members of the cytokine receptor superfamily, such as the EpoR and prolactin receptor (PrlR), function as ligand-induced or ligand-stabilized homodimers (1). Ligand binding triggers auto- or trans-phosphorylation of a Janus kinase (JAK) bound to the receptor cytosolic domain, activating JAK kinase activity (2). JAK substrates include the receptors themselves, signal transducers and activators of transcription (STAT) proteins, and a variety of other cytosolic signaling molecules (3).
The relative orientation of the EpoR extracellular (EC) domains in a receptor dimer is directly related to the efficiency of signaling through the cytoplasmic (CT) domain (4–7). The EpoR can be productively activated by several means in additional to binding of Epo, its normal ligand. These include small Epo mimetic peptides (8), bivalent monoclonal antibodies directed to the EpoR (9), and an R129C point mutation in the EC domain that results in a disulfide bond connecting two receptor monomers (1, 10). Dimerization of EC domains is not sufficient for signaling because nonpermissive orientations of the dimerized EC domains have been identified (5).
A cornerstone in understanding signaling by EpoR is the oligomerization state of the full-length receptor on the cell surface before ligand binding. One model is that of two monomeric receptors brought together into a dimer after the binding of Epo, with signaling being the result of the close proximity of the two receptor polypeptides. However, the EpoR may be present in the membrane as a preformed dimer or higher oligomer with ligand binding triggering a specific conformational change that activates the receptors. The model of ligand-induced dimerization is in accord with the ability of bivalent monoclonal antibodies, small dimerized peptides, and the R129C mutation to activate the EpoR (1, 8–10); however, these agents could also shift an already dimeric/oligomeric EpoR from an inactive to an active conformation.
The crystal structure of the soluble truncated EC domain of the human EpoR in its unliganded form unexpectedly revealed a preformed dimer with a geometry different from that of the Epo-bound receptor (6, 7). That the unliganded receptor is a dimer was supported by an in vivo fragment complementation assay performed on a truncated receptor containing the EC and transmembrane (TM) domains of the mouse EpoR fused to fragments of the dihydrofolate reductase enzyme (11). However, it is not clear whether the dimers observed in the dense crystalline state occur at the much lower receptor densities present on the plasma membrane. The presence of the TM and CT domains may also alter the oligomeric interactions of the intact receptors. Although no dimerization can be detected between EC domains in solution, it was suggested that simple membrane anchorage (not to mention possible interactions between TM domains) might significantly enhance low-affinity interactions that are undetectable in solution (6), as might be expected on entropic grounds.
We therefore studied the oligomerization state of the full-length EpoR situated in its natural environment, the plasma membrane of living cells. Our findings demonstrate that a high proportion of the murine EpoR expressed at the surface of 293-derived BOSC cells form dimers or higher oligomers in the absence of ligand and that the extent of receptor oligomerization is not affected significantly by Epo binding. Using chimeric constructs where the EC, TM, and/or CT domains of the EpoR were swapped with those of the PrlR, we found that the EpoR TM domain is essential for the ligand-independent oligomerization and is sufficient to enable the PrlR to associate with the EpoR. Although we cannot rule out a weak interaction between the EpoR EC domains not detected by our assay, our findings clearly demonstrate that the TM domain of the murine EpoR is endowed with a powerful oligomerizing ability and do not support a role for the EC or CT domains in ligand-independent oligomerization of the EpoR.
Materials and Methods
Generation of EpoR Mutant Plasmids and Cell Lines.
The murine EpoR cDNA was cloned in the pMX-IRES-GFP 1.1 bicistronic retroviral vector upstream of the internal ribosome entry site (IRES) as described (12, 13). The level of green fluorescent protein (GFP) expression from these vectors is proportional over a 50-fold range to the level of expression of the protein encoded by the cDNA placed upstream of IRES (13). DNA sequences encoding the hemagglutinin (HA; YPYDVPDY) or Myc (EQKLISEEDL) epitope tags were inserted by PCR just upstream of the receptor sequence 31PDPKFE36, five residues downstream of the signal peptidase cleavage site predicted by the signalp program (14). The rabbit PrlR cDNA and the cDNA coding the CHI PrlR-EpoR chimeric construct cloned in mouse stem cell virus (MSCV) (15) were tagged by replacing the signal peptide with a signal peptide from pFLAG-CMV-1 (Kodak) followed by the sequence encoding two Flag epitopes. The tagged receptors were cloned in pMX-IRES-GFP 1.1. In the PEE construct, the PrlR EC domain, ending in 206FTMKD210, is followed by the EpoR TM (starting with Leu226) and CT segments of the EpoR, as described for the original PrlR–EpoR chimeric construct provided by Isabelle Dusanter-Fourt (Hôpital Cochin, Paris).
To generate chimeric EpoR–PrlR constructs SalI (G/TCGAC) and BspEI (T/CCGGA) sites flanking the TM of the EpoR cDNA were introduced, thereby creating two point mutations in the EpoR, Leu223 → Val and His249 → Gly. These mutations had no effect on the biological activity of the receptor, which was functionally indistinguishable from the wild-type receptor in biological assays (see Fig. 5). This construct is designated EEE. The chimeric constructs are named according to the receptors (P for PrlR and E for EpoR) from which the EC, TM, and CT domains are derived. In the PEP construct, the junction between the PrlR EC domain and the EpoR TM domain is similar to that in PEE; the junction between the EpoR TM and PrlR CT domain is represented by Gly249 of EpoR followed by the predicted CT domain of the PrlR, starting with 237SMVT240. In the PPE construct, the PrlR TM domain ending in 232VALK235 is fused to the CT domain of the EpoR starting with 248SG249. In the EPE construct, the EC domain of EpoR SalI–BspEI ending in 223VD224 is fused to the TM of PrlR (starting with 213VWI215 and ending with 232VALK235 and with the CT domain of the EpoR starting with 248SG249. For generation of the EpoR–glycophorin A (GpA) chimeras (EGE), double-stranded DNA oligonucleotides encoding the different GpA TM mutants were cloned into the SalI–BspEI sites flanking the EpoR TM domain. All constructs were verified by sequencing.
Figure 5.

EpoR can tolerate replacement of its TM with that of GpA. TM domains (depicted over orange background) and flanking amino acid residues of the EGE chimeric molecules are shown. DNA oligonucleotides coding for the GpA TM domain or the indicated deletion mutants (deleted residues are crossed in red) were cloned in the EpoR EEE construct, which contains the two point mutations Leu223 → Val and His249 → Gly flanking the TM domain. Activity is defined by percent of proliferation of Ba/F3 cells expressing equal numbers of wild-type or mutant EpoRs, as revealed by similar GFP fluorescence, measured 4 days after plating in medium supplemented with Epo at 1 unit/ml. A representative experiment in which samples were counted in duplicate is shown. No activity was detected in the absence of Epo.
Immunofluorescence Copatching of Cell-Surface Receptors.
To measure oligomerization of EpoR mutants directly at the cell surface, we used antibody-mediated immunofluorescence copatching of epitope-tagged receptors, as described (16–18). BOSC23 cells grown on glass coverslips were transiently cotransfected by using calcium phosphate with HA-EpoR and Myc-EpoR, Flag-PrlR, chimeric constructs of the two receptors, or other control cDNAs in mammalian expression vectors. After 48–72 h, the cells were treated with normal goat IgG [200 μg/ml, 45 min, 4°C, in Hanks' balanced salt solution containing 20 mM Hepes (pH 7.4) and 1% BSA] and then labeled at 4°C (to avoid internalization and enable exclusive cell-surface labeling) in the same buffer with the following primary anti-tag IgG antibodies (30 μg/ml, 45 min): rabbit HA.11 against the HA tag (Babco, Richmond, CA) and mouse 9E10 anti-Myc (Babco) or mouse M2 anti-Flag (Sigma) antibodies. This labeling was followed by labeling/patching with secondary IgG, Cy3-conjugated goat anti-rabbit (GαM) and FITC-conjugated goat anti-mouse (GαM) antibodies, both from Jackson Immunoresearch, at 20 μg/ml for 45 min. After washing, the cells were fixed in methanol (5 min, −20°C) and acetone (2 min, −20°C), thereby denaturing GFP and eliminating its fluorescence, and mounted in SlowFade (Molecular Probes). Fluorescence digital images were recorded with a charge-coupled device camera as described (18). The FITC and Cy3 images were exported in TIFF format to PHOTOSHOP (Adobe Systems, Mountain View, CA) and superimposed. The numbers of red, green, and yellow (superimposed red and green) patches were counted on the computer screen, in each case counting at least 100 patches per cell on 10–15 cells.
Infection of Ba/F3 Cells and Epo-Dependent Proliferation Assays.
For infection of Ba/F3 cells with GFP-coding bicistronic retroviruses, high-titer replication-defective retroviral supernatants were generated as described (13). Viral supernatants were used to infect IL-3-dependent Ba/F3 cells growing in RPMI medium 1640 supplemented with 10% FCS, streptomycin (100 μg/ml), penicillin (100 units/ml), and 5% supernatant of WEHI cell line as a source of IL-3. Cells were assayed by flow cytometry 48 h after infection to measure infection efficiency by GFP expression. Populations of cells expressing GFP above a predetermined level (the top 1% of the population) were sorted by flow cytometry. Cells expressing wild-type or mutant EpoRs were washed extensively in RPMI and then placed in medium containing various amounts of human Epo (provided by Amgen Biologicals). Cell numbers were counted after 3–6 days with a Coulter counter.
Western Immunoblotting and Immunoprecipitation.
Western blot analysis of transfected BOSC cell lysates was performed as described (12). For analysis of the tyrosine phosphorylation status of the EpoR, BOSC cells were transiently transfected within 24 h after seeding with EpoR cDNA in the pMX-IRES-GFP 1.1 vector by the calcium phosphate procedure. The transfection mixture was removed after 10 h, and the cells were incubated in DMEM containing 10% FCS. Medium was changed 38–40 h after transfection, and cells were treated with Epo (100 units/ml) for 7 min or not treated. Cells were placed on ice and rapidly lysed in 1% Nonidet P-40 immunoprecipitation buffer as described (19). Lysates were immunoprecipitated with C187 antiserum, which recognizes the C terminus of the EpoR. Samples were separated by SDS/PAGE on 8.5%–12.5% gradient gels, transferred to nitrocellulose membranes, and incubated with anti-phosphotyrosine monoclonal antibody (4G10, Upstate Biotechnology) followed by peroxidase-coupled anti-mouse antibodies (Amersham Pharmacia), which were detected by enhanced chemiluminescence (ECL).
Results
Cell-Surface EpoRs Form Oligomers in the Absence of Ligand but Require Epo for Signaling.
To detect oligomerization of the intact murine EpoR at the surface of live cells, we used the immunofluorescence copatching method that we have used to demonstrate homo- and heterooligomerization of transforming growth factor β (TGF-β) family receptors (16–18, 20). In this method, as detailed (18), two receptors bearing different epitope tags at their EC termini are coexpressed at the surface of live cells. One tagged receptor is forced into micropatches by a double layer of bivalent IgGs using a fluorescent secondary antibody. The coexpressed receptor, which carries a different tag, is patched and labeled by primary antibodies from another species and secondary antibodies coupled to another fluorophore. Receptors residing in mutual oligomers will be swept into the same micropatches. If one uses red (e.g., Cy3) and green (FITC) fluorophores, mutual patches appear yellow when the two images are overlapped.
Fig. 1 shows typical results of copatching experiments aimed at analyzing the oligomerization state of the murine EpoR. The averaged results of such measurements on many cells are depicted in Fig. 2. BOSC23 cells were cotransfected with HA-EpoR and Myc-EpoR, and surface receptors were patched by using specific antibodies with different fluorophores. The images reveal a significant amount of copatching (yellow patches) in the absence and presence of Epo ligand (Fig. 1 A and B). In contrast, control experiments to examine the level of background nonspecific interactions of HA-EpoR with other receptors [Flag-PrlR, Myc-TGF-β receptor type II, or Flag-thrombopoietin receptor (TpoR)] yielded much lower extents of copatching (Figs. 1 and 2). This level presumably represents background copatching from the cumulative contribution of factors other than specific oligomeric interactions (e.g., accidental overlap of patches, colocalization due to mutual localization in certain subcellular membrane domains, or nonspecific interactions) to the copatching.
Figure 1.
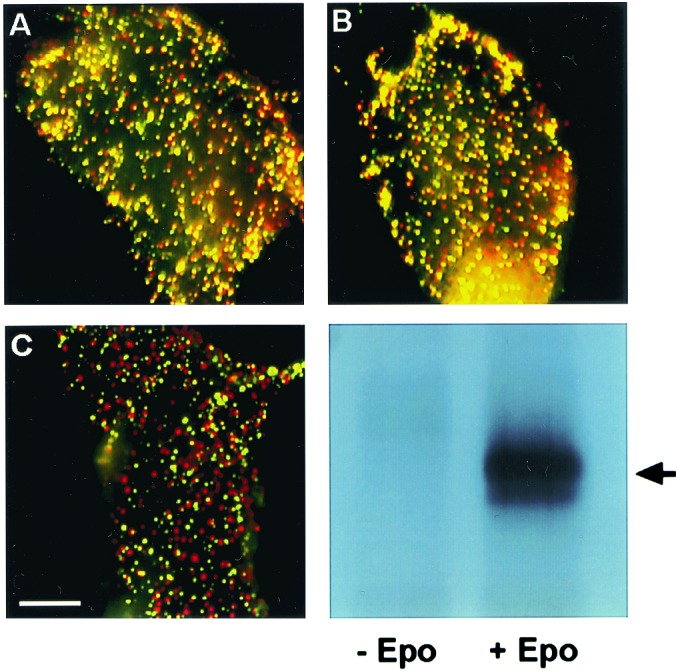
Immunofluorescence copatching to explore homomeric EpoR complexes at the cell surface. BOSC cells were cotransfected transiently with HA-EpoR plus Myc-EpoR (A and B) or with HA-EpoR plus Flag-PrlR (C). Live cells were labeled consecutively with a series of antibodies at 4°C to mediate patching and fluorescent labeling. This labeling protocol results in HA-EpoR labeled by Cy3 (red) and Myc-EpoR or Flag-EpoR labeled by FITC (green). Mutual patches containing both red- and green-labeled receptors appear yellow when the two fluorescent charge-coupled devise images are overlapped. (Bar, 20 μm.) (A) No ligand added. HA-EpoR (red) and Myc-EpoR (green) exhibit a high degree of copatching (yellow). (B) Same as A, but the cells were incubated with Epo at 100 units/ml. The ligand was added with the normal goat IgG before copatching and was retained during successive incubations. Only a minor increase in copatching is observed. (C) No ligand added. HA-EpoR (red) shows a low degree of copatching with Flag-PrlR (green), which is similar to the background level observed for HA-EpoR patching with unrelated receptors (see Fig. 2). (D) As in A, BOSC cells were transfected with EpoR cDNA cloned in pMX-IRES-GFP. At 48 h after transfection, cells were stimulated with Epo at 100 units/ml for 7 min at 37°C or left unstimulated. Cells were lysed on ice in 1% Nonidet P-40 buffer, immunoprecipitated with anti-EpoR antibodies, and analyzed by Western blotting with anti-phosphotyrosine monoclonal antibodies 4G10. The arrow indicates EpoR.
Figure 2.

Quantification of copatching between EpoR and various chimeric and mutant receptors at the surface of live cells. Immunofluorescence copatching experiments were performed on BOSC cells expressing various pairs of differently tagged receptors, as described in Figs. 1 and 3. Superimposed red and green images were analyzed by counting the numbers of green (G), red (R), and yellow (Y) patches (counting at least 100 patches per cell on 10–15 cells in each sample). The percent copatching (percentage of a given tagged receptor in mutual patches with the other receptor) is given by 100 × [Y/(Y + R)] for the red-labeled receptors and by 100 × [Y/(Y + G)] for the green-labeled receptors (18). Because the values of Y/(Y + R) and Y/(Y + G) were very close for each receptor pair, only one value (mean ± SEM) is depicted for each pair. In all cases (except for the bar marked with an asterisk), HA-EpoR was coexpressed with the tagged receptor indicated below each bar. All of the copatching experiments shown were performed in the absence of ligand, except the experiment labeled “+”, where Epo was included (for conditions, see Fig. 1). TGFβRII, TGF-β type II receptor.
As shown in Figs. 1 and 2, the average percentage of HA-EpoR found in mutual patches with coexpressed Myc-EpoR (and vice versa) was 53%, increasing only slightly (to 61%) in the presence of Epo. This percentage is very close to the statistical prediction of 66.6% (2/3) copatching expected for a pure homodimeric population; as discussed (18), this is due to the fact that dimers containing two receptors with the same tag may also form but would not be swept into mutual patches with receptors carrying the other tag. The fraction of same-tag complexes expected for a dimer is 1/3, leaving 2/3 of the dimers containing two different tags. Thus, 53% copatching would reflect 80% (53% divided by 66.6%) of the population being in preformed dimers. Even when the background copatching level (averaging 25% for all control pairs) is subtracted, one is left with copatching levels of 28% and 36% (without and with ligand, respectively), suggesting that at least 42%–54% of the EpoR reside in dimers. Although these studies do not exclude the existence of EpoR trimers or higher oligomers, their percentage cannot be high, because such higher complexes would give rise to a much higher degree of copatching (6/7, equivalent to 85.7%, for trimers; 14/15, equivalent to 93.3%, for tetramers).
To examine whether the preformed EpoR complexes are constitutively active, in the same transfected cells used for copatching studies, we examined the tyrosine phosphorylation status of EpoR in the absence or presence of Epo. We transfected BOSC cells with murine EpoR cDNA and used immunoprecipitation of the EpoR with an antibody recognizing the C terminus of the EpoR, followed by Western blotting with anti-phosphotyrosine antibodies. As shown in Fig. 1D, the EpoR was tyrosine-phosphorylated only when the cells were incubated with Epo.
EpoR Oligomerization Is Mediated by Its TM Domain.
To investigate the role of the different EpoR domains (EC, TM, and CT) in the ligand-independent oligomerization of the receptors at the cell surface, we used a series of chimeric receptors, taking advantage of the fact that the EpoR does not copatch with the PrlR above the background level. We examined the ability of PrlR chimeric constructs containing specific EpoR domains to copatch with the EpoR; the three letter names of the constructs denote the origin of the sequences, so that E stands for EpoR and P stands for PrlR. Construct PEE, for instance, consists of the EC domain of PrlR and of the TM and CT domains of EpoR. The results (Figs. 2 and 3) clearly demonstrate that ligand-independent oligomerization of the EpoR is driven mainly by the TM domain, as shown by the high degree of copatching measured between HA-EpoR and Flag-PEE or Flag-PEP (Figs. 2 and 3 A and B). The latter is a chimeric receptor containing only the EpoR TM domain flanked by the PrlR EC and CT domains. Importantly, the extent of copatching of these chimeras with the HA-EpoR was similar to that of two differently tagged EpoRs (Figs. 1 and 2).
Figure 3.

Immunofluorescence copatching demonstrates that the EpoR TM domain enables the PrlR to copatch with the EpoR. BOSC cells were cotransfected with HA-EpoR plus Flag-PEE (A), HA-EpoR plus Flag-PEP (B), HA-EpoR plus Myc-EPP (C), or HA-EPP plus Flag-PPE (D). Labeling with antibodies and copatching were as described in Fig. 1. (Bar, 20 μm.) (A) HA-EpoR (red) and Flag-PEE (green) show a high degree of copatching (yellow). (B) HA-EpoR (red) and Flag-PEP (green) exhibit a high copatching level. (C) HA-EpoR (red) and Myc-EPP (green) exhibit only a background level of copatching. (D) HA-EPP (red) and Flag-PPE (green) do not undergo copatching above the background level.
Based on previous x-ray studies that showed that, under high salt conditions, the soluble EC domain of the human EpoR crystallized as a dimer (6), the EC domain of the EpoR would have been predicted to promote receptor association. However, HA-EpoR exhibited only background copatching levels with Myc-EPP (Figs. 2 and 3C), suggesting that the EpoR EC domain is not sufficient to mediate EpoR oligomerization at the cell surface.
These differences could be due to dissimilarities between the human and mouse EpoR or could reflect differences between the receptor interactions when crystallized from a high salt solution and when expressed as full-length receptors at low densities at the plasma membrane. However, we cannot exclude the possibility that the presence of a foreign TM domain (that of the PrlR) causes steric hindrance that weakens the interactions between the EpoR EC domains. At any rate, the positive demonstration of the strong oligomerizing capability of the EpoR TM domain suggests that, in the cell membrane, such interactions play a major role in EpoR dimerization.
To explore the possible role of the EpoR CT domain in EpoR oligomerization, we studied copatching of HA-EpoR with Flag-PPE. Fig. 2 demonstrates that this pair exhibits only a marginal increase in the level of copatching above the background level (e.g., that of HA-EpoR with Flag-PrlR). Thus, interactions between the EpoR CT domains contribute little to oligomerization of the receptors at the cell surface. Collectively, the copatching data show that neither the EC nor the CT domains of the EpoR are capable of mediating homooligomerization of the EpoR at the cell surface and point to the TM sequence of the receptor as responsible for this phenomenon. Interestingly, the murine EpoR TM domain has been shown to self-associate when placed in the context of a chimeric bacterial protein (21).
Replacement of the EpoR-Dimerizing TM Domain Results in Functional Receptors.
Our results suggest that dimerization per se of the EpoR is not sufficient for signaling. However, homomeric interactions of EpoR TM domains could modulate the response to the ligand in a more subtle manner. To explore this possibility, we compared the response to Epo of cell lines expressing either the EpoR or a mutant where the TM region was swapped with that of the PrlR (EPE). The PrlR TM domain fails to promote oligomerization not only with receptors containing the EpoR TM domain (as shown by no copatching for the pair HA-EpoR/Flag-PrlR; Figs. 1 and 2) but also with receptors containing the same PrlR TM domain (e.g., HA-EPP/Flag-PPE; Figs. 2 and 3D). Pools of Ba/F3 cells were infected by bicistronic retroviral vectors encoding GFP and either EEE or the EPE mutant. The level of GFP expression in individual cells correlates with that of the EpoR over a 50-fold range (13). Infected Ba/F3 cells were cultured for several days in IL-3 and then were sorted for those expressing the top 1% of GFP. These pools, which expressed the same amount of EpoR (data not shown), were cultured in IL-3 for 3 more days, washed to remove IL-3, and then grown in Epo. Fig. 4 shows that over a wide range of Epo concentrations (0.001–20,000 units/ml), the two receptors supported growth in Epo, although there were significant differences at low Epo concentrations (i.e., 0.05 unit/ml).
Figure 4.

Epo dose-dependent proliferation of Ba/F3 cells expressing wild-type EpoR (EEE) and EPE. Ba/F3 cells growing in IL-3 were infected with retroviruses encoding EEE and EPE receptors. Cells were sorted after 3 or 4 days of proliferation in IL-3 for the top 1% expression of GFP. Pools of cells expressing similar levels of GFP were washed extensively to remove IL-3 and then plated in triplicate in 24-well plates at 1.25 × 105 cells per ml in medium supplemented with Epo (units/ml) as indicated. Cell numbers per ml (mean ± SEM) were recorded after 3 days in culture.
Other homodimerizing receptors, such as PrlR, manifest self-antagonism at high ligand concentration. It has been difficult to obtain self-inhibition for Epo; even at Epo concentrations as high as 40 μM, complete Epo self-inhibition could not be demonstrated (22). Self-inhibition of the growth hormone (GH) receptor (GHR) is thought to be caused by binding of GH to all of the monomeric cell surface GHRs, leaving no unliganded receptors to bind to the GH–GHR complex (23). If the EpoR formed dimers even in the absence of Epo, then self-inhibition at high Epo concentrations would not be expected to occur.
EpoR Can Signal When Its TM Domain Is Replaced with That of GpA.
Because our data show that the EpoR TM domain is promoting oligomerization of intact EpoR in living cells without activating the receptor, we investigated the effect of replacing the EpoR TM domain with a sequence known to mediate dimerization, that of GpA, which undergoes strong dimerization via the LIXXGVXXGVXXT motif in its TM sequence (24, 25). Not only has the EpoR TM sequence been found to mediate oligomerization of a chimeric bacterial protein (21) but also recent studies suggest that this oligomerization activity is higher than that of the GpA TM domain (William P. Russ and Donald M. Engelman, personal communication). The chimeric EGE receptor, which contains the GpA TM domain (depicted in Fig. 5) was expressed in Ba/F3 cells by retroviral infection, and the ability of Epo to promote survival and proliferation of these cells was examined (Fig. 5). EGE exhibited normal biological activity in response to Epo although expressing cells did not proliferate in the absence of Epo. This observation suggests that ligand binding induces very specific conformational changes that cannot be attained simply by the introduction of a dimerizing sequence into the TM domain. This result with EGE contrasts with findings on certain receptor tyrosine kinases, because both the insulin and neu (ErbB-2) receptors lost biological activity when their TM sequence was replaced with that of GpA (26, 27). Presumably such receptor dimers become locked in a nonproductive conformation. Therefore, fundamental differences exist between the oligomerization requirements for EpoR and for certain tyrosine kinase receptors.
Because previous studies with insulin receptor- or neu-GpA chimeras did not explore a possible orientation or steric-dependent effect of the GpA TM domain, we deleted (shown as crossed residues, Fig. 5) one, two, or three of the GpA TM residues. None of these mutants was constitutively active and all supported normal Epo-dependent proliferation of Ba/F3 cells. Thus, irrespective of the phase of the GpA TM domain α-helix, signaling by these receptors required Epo. The small quantitative differences among wild-type EGE; EGE ΔI243; EGE ΔI243, S244; and EGE ΔI243, S244, Y245 do not reflect different levels of surface expression, as 125I-labeled Epo binding assays revealed similar expression of these constructs (data not shown). These differences may reflect a requirement for a certain orientation of the TM domain relative to the CT domain for optimal signaling, as we have recently shown for the EpoR TM domain (28).
Discussion
Our main result is that a major portion of the murine EpoR population appears at the cell surface as preformed oligomers, most likely dimers. This is in accord with a recent study that showed that a fusion protein containing the EC and TM domains of the murine EpoR fused to dihydrofolate reductase fragments can oligomerize in the absence of Epo (11). However, whereas this report proposed that the dimerizing interactions occur via the EC domains, our findings demonstrate that, for full-length receptors expressed at the cell surface, dimerization is mediated mainly by the TM domains. EpoR dimers formed in the absence of ligand are in an inactive conformation and cannot signal activation of JAK2 unless Epo is added. This observation is in agreement with previous studies that showed that the EpoR TM domain induces homooligomerization of a chimeric bacterial protein (21). Furthermore, the EpoR can tolerate the presence of a highly dimerizing TM domain, like that of GpA (Fig. 5), without constitutive activation or inhibition of activity, which is different from tyrosine kinase receptors such as neu and insulin receptors (26, 27). We propose a model where Epo binding induces a precise conformational change that is transduced to the CT domain and leads to receptor activation. This model is in line with crystallographic studies that found different conformations for the liganded and unliganded EC domains of the EpoR (4, 6) and with identification of subtle changes in the orientation of the EpoR EC domains that disrupt signaling (5).
Our work suggests that the dimerizing capacity of the EpoR TM domain maintains unliganded receptors in an inactive state irrespective of the levels of expression on the cell surface. What is the possible utility of such dimers? One possibility is that, because TM-mediated dimerization locks the receptor in an inactive state, spontaneous receptor activation in the absence of ligand is prevented. Another possibility is that a preformed dimer allows rapid activation of the receptor at low Epo concentrations, especially at the very low density of cell-surface EpoRs found on hematopoietic cells, 200-1000 surface receptors per cell (29). Studies where the TM residues of the EpoR were mutagenized showed that many TM sequences are apparently compatible with EpoR signaling, as far as proliferation of Ba/F3 cells is concerned (ref. 12 and data not shown). Therefore, it is unlikely that a highly specific TM interaction is absolutely required for EpoR signaling. However, when the EpoR TM domain was replaced with that of the PrlR, we could detect small but significant changes in the response to low doses of Epo (Fig. 4). It is therefore possible that in erythroid progenitors, at very low levels of EpoR expression (29), the dimerizing/oligomerizing activity of the EpoR TM domain becomes relevant for rapid receptor recruitment, leading to receptor activation and eventual protection of these cells against apoptosis.
We do not know the extent to which our findings can be generalized to other homomeric cytokine receptors. We have demonstrated that the PrlR TM domain cannot mediate formation of oligomers with EpoR constructs but have not yet tested the ability of two differently tagged wild-type PrlRs to form copatches. Studies of the properties of GH mutants are most consistent with the notion that GH binds first to a monomeric GHR on the cell surface and then this GH–GHR complex forms a stable interaction with a second cell surface GHR (23). However, these results are not inconsistent with a model in which GH binds to a preformed GHR dimer and alters its conformation. Finally, it is likely that preformed cytokine receptor dimers exist on the cell surface in equilibrium with monomeric receptors and that ligand binding both stabilizes the dimer and causes a change in its conformation. This prediction would be in accord with our observation that Epo binding causes a small but significant increase in the fraction of cell surface EpoRs in dimers or higher oligomers.
Acknowledgments
We thank Dr. Xuedong Liu for stimulating discussions, Stream Wang and Yohan Royer for excellent technical assistance, and Glenn Paradis, MIT/Center for Cancer Research Central Flow Cytometry Laboratory, for invaluable help with flow cytometry sorting and analysis. This research was supported by Grant HL 32262 from the National Institutes of Health and by a grant from Amgen Corporation to H.F.L. S.N.C. held fellowships from the Anna Fuller Fund and The Medical Foundation/Charles A. King Trust. This work is in partial fulfillment of the requirements for a Ph.D. thesis by Tzvia Keren, to be submitted to the Senate of Tel Aviv University.
Abbreviations
- Epo
erythropoietin
- EpoR
Epo receptor
- TGF-β
transforming growth factor β
- TM
transmembrane
- PrlR
prolactin receptor
- JAK
Janus kinase
- EC
extracellular
- CT
cytoplasmic
- GFP
green fluorescent protein
- HA
hemagglutinin
- GH
growth hormone
- GHR
GH receptor
References
- 1.Watowich S S, Wu H, Socolovsky M, Klingmuller U, Constantinescu S N, Lodish H F. Annu Rev Cell Dev Biol. 1996;12:91–128. doi: 10.1146/annurev.cellbio.12.1.91. [DOI] [PubMed] [Google Scholar]
- 2.Witthuhn B A, Quelle F W, Silvennoinen O, Yi T, Tang B, Miura O, Ihle J N. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- 3.Darnell J E, Kerr I M, Stark G. Science. 1994;264:1415–1420. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 4.Livnah O, Stura E A, Johnson D L, Middleton S A, Mulcahy L S, Wrighton N C, Dower W J, Jolliffe L K, Wilson I A. Science. 1996;273:464–471. doi: 10.1126/science.273.5274.464. [DOI] [PubMed] [Google Scholar]
- 5.Livnah O, Johnson D L, Stura E A, Farrell F X, Barbone F P, You Y, Liu K D, Goldsmith M A, He W, Krause C D, et al. Nat Struct Biol. 1998;5:993–1004. doi: 10.1038/2965. [DOI] [PubMed] [Google Scholar]
- 6.Livnah O, Stura E A, Middleton S A, Johnson D L, Jolliffe L K, Wilson I A. Science. 1999;283:987–990. doi: 10.1126/science.283.5404.987. [DOI] [PubMed] [Google Scholar]
- 7.Syed R S, Reid S W, Li C, Cheetham J C, Aoki K H, Liu B, Zhan H, Osslund T D, Chirino A J, Zhang J, et al. Nature (London) 1998;395:511–516. doi: 10.1038/26773. [DOI] [PubMed] [Google Scholar]
- 8.Wrighton N C, Farrell F X, Chang R, Kashyap A K, Barbone F P, Mulcahy L S, Johnson D L, Barrett R W, Jolliffe L K, Dower W J. Science. 1996;273:458–464. doi: 10.1126/science.273.5274.458. [DOI] [PubMed] [Google Scholar]
- 9.Elliott S, Lorenzini T, Yanagihara D, Chang D, Elliott G. J Biol Chem. 1996;271:24691–24697. doi: 10.1074/jbc.271.40.24691. [DOI] [PubMed] [Google Scholar]
- 10.Yoshimura A, Longmore G, Lodish H F. Nature (London) 1990;348:647–649. doi: 10.1038/348647a0. [DOI] [PubMed] [Google Scholar]
- 11.Remy I, Wilson I A, Michnick S W. Science. 1999;283:990–993. doi: 10.1126/science.283.5404.990. [DOI] [PubMed] [Google Scholar]
- 12.Constantinescu S N, Liu X, Beyer W, Fallon A, Shekar S, Henis Y I, Smith S O, Lodish H F. EMBO J. 1999;18:3334–3347. doi: 10.1093/emboj/18.12.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Constantinescu S N, Sun Y, Bogan J S, Hirsch D, Weinberg R A, Lodish H F. Anal Biochem. 2000;280:20–28. doi: 10.1006/abio.2000.4478. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Socolovsky M, Dusanter-Fourt I, Lodish H F. J Biol Chem. 1997;272:14009–14012. doi: 10.1074/jbc.272.22.14009. [DOI] [PubMed] [Google Scholar]
- 16.Henis Y I, Moustakas A, Lin H Y, Lodish H F. J Cell Biol. 1994;126:139–154. doi: 10.1083/jcb.126.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilboa L, Wells R G, Lodish H F, Henis Y I. J Cell Biol. 1998;140:767–777. doi: 10.1083/jcb.140.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis Y I, Knaus P. Mol Biol Cell. 2000;11:1023–1035. doi: 10.1091/mbc.11.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klingmüller K, Lorens U, Cantley L C, Neel B G, Lodish H F. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- 20.Wells R G, Gilboa L, Sun Y, Liu X, Henis Y I, Lodish H F. J Biol Chem. 1999;274:5716–5722. doi: 10.1074/jbc.274.9.5716. [DOI] [PubMed] [Google Scholar]
- 21.Gurezka R, Laage R, Brosig B, Langosch D. J Biol Chem. 1999;274:9265–9270. doi: 10.1074/jbc.274.14.9265. [DOI] [PubMed] [Google Scholar]
- 22.Schneider H, Chaovapong W, Matthews D J, Karkaria C, Cass R T, Zhan H, Boyle M, Lorenzini T, Elliott S G, Giebel L B. Blood. 1997;89:473–482. [PubMed] [Google Scholar]
- 23.Cunningham B C, Ultsch M, De Vos A M, Mulkerrin M G, Clauser K R, Wells J A. Science. 1991;254:821–825. doi: 10.1126/science.1948064. [DOI] [PubMed] [Google Scholar]
- 24.Lemmon M A, Flanagan J M, Treutlein H R, Zhang J, Engelman D M. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 25.MacKenzie K R, Prestegard J H, Engelman D M. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 26.Gardin A, Auzan C, Clauser E, Malherbe T, Aunis D, Cremel G, Hubert P. FASEB J. 1999;13:1347–1357. doi: 10.1096/fasebj.13.11.1347. [DOI] [PubMed] [Google Scholar]
- 27.Burke C L, Lemmon M A, Coren B A, Engelman D M, Stern D F. Oncogene. 1997;14:687–696. doi: 10.1038/sj.onc.1200873. [DOI] [PubMed] [Google Scholar]
- 28.Constantinescu S N, Huang L J, Nam H, Lodish H F. Mol Cell. 2001;7:377–385. doi: 10.1016/s1097-2765(01)00185-x. [DOI] [PubMed] [Google Scholar]
- 29.D'Andrea A D, Zon L I. J Clin Invest. 1990;86:681–687. doi: 10.1172/JCI114763. [DOI] [PMC free article] [PubMed] [Google Scholar]