

Abstract
Hydrogen deuterium exchange coupled to mass spectrometry (HDX-MS) has emerged as a technique for studying glycoproteins, which are often refractory to classical methods. Glycan chains are generally assumed to exchange protons very rapidly making them invisible to this technique. Here we show that under conditions commonly used for HDX-MS, acetamido groups within glycan chains retain a significant amount of deuterium. Using mono and polysaccharide standards along with glycopeptides from a panel of glycoproteins, we demonstrate that N-acetyl hexosamines, along with modified Asn sidechains are responsible for this effect. Model compounds for sialic acid also displayed similar exchange kinetics, but terminal sialic acids in the context of an entire glycan chain did not contribute to deuterium retention. Furthermore, the presence of sialic acid appears to enhance the exchange rate of the nearby N-acetyl glucosamines. The ability to detect deuterium exchange at the glycan level opens the possibility of applying HDX-MS to monitor glycan interactions and dynamics.
Keywords: Hydrogen/deuterium exchange, carbohydrate, glycan, glycopeptide, glycoprotein, mass spectrometry, Fetuin, alpha-1-acid glycoprotein, orosomucoid, HIV Envelope gp120, sialic acid, N-acetyl glucosamine
Introduction
Protein glycosylation is one of the most prominent post-translational modifications in eukaryotic systems. These modifications are found in roughly half of the proteins in humans,1 and both N-linked (Asn) and O-linked (Ser/Thr) glycosylation are now understood to modulate enzymatic activity and key immunological functions.2–4 Interest in understanding glycan contributions to glycoprotein structure and function has risen in parallel with the development of biopharmaceutical glycoproteins.5 Our understanding of the role of glycans in modulating glycoprotein structure and function has been limited because the varying degrees of modification, microheterogeneity (different glycoforms even at a single site) and the relatively high flexibility of these regions render glycoproteins refractory to most structure determination methods.6
Hydrogen/deuterium exchange with mass spectrometry (HDX-MS) has emerged as a powerful solution-phase technique for studying challenging proteins, and has recently been applied to examine conformational dynamics of glycoproteins.7–9 The primary focus of most studies has been to monitor the exchange of backbone amide protons with deuterium in solvent. This method can provide detailed sequence-specific information about local structural order/disorder and solvent accessibility. Due to faster exchange rates compared to backbone amides, deuterium at sidechain positions is largely lost before detection.10 Recent applications of HDX-MS to study glycoproteins have assumed the consequences of glycan exchange to be negligible.9, 11 However, neither the intrinsic exchange kinetics within glycopeptides nor the contribution of glycans to the overall level of deuterium retention has been addressed.
N and O-linked glycans contain N-acetyl hexose (HexNAc) and sometimes sialic acid (NeuAc), both of which have an acetamido group (NH-CO-CH3) (Fig. 1). All N-linked glycans (high mannose, hybrid, or complex type depending on the composition of the antennae) possess a conserved pentasaccharide core with two N-acetyl glucosamines (GlcNAc).2 An additional acetamido group is formed on the sidechain of the modified Asn. If the hydrogen exchange kinetics of these acetamido groups is slow under quench conditions (pH 2.5 and ~0°C), then a significant amount of deuterium will be retained and detected by HDX-MS. Using model compounds, full glycan chains, and several glycoproteins, we show that glycans can in fact retain deuterium on a sufficiently long time scale, and that HDX-MS can also detect exchange at glycan positions.
Figure 1.

Chemical structure of a common complex type biantennary glycan structure shown as Haworth projections. Core pentasaccharide structure is highlighted in light grey, and acetamido groups are highlighted with dark grey ovals. Simple representations of glycan chains are shown in the lower right for this specific complex glycan (upper), and a high mannose type glycan (lower), with GlcNAc (grey squares), mannose (grey circles), galactose (white circles), and terminal sialic acids (grey diamonds). α(1–6) core fucose (fucosylation potentially also occurring antennae GlcNAcs) is shown as the grey triangle. Model compounds Sialyllactose (*without the N-acetyl group on the glucose) and GlcNAc-Asn representing portions of these chains used in this study are labeled. Triantennary and tetraantennary complex glycans would contain additional GlcNAc-Gal-NeuAc branches stemming from the terminal mannose moieties in the core pentasaccharide.
Methods
Reagents
D2O (99.96%) was purchased from Cambridge Isotope Labs (Andover, MA). α(2–3) and α(2–6) sialyllactose were obtained from Carbosynth (Compton, UK). Bovine Fetuin and human α1-acid glycoprotein (α1AGP), GlcNAc, NeuAc, LacNAc, and GlcNAc-Asn were from Sigma-Aldrich. Endoglycosidases H, F2, peptide N-glycanase and exoglycosidases β-(1–3,4) galactosidase, and β-N-Acetylhexosaminidase (jack bean) were from Prozyme (Hayward, CA). Neuraminidase was from New England Biolabs (Ipswitch, MA). Crystallized pepsin was purchased from Worthington Biochemical (Lakewood, NJ). Methanol, Acetonitrile, Chloroform and Acetone (>99.9% or optima grade) were all from Fisher scientific. Recombinant gp120 (isolate SF162) expressed in CHO cells12 was a kind gift of Dr Shiu-Lok Hu.
Preparation of glycan chains
Full glycan chains were cleaved from bovine Fetuin or Bovine RNAseB with Endoglycosidase H (EndoH) or Peptide N-Glycanase (PNGaseF) digestion according to manufacturer’s directions. Glycan chains were purified by acetone precipitation.13 Complex glycans from Fetuin were further digested with Neuraminidase, β(1–3,4)Galactosidase and β-N-Acetylhexosaminidase according to manufacturer’s protocols, and repurified by acetone precipitation. A final chloroform-water extraction (1:1) was used to remove any remaining detergent and glycans were dried to completion by speedvac.
UV
Absorbance shift at 220nm was used to track deuterium exchange kinetics on a Cary 50 UV-Vis spectrophotometer (Varian, Santa Clara, CA) as described previously.14 10–20mg/mL glycan standards (50mM NaPO4, pH 2.5) were rapidly diluted 10 fold into deuterated buffer and absorbance at 220nm was monitored at 1 sec intervals for 8 minutes at 20°C. Small additions of 1M HCl (1–2μLs) were included in the dilutions to ensure that the final pH after mixing was 2.50. The absorbance shift as a function of time was fit to a single exponential decay to obtain the exchange rate. All experiments were run in triplicate, with errors reported as the standard deviation. Rates were scaled to 0°C using equations described in Bai et al 10 with an activation energy of 19 kcal/mol (this value was also close to that estimated from comparing exchange rates at 22°C and 0°C by MS which yielded 19.070 kcal/mol).
NMR
NMR experiments were performed on a Varian Unity-Inova 500 MHz spectrometer equipped with a triple-resonance probe, z-axis pulsed-field gradient coil, and a Standard Liquid Variable Temperature (VT) Controller, model VTC4 set to 3.7°C. Probe temperature was calibrated using neat methanol according to manufacturer’s directions. Glycan standards (~10mM) were diluted 10 fold into deuterated buffer as described for the UV measurements. Immediately after mixing, a total of 170 individual spectra (8 scans, 4k complex points, sw = 6000 Hz, d1 = 1 sec) separated by a pre-acquisition delay of 0.583 sec were collected with WATERGATE solvent suppression.15 Dead time between mixing and acquisition was around 40 sec. Spectra were processed in NMRPipe,16 and peak areas for the NH resonances were fit as a function of time to a single exponential decay. Error is reported as the standard deviation from duplicate runs. For assignment purposes, spectra were acquired at a resolution of 32k points in the time domain (16k complex) and with 64 accumulations each (sw = 6000 Hz, d1 = 3 s), with Sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) as the internal chemical shift reference. For GlcNAc and LacNAc the α anomer was identified as the more predominant upfield doublet at (8.28ppm) as assigned previously.17 Identification of the acetamido group in GlcNAc-Asn was based on the very close chemical shift of this resonance to that seen for the β anomer in isolated GlcNAc (8.36ppm) (Fig. S-1). The second peak at 8.9ppm was therefore assigned as the Asn sidechain group.
Mass Spectrometry
Glycan standards at 10–20mg/mL were diluted into 90% D2O with 0.2% formic acid (FA) pH 2.5, and incubated several hours to allow for full deuteration. Full glycan chains were resuspended in 100μL deuterated 0.2%FA pH 2.5 (final D2O 96%) and incubated at 85°C for 10min. The solutions were then rapidly diluted 100 fold into cold (~0°C) aqueous 0.2%FA and infused into a Waters Synapt mass spectrometer. Small additions (~1–2μLs) of 1M HCl were included in the dilutions for a final pH of 2.50. Infusions were performed by loading the sample on a 500μL steel injection loop immersed in melting ice, and pumping cold 0.2% FA in 0.9% D2O (to match the final deuterium content in the sample) at a rate of 15μL/min for 30 minutes. Source and desolvation temperatures of 80°C and 150°C were used to minimize exchange during ionization.8 Exhaustively deuterated α(2–6) sialyllactose and Leu-enkephalin (Sigma-Aldrich), used for estimating back-exchange, were prepared by a 30 min incubation at 85°C in 100mM ammonium bicarbonate (pH 8), 90% D2O and subsequent quench to pH 2.5 and 0°C.
Centroids were calculated by integrating the area of all peaks within the mass envelope at each time point using the MassLynx software package (Waters) together with custom excel macros. Exchange rates were calculated by fitting the change in the centroid to an exponential decay. For larger compounds an early fast rate was also observed (Fig. S-2), and data were fit using a double exponential decay. Error is reported as the standard deviation between observable adducts and duplicate runs. Exchange rates of antennae GlcNAcs were approximated by including rates and mass shifts from the pentasaccharide core in fitting the data for the larger glycoforms.
Glycopeptide preparation
Bovine Fetuin, human α1AGP, and HIV gp120 (20–40μg at 1–2mg/mL) were denatured by incubation at 60°C for 30 minutes in 50mM Tris pH 8.0, 4M Urea, 20mM DTT. Denatured proteins were diluted into deuterated PBS (50mM NaPO4 pH 6.0), incubated at 37°C for 5 hours. 25U Neuase, or 0.3mU EndoH and 0.1mU EndoF2 were included in this first incubation to generate deglycosylated forms. All samples were subsequently adjusted to pH 8.0 and incubated overnight at 37°C. 2mU PNGaseF was included in this second incubation to obtain the fully deglycosylated standards. Identical additions of equivalent buffer (no enzyme) were added to untreated samples, therefore full, trimmed, and deglycosylated glycoproteins were prepared under identical deuteration conditions. Samples were quenched by addition of ice cold 0.2%FA with 500mM TCEP, and digested with 0.05mg/mL solution of pepsin (Worthington Labs) for 5 min on ice. 100μL aliquots were frozen in liquid nitrogen and stored at −80°C for up to several weeks before analysis.
Glycopeptide MS analysis
Frozen aliquots were thawed on ice for 5 min and injected onto a C18 column (1X50mm 3μm Zorbax SB300, Agilent, Santa Clara, CA) with a Waters Acquity solvent delivery system. The column, loop, lines, and syringe were all kept on melting ice to ensure minimal back-exchange. Peptides were loaded for 3 minutes and eluted with linear gradient of 10–50%B (A: 5% ACN, 0.05%TFA, B: 80%ACN, 0.05%TFA) over 12 minutes. Since deglycosylated peptides elute later, a variable delay was introduced to match the elution times of various glycoforms (1 min delay for fully glycosylated, 15 sec for Neuraminidase, 30 sec for EndoH/F2, and no delay for PNGaseF treated samples). Errors were obtained from comparisons of distinct charge states as well as duplicate runs, which were staggered to minimize systematic errors. No back-exchange correction was applied to the data.
Glycopeptides mass envelopes were centroided using HX-express.18 Glycoforms of high mannose containing peptides were averaged as no significant changes were observed between them. Peptic glycopeptides were identified using a combination of exact mass, MS/MS, and confirmed by the mass shifts upon glycosidase treatments. Core and antenna fucosylation were distinguished by collision-induced dissociation (CID) MS/MS.19 Theoretical exchange rates of backbone amides were estimated using previously described methods.10
Results/discussion
Model compounds
Initial analysis focused on measuring the intrinsic exchange rates of several commercially available carbohydrate standards. N-acetyl glucosamine (GlcNAc) and sialic acid (NeuAc) are two prevalent glycan moieties with acetamido protons that could potentially exchange at intermediate rates under quench conditions used for HDX-MS (0°C pH 2.5). Three different methods were used to quantify the exchange rates of these standards (Fig. 2). UV absorbance shifts upon deuteration were monitored as a change in the absorbance at 220nm. Similar exchange-in measurements by mass spectrometry resulted in large mass shifts (due to deuteration of the fast exchanging hydroxyl groups), which lead to over-sensitivity to subtle changes in solvent deuterium content. Instead, mass spectrometry was used to monitor the exchange-out of deuterium to avoid this source of error. Lastly, NMR spectroscopy was used to monitor the disappearance of HN resonances upon deuteration. The exchange rates measured using the different methods were in good agreement (Table 1).
Figure 2.
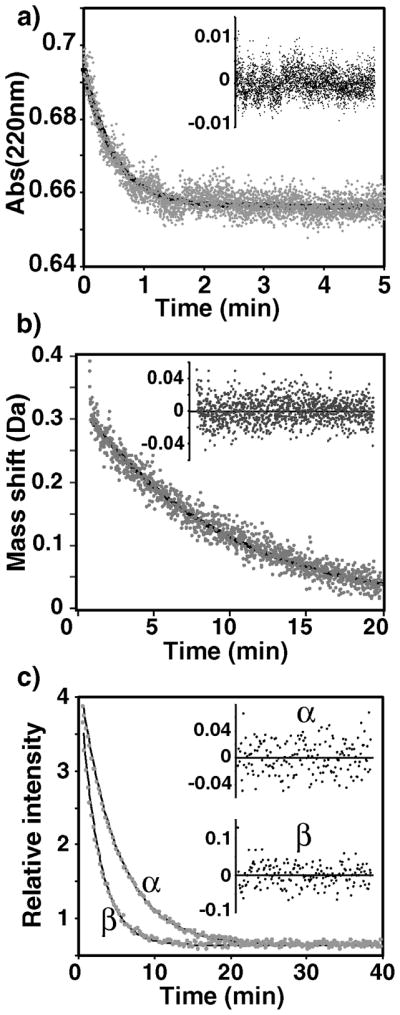
Exchange kinetics measurement of GlcNAc using UV at 22°C (a), MS at 0°C (b) and NMR at 3.7°C (c) with insets showing the residual plots for single exponential fits. The α/β anomeric forms were resolved by NMR.
Table 1.
Rates measured for all model compounds using UV, MS and NMR. Where applicable, the rates for α/β anomers resolved by NMR are listed. The GlcNAc vs. the Asn sidechain acetamido groups in GlcNAc-Asn are labeled next to their values. Rates from UV and NMR, obtained at higher temperatures, were scaled to normalize all data to 0°C.10
| Compound | UV (min−1) | MS (min−1) | NMR (min−1) |
|---|---|---|---|
| NeuAc | 0.083 +/−0.006 | 0.095 +/−0.004 | 0.079 +/−0.001 |
| α(2–3)Sialyllactose | 0.089 +/−0.005 | 0.095 +/−0.025 | 0.125 +/−0.004 |
| α(2–6)Sialyllactose | 0.093 +/−0.010 | 0.119 +/−0.022 | 0.115 +/−0.002 |
| GlcNAc | 0.122 +/−0.008 | 0.105 +/−0.005 | 0.236 +/−0.031 (β) 0.121 +/−0.011 (α) |
| LacNAc | 0.114 +/−0.004 | 0.099 +/−0.005 | 0.219 +/−0.014 (β) |
| 0.074 +/−0.019 (α) | |||
| GlcNAc-Asn | 0.221 +/−0.018 | 0.265 +/−0.005 | 0.428 +/−0.013 (GlcNAc) 0.171 +/−0.002 (Asn) |
The acetamido proton of NeuAc exchanged at a rate of ~0.1min−1. Both α(2–3) and α(2–6) sialyllactose displayed similar rates indicating a minimal influence from the additional carbohydrate groups. The acetamido group on GlcNAc had similar rates, but unlike NeuAc, the α/β anomers were resolved by NMR and displayed different exchange rates. The higher populated α anomer showed a rate similar to NeuAc, but the β anomer exchanged more than two-fold faster (Fig. 2c). This anomeric effect was previously observed in studies of hyaluronic acid oligomers.20 Both UV and MS measurements were unable to resolve anomers, and therefore the fits reflect the combined rates of both. A modest decrease was observed in LacNAc (Gal-β(1–4)-GlcNAc) indicating that the β(1–4) linkage of galactose, common in complex N-linked glycans, has only a minor effect on the exchange rate.
GlcNAc-Asn was used as a model compound to probe the exchange kinetics of the acetamido group at the modified Asn sidechain. UV and MS were unable to resolve the two acetamido groups but NMR was able to resolve the glycan N-acetyl and the Asn sidechain groups (Fig. S-1). Surprisingly, both showed fast rates (Table 1), with the GlcNAc group exchanging 4 times faster than in isolated GlcNAc. The positive charge at the Asn, in close proximity to both groups, likely results in accelerated exchange. A similar effect is seen in peptide amide exchange rates near the amino terminus.10 Unfortunately this effect makes GlcNAc-Asn a poor compound to measure the intrinsic exchange rate of the Asn acetamido group.
In some cases, most notably in the sialyllactose, the mass spectrometry data also showed a rapidly exchanging (>1 min−1) component and required a double exponential fit (Fig. S-2). Since these compounds (i.e. sialyllactose) only contain a single acetamido group, the first fast rate may reflect some residual deuterium at the many hydroxyl groups. This is surprising since the rate of hydroxyl proton exchange is expected to be upwards of 100 fold faster, 21 thus, all deuterium is expected to be lost even within a few seconds. Hydrogen bond networks within these groups may sufficiently slow the exchange to account for the observed fast component.
Full glycans
Next we sought to probe the exchange rates of the acetamido groups within a full N-linked glycan chain. Complex sialylated bi and triantennary chains from Fetuin,22 and various high mannose chains from RNAseB 4 were isolated by endoglycosidase digestion. To compare the contributions of the different glycan moieties, complex chains were treated with exoglycosidases to yield a panel of glycans trimmed to various degrees. Due to the limited amount of material along with significant heterogeneity, mass spectrometry was the only feasible approach for monitoring the exchange kinetics under quench conditions. While this approach can not resolve the rate at each site, it serves to compare the effects and contributions of the various glycan groups.
Mass spectrometry reveals not only the exchange rates, but the shift in the mass envelope is also indicative of the number of sites associated with the exchange. Assuming that all acetamido groups can retain deuterium, the observed mass shifts for glycan chains were much lower than expected (~0.3Da vs. ~1.0Da/acetamido group) (Fig. 3, Table S-1). This effect was also seen with small compounds containing only a single acetamido group (Fig. S-2). One possibility is that the prior incubation in D2O was insufficient for achieving full deuteration. However, experiments with exhaustively deuterated samples showed identical mass shifts (Fig. S-3a). The use of higher ionization temperatures significantly decreased the observed mass shifts (Fig. S-3), therefore we attribute this to the loss of deuterium to solution and possibly gas-phase back-exchange occurring during electrospray ionization.23 Peptide backbone amides also showed source temperature dependent loss of deuterium content, but to a lesser extent, as assessed from a deuterated sample of Leu-enkephalin (Fig. S-3b). As the intrinsic exchange rate of glycans is roughly ten fold faster than backbone amides,10 this effect is more pronounced, resulting in >50% back-exchange.
Figure 3.

Exchange rates and mass shifts for the ensemble of glycan chains. a) Mass shifts as a function of time are plotted for pentasaccharide core (dark grey), full triantennary (light grey), and asialo triantennary (black) glycans. Black lines show the double exponential fit for each species. Diagrams of each glycan are included showing GlcNAc (grey squares), mannose (grey circles), galactose (white circles), and sialic acids (diamonds). b) Exchange rates (top) and mass shifts (bottom) are shown for single core GlcNAc high mannose (1), dual core GlcNAc high mannose (2), tri-sialylated triantennary (3), bi-sialylated biantennary (4), asialo triantennary (5), asialo biantennary (6), asialo agalacto triantennary (7), asialo agalacto biantennary (8), and pentasaccharide core (9). Diagrams of each glycan above each bar are drawn as described in a).
Based on relative mass shifts it is evident that the GlcNAc acetamido groups are contributing to the measured deuterium content within glycan chains. The full high mannose glycan (containing 2 GlcNAcs) shows a larger shift than the single GlcNAc form, consistent with two groups retaining deuterium (Fig. 3b). The mass shift for complex type glycans is significantly higher, consistent with additional antennae GlcNAcs and NeuAcs contributing to deuterium retention. All triantennary glycans show a shift nearly 5 times that of the single GlcNAc containing high mannose glycan. As expected, the fully trimmed pentasaccharide core (Fig. 3b) shows a shift nearly identical to that seen for the two GlcNAc high mannose glycan. Together this shows that GlcNAc moieties retain deuterium on a time scale relevant for HDX-MS.
Terminal sialic acid showed two unexpected trends in these studies. Although NeuAc exchanged with similar kinetics to GlcNAc, in the full chains their contribution to deuterium retention was negligible. Additionally, the presence of NeuAc appears to accelerate the exchange of the nearby GlcNAcs. This effect was roughly two fold in the case of triantennary glycans and a similar effect was seen for biantennary glycans, to a lesser degree. Removal of the branched galactose moieties had almost no effect on the exchange rates. Based on comparisons to the pentasaccharide core, the rates for the antennae GlcNAcs are approximately 0.056 +/−0.004min−1.
Due to lack of linkage and anomeric effects, the exchange rates measured for the reducing end GlcNAc in the cleaved glycans do not accurately represent the rates in the context of the glycoprotein. In the single GlcNAc containing high mannose chain, the rate was slightly slower than in LacNAc (0.068 +/−0.031min−1 vs. 0.099 +/−0.005min−1). Comparison of the single and double GlcNAc high mannose glycoforms indicates that the outer stem GlcNAc exchanges faster. Assuming that the reducing end GlcNAc exchanges at similar rates in both high mannose species and the pentasaccharide core, a rough estimate of 0.24 +/−0.16min−1 is approximated for the outer stem GlcNAc (which is locked in the β form). This value is slightly larger than seen for the β anomer of LacNAc (0.219 +/−0.014min−1).
Glycopeptides
Deuterium retention at glycan positions was next examined at the glycopeptide level. Bovine Fetuin, human α1AGP, and HIV Envelope glycoprotein (gp120) were exhaustively deuterated, digested with pepsin and the deuteration of the resulting glycopeptides was analyzed by LC-MS. Fully glycosylated fragments were compared to trimmed (Neuase), or deglycosylated (PNGaseF, or EndoH & EndoF2) fragments. Due to the large change in hydrophobicity of glycopeptides upon deglycosylation, the LC retention times showed moderate shifts. A delay was incorporated into the gradient for the earlier eluting species to match the elution times and thus all glycoforms experienced equal back-exchange time.
Peptic digestion of Fetuin yielded four observable glycopeptides covering an N-linked glycosylation site at position 138. All of the fragments showed a marked increase in deuterium content (~2.3Da) compared to the deglycosylated forms (Fig. 4a). Therefore, while the deglycosylated fragments show deuterium content consistent with the expected number of amides, the glycopeptic fragments clearly incorporate more deuterium. In some cases the mass shift (without any back-exchange correction) was more than theoretically possible when only accounting for backbone amides. The time for back-exchange was ~11–16min, therefore these additional glycan sites exchange slowly enough to be observed by this method. The effect of terminal sialic acids on the glycan chain was very slight. On average, sialic acids contributed to less than 2% change in deuterium content compared to the desialylated samples (Fig. 4a).
Figure 4.
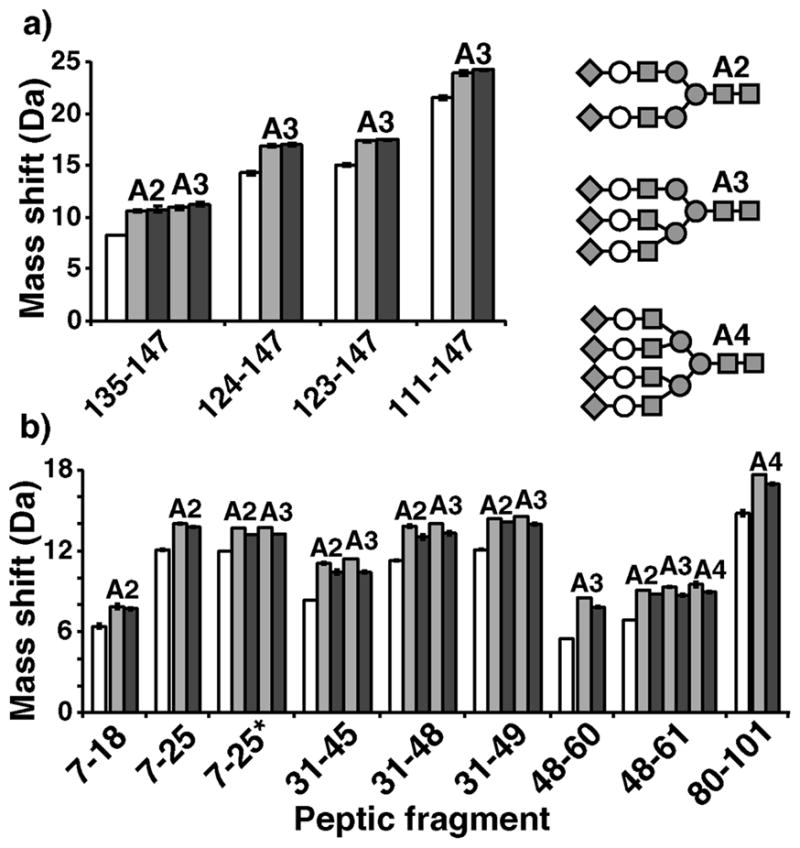
Glycopeptide deuteration data on Fetuin (a) and human α1AGP (b). Mass shifts from after exhaustive deuteration are shown for various deglycosylated (PNGaseF treated) (white), asialo (Neuase treated) (light grey), and fully sialylated (untreated) (dark grey) peptic fragments of each protein. Labels above bars indicate degree of branching: biantennary (A2), triantennary (A3), and tetraantennary (A4). *Fragment 7–25 of α1AGP displayed two variants containing either Arg or Gln at position 20.
Pepsin digestion of α1AGP generated 9 visible glycopeptides covering 4 N-linked glycan sites. Bi, tri, and tetraantennary complex glycans were observable in several fragments allowing for direct comparisons of distinct glycoforms. In all cases a drastic increase in deuterium content was attributed to the glycan chains (Fig. 4b). The asialo forms had a modest but consistent increase in deuterium content. The mass increase per NeuAc removed was 0.18 +/−0.013Da. Comparison of the bi, tri and tetraantennary asialo forms showed each additional antenna GlcNAc contributing 0.19 +/−0.11Da, indicating that deuterium was also retained on these outer antennae sites. A fucosylated glycoform was observed in 6 of the fragments, predominantly at antennae sites (see methods). These additional fucose groups had no effect on the observed mass shift (average difference was 0.02 +/−0.09Da, with no apparent trend).
HIV Envelope gp120 served as a good model glycoprotein as it contains over 20 N-linked glycosylation sites, with both high mannose and complex type structures. Expression of this protein in Chinese Hamster Ovary (CHO) cells yields predominantly sialylated bi and triantennary complex glycans with core fucosylation.24 For this study deglycosylation with EndoH and EndoF2 was used to generate fragments containing only the core GlcNAc (in some cases with a core fucose). This enabled measuring the role of only the core GlcNAc and modified Asn sidechain. Peptic digestion of gp120 provided glycopeptides covering 8 N-linked glycosylation sites (4 high mannose and 4 complex type structures). Some fully deglycosylated fragments were not generated due to altered pepsin specificity for the PNGaseF treated form. In these cases the fully glycosylated forms could only be compared to the EndoH/F2 treated forms. Once again, the difference between PNGaseF treated and native fragments demonstrates that a significant amount of deuterium is attributed to the glycans. For the high mannose containing fragments, the EndoH treated fragment shifts nearly equal to that of the fully glycosylated form. This indicates that the modified Asn sidechain and core GlcNAc are the predominant factors in glycan deuterium retention, whereas the second stem GlcNAc has little effect (Fig. 5a). A comparison of the PNGaseF and EndoF2 treated complex type glycopeptides showed similar deuterium retention on the core GlcNAc and Asn sidechain acetamido groups (Fig. 5b). Significantly more deuterium is seen in the full complex glycoform compared to the EndoF2 treated form, revealing that the antennae GlcNAc groups also contribute. Comparison of these glycopeptides to the less abundant non-core fucosylated glycoforms showed no difference in deuterium content (less than 0.03Da).
Figure 5.

Glycopeptide data from HIV Env gp120. a) Deuterium uptake for fully deglycosylated (PNGaseF) (white), mostly deglycosylated (EndoH/F2) (light grey) and untreated (dark grey) high mannose type glycopeptides. Unlike the first two fragments, 314–346 contains two high mannose N-linked glycans. Above diagrams display the predominant glycoform observed. b) Deuterium uptake for complex type glycopeptides with same coloring as in a). In some cases, the fully deglycosylated form was not observed. The predominant glycoform was core fucosylated mono-sialylated biantennary type (*except for fragment 268–281, which was bi-sialylated). Fragment 293–313 contained a high mannose type in addition to a complex type glycan. c) Comparisons of unmodified and O-glycosylated fragment 475–500. The modified form contained a HexNAc1Hex1NeuAc1 at T490. An asialo (-NeuAc) form of the fragment was also prepared and examined.
O-linked glycan effects
Peptic digestion of gp120 also yielded a fragment with an O-linked glycosylation (HexNAcHexNeuAc) at T490. Relative to the nonglycosylated peptide, this fragment had a mass shift increase of 1.16Da, indicating that moieties in O-linked glycans also contribute to deuterium retention. A desialylated form was also examined to test solely the contribution of the core HexNAc (presumably GalNAc).2 Surprisingly, an even larger increase in deuteration was observed (1.37Da). This agrees with our observations that terminal sialic acids do not contribute to deuterium retention and accelerate the exchange of the nearby GlcNAc groups. The larger than expected mass shift attributed to the single HexNAc suggests that the glycosylated form of the peptide may have decreased back-exchange rates for backbone amides, possibly due to weak glycan interactions.
Implications for examining glycans by HDX-MS
Examination of glycan chains together with a pool of glycoprotein fragments demonstrates that GlcNAc moieties can retain deuterium on a long enough time scale to measure by HDX-MS. Based on full glycan exchange data, an estimate for the exchange rates under quench conditions of the second stem and antennae GlcNAcs are ~0.24min−1 and 0.056min−1, respectively. Exchange rates for core GlcNAc and modified Asn sidechain acetamido groups are slower, estimated in the range of 0.03min−1. Exact prediction of deuterium retention at the glycans is complicated by higher back-exchange rates (especially during ionization) and secondary effects possibly due to glycan-peptide interactions. Fucosylation (at both core and antennae) appears to have no effect on these exchange rates. All of the data confirm that sialic acid groups exchange too quickly leaving no measurable deuterium by the time of detection. In most cases the presence of terminal sialic acids led to overall faster exchange of associated GlcNAcs. The single exception was seen with glycopeptides from Fetuin, which didn’t display the increased back-exchange from sialic acids. It is possible that the difference is caused by this particular glycan (at residue 138) having a unique structure (β(1–4)Gal vs. β(1–3)Gal or α(2–3)NeuAc vs. α(2–6)NeuAc)22 compared to the other samples. Regardless, it is clear that sialic acids do not contribute to any observable deuterium retention. This raises the possibility that different branching patterns and further modifications (sulfation, phosphorylation), may have significant effects on glycan exchange rates.
A previous study of a Fetuin glycopeptide proposed that three terminal sialic acids form a hydrogen bond network resulting in significant protection for two of the acetamido groups.20 In contrast, our current results exhibit very fast exchange at all terminal sialic acid acetamido groups. The low pH used in the current study (pH 2.5) favors protonation of sialic acids (pKa 2.6) 25 possibly disrupting the proposed hydrogen bonding network. Another key difference is that the previous study focused on the glycan at position 158, which may contain different glycoforms than our current pool.
It is now appreciated that subtle changes in glycan structures can have large biological effects.2, 5 Glycosylation is present in 40% of all currently approved biopharmaceutics and much effort is devoted to monitoring both protein and glycan structures to ensure authenticity and consistent production. The precision and sensitivity of HDX-MS makes it a very promising approach for this application,26, 27 especially in light of the current data indicating that the associated glycans can also be monitored. Although glycans exchange roughly 10 times faster under quench conditions, they may be highly protected under native conditions, resulting in slow exchange during deuterium labeling.20, 28 Even if glycan groups are highly accessible under native exchange conditions, their contribution to deuterium content should be considered when interpreting the exchange of glycopeptides. Calculation of protection factors based on intrinsic peptide exchange rates should take into account any glycan acetamido groups in glycopeptide fragments.7 This effect may also hold true for other protein modifications introducing acetamido groups (i.e.: acetylation).
Ideally, one would like to resolve the glycan deuterium content from that of the peptide backbone, but that is currently beyond the scope of this methodology. Although MS/MS by CID is extremely efficient for fragmentation of the glycan chain from the peptide, the scrambling effect will likely sacrifice all positional information.29 Electron transfer dissociation under gentle ionization has been shown to minimize scrambling.30 This may prove to be a very valuable tool as it can potentially monitor deuterium exchange at the glycan positions, and extend HDX-MS to monitor specific glycan dynamics.
Conclusions
Under conditions commonly used for hydrogen/deuterium exchange mass spectrometry, several of the carbohydrate acetamido groups in glycopeptides retain a significant amount of deuterium. Interpretation of HDX-MS data involving quantification of protected amides or calculation of protection factors for glycosylated fragments should account for this effect. The fact that deuterium is retained on certain glycan groups raises the possibility of extending HDX-MS studies beyond the peptide backbone to directly monitor glycan interactions and dynamics.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R00GM080352 and UAB CFAR grant (P30AI027767). We thank Dr Shiu-Lok Hu for generously providing HIV envelope gp120. We would like to acknowledge Dr. Katalin Medzihradszky, Dr. Jonathan Bones, Dr. John Engen and Dr. Andras Guttman for insightful discussion and advice.
Abbreviations
- HDX-MS
hydrogen/deuterium exchange coupled to mass spectrometry
- QTOF
quadrupole time of flight
- NMR
nuclear magnetic resonance
- GlcNAc
N-acetyl glucosamine
- LacNAc
N-acetyl lactosamine
- GlcNAc-Asn
GlcNAc-β(1-N)Asparagine
- NeuAc
sialic acid
- α1AGP
alpha-1 acid glycoprotein
- PNGaseF
peptide N-glycanase F
- EndoH
Endoglycosidase H
- EndoF2
Endoglycosidase F2
- ACN
acetonitrile
- TFA
trifluoroacetic acid
- FA
formic acid
- DTT
dithiothreitol
- TCEP
tris(2-carboxyethyl)phosphine
- HPLC
high performance liquid chromatography
Footnotes
Additional information as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brooks SA. Mol Biotechnol. 2009;43:76–88. doi: 10.1007/s12033-009-9184-6. [DOI] [PubMed] [Google Scholar]
- 2.Marino K, Bones J, Kattla JJ, Rudd PM. Nat Chem Biol. 2010;6:713–723. doi: 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- 3.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 4.Rudd PM, Joao HC, Coghill E, Fiten P, Saunders MR, Opdenakker G, Dwek RA. Biochemistry. 1994;33:17–22. doi: 10.1021/bi00167a003. [DOI] [PubMed] [Google Scholar]
- 5.Walsh G. Nat Biotechnol. 2010;28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 6.Petrescu AJ, Petrescu SM, Dwek RA, Wormald MR. Glycobiology. 1999;9:343–352. doi: 10.1093/glycob/9.4.343. [DOI] [PubMed] [Google Scholar]
- 7.Englander SW. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan CR, Engen JR. Curr Protoc Protein Sci. 2009;Chapter 17(Unit 17):16, 11–17. doi: 10.1002/0471140864.ps1706s58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong L, Huang CC, Coales SJ, Molnar KS, Skinner J, Hamuro Y, Kwong PD. J Virol. 2010;84:10311–10321. doi: 10.1128/JVI.00688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bai Y, Milne JS, Mayne L, Englander SW. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Anal Chem. 2009;81:5966. doi: 10.1021/ac9009287. [DOI] [PubMed] [Google Scholar]
- 12.Scandella CJ, Kilpatrick J, Lidster W, Parker C, Moore JP, Moore GK, Mann KA, Brown P, Coates S, Chapman B, et al. AIDS Res Hum Retroviruses. 1993;9:1233–1244. doi: 10.1089/aid.1993.9.1233. [DOI] [PubMed] [Google Scholar]
- 13.Verostek MF, Lubowski C, Trimble RB. Anal Biochem. 2000;278:111–122. doi: 10.1006/abio.1999.4433. [DOI] [PubMed] [Google Scholar]
- 14.Englander JJ, Calhoun DB, Englander SW. Anal Biochem. 1979;92:517–524. doi: 10.1016/0003-2697(79)90693-6. [DOI] [PubMed] [Google Scholar]
- 15.Piotto M, Saudek V, Sklenar V. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 16.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 17.Blundell CD, DeAngelis PL, Day AJ, Almond A. Glycobiology. 2004;14:999–1009. doi: 10.1093/glycob/cwh117. [DOI] [PubMed] [Google Scholar]
- 18.Weis DD, Engen JR, Kass IJ. J Am Soc Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Hincapie M, Rejtar T, Karger BL. Anal Chem. 2011;83:2029–2037. doi: 10.1021/ac102825g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oberholtzer JC, Englander SW, Horwitz AF. Biochemistry. 1981;20:4785–4792. doi: 10.1021/bi00519a040. [DOI] [PubMed] [Google Scholar]
- 21.Liepinsh E, Otting G. Magn Reson Med. 1996;35:30–42. doi: 10.1002/mrm.1910350106. [DOI] [PubMed] [Google Scholar]
- 22.Guttman A. Nature. 1996;380:461–462. doi: 10.1038/380461a0. [DOI] [PubMed] [Google Scholar]
- 23.Katta V, Chait BT. J Am Chem Soc. 1993;115:6317–6321. [Google Scholar]
- 24.Raska M, Takahashi K, Czernekova L, Zachova K, Hall S, Moldoveanu Z, Elliott MC, Wilson L, Brown R, Jancova D, Barnes S, Vrbkova J, Tomana M, Smith PD, Mestecky J, Renfrow MB, Novak J. J Biol Chem. 2010;285:20860–20869. doi: 10.1074/jbc.M109.085472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zubay G, Strominger J, Zubay G, editors. McMillan Pub. New York: 1988. pp. 131–153. [Google Scholar]
- 26.Houde D, Berkowitz SA, Engen JR. J Pharm Sci. 2011;100:2071–2086. doi: 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houde D, Peng Y, Berkowitz SA, Engen JR. Mol Cell Proteomics. 2010;9:1716–1728. doi: 10.1074/mcp.M900540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oberholtzer JC, Englander SW, Horwitz AF. FEBS Letters. 1983;158:305–309. [Google Scholar]
- 29.Ferguson PL, Konermann L. Anal Chem. 2008;80:4078–4086. doi: 10.1021/ac8001963. [DOI] [PubMed] [Google Scholar]
- 30.Zehl M, Rand KD, Jensen ON, Jorgensen TJ. J Am Chem Soc. 2008;130:17453–17459. doi: 10.1021/ja805573h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.