

Abstract
5-(Z)-Alkylidene-2-thioxo-1,3-thiazolidin-4-ones (rhodanine derivatives) were prepared by reaction of in situ-generated dithiocarbamates with recently reported racemic α-chloro-β,γ-alkenoate esters. This multicomponent sequential transformation performed in one reaction flask represents a general route to this medicinally valuable class of sulfur/nitrogen heterocycles. Using this convergent procedure, we prepared an analog of the drug epalrestat, an aldose reductase inhibitory rhodanine.
Sequentially linking several different components in one reaction vessel has been studied intensively as a rapid way to increase molecular complexity while avoiding costly and environmentally unfriendly isolation and purification of intermediates.1–4 Such efficient multicomponent reactions, like the Ugi reaction,5 often produce privileged scaffolds of considerable medicinal value. Rhodanines (2-thioxo-1,3-thiazolidin-4-ones) are 5-membered ring sulfur/nitrogen heterocycles some of which have antimalarial, antibacterial, antifungal, antiviral, antitumor, anti-inflammatory or herbicidal activities.6 5-Arylidene rhodanines are common,7–14 whereas 5-alkylidene rhodanines are rare.15–17 In one direct comparison, 5-alkylidene rhodanine A was more potent as a class C β-lactamase inhibitor than the corresponding vinylogous 5-arylidene rhodanine B (Figure 1).16
Figure 1.

5-Alkylidene rhodanine A and vinylogous 5-arylidene rhodanine B
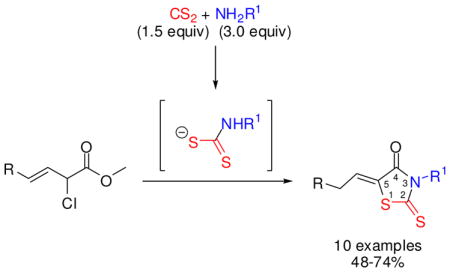
Three-component, one-flask preparation of 5-arylidene rhodanines from aryl propiolates and dithiocarbamates has been reported18 as well as three-component preparation of 5-hydrazinoalkylidene rhodanines,19 5-acyl rhodanines,20 5-carboxymethyl rhodanines,21 and 2-amino 4-thiazolones22 and also two-component rhodanine syntheses.23 5-Alkylidenation of rhodanine-3-acetic acid at 75–100 °C has been achieved.15 5-Alkylidenation of 3-NH rhodanine, however, proceeds in only 11–19% yields.17 We report here a synthetic approach to these heterocycles involving three-component, one-flask, sequential 1+2+2 atom construction of 5-membered ring 5-alkylidene rhodanines 3–5 (Scheme 1). Our multicomponent protocol involves amine addition to carbon disulfide followed in situ by exclusive SN2 displacement of chloride from recently reported α,β,γ-trifunctional ester allylic chloride 2, carbon-carbon double bond isomerization into conjugation with the ester group (β,γ-enoate → α,β-enoate), and finally 5-exo-trig24 cyclization to produce 5-(Z)-alkylidene rhodanines 3–5 in 48–74% yields with diverse R and R1 groups (Scheme 1).
Scheme 1.


Formation of 5-(Z)-alkylidene rhodanines 3–5
We recently described gram scale synthesis and various transformations of versatile and diverse α-chloro-β,γ-alkenoate esters 2, with R groups including benzyl, n-pentyl, cyclohexyl, 9-nonenyl, and 4-benzyloxybutyl, prepared from γ-selenyl α,β-enoate esters 1 (Scheme 1).25–27 A new transformation of multifunctional enoate esters 2, occurring in 2-propanol solvent under mild (25 °C) conditions, produces various 5-alkylidene rhodanine derivatives typically favoring Z over E olefins by 30:1 to 7:1 ratios; routine silica gel column chromatography afforded the pure 5-(Z)-alkylidene rhodanines in the yields shown in Scheme 1. The (Z)-geometry of the 5-alkylidene rhodanines 3–5 is consistent with literature analogies28–31 and was confirmed by 1H NMR spectroscopy. The 1H NMR spectra of the crude reaction mixtures contained two distinct triplets for the single olefinic proton, the major triplet at 6.98–7.02 ppm and the minor triplet at 6.47–6.52 ppm. These 1H NMR data are consistent with the Z-isomer olefinic proton being further downfield then the E-isomer olefinic proton, because of deshielding from the carbonyl group.28–31
The second step in this one-flask process is an SN2 displacement of chloride by in situ-generated dithiocarbamate; in a closely related system we have isolated some of the uncyclized SN2 product with β,γ-unsaturation. Next, probably excess amine induces migration of the β,γ-carbon-carbon double bond into conjugation with the ester group, forcing the dithiocarbamate nitrogen atom and the ester carbonyl group into close proximity, thereby assisting cyclization into lactams 3–5.
We have successfully achieved synthesis of rhodanine 3e on a 250 mg scale. Various simple as well as functionalized (olefinic, alcoholic, acetal, and furyl) commercial primary amines (although not less nucleophilic anilines)32 participated in forming the rhodanines 3–5 shown in Scheme 1. Several amines failed to produce rhodanines via Scheme 1, including propargylamine, aminoacetonitrile, 2-(aminomethyl)pyridine, 6-aminohexanoic acid, glycine, and glycine ethyl ester.
Primary amine orthoester 633 was utilized to give fragile rhodanine orthoester 7 which was treated immediately with methanol and a catalytic amount of p-toluenesulfonic acid (Scheme 2). In this three-component, one-flask protocol, the desired desmethyl-dihydro-epalrestat methyl ester 8 was produced chromatographically pure in 48% yield as a close analog of the aldose inhibitory drug epalrestat (9).34,35 Short syntheses of carboxylic acid analogs of methyl ester 8, although not of epalrestat, have been reported.15
Scheme 2.

Synthesis of desmethyl-dihydro-epalrestat methyl ester 8
In conclusion, convergent syntheses of N-alkyl 5-(Z)-alkylidene rhodanine derivatives have been achieved using recently reported racemic α-chloro-β,γ-alkenoate ester 2 building blocks.27 The formation of these rhodanine derivatives involves a three-step, one-flask protocol that provides quick access to biologically valuable sulfur-nitrogen heterocycles. The method is mild and versatile, working successfully with a variety of simple as well as functionalized primary amines and with several different α-chloro esters 2,26,27 allowing preparation of a small library of new N-alkyl 5-alkylidene rhodanines.
Experimental Section
General Experimental Methods
1H and 13C NMR spectra were recorded at 400 and 100 MHz respectively, using the residual solvent peak as the internal standard. FAB mass spectra were obtained using a double focusing magnetic sector mass spectrometer equipped with a Cs ion gun (28kV @ 2uA), an off-axis electron multiplier and an MSS data system. The resolution of the instrument was set at 10,000 (100ppm peak width). Samples were mixed with m-nitrobenzyl-alcohol matrix deposited on the target of a direct insertion probe for introduction into the source. Spectra were acquired under control of the data system. Nominal mass scan spectra were acquired with a mass scan range of 10–1000 amu using a magnet scan rate of 25 sec/dec. For accurate mass measurements, a narrower mass scan range was employed, with the matrix containing 10% PEG or PEGMME mass calibrant. Fourier Transform-Infrared (FT-IR) experiments were obtained from 4000 to 600 cm−1. Microwave reactions were run sealed tubes in a Biotage Initatior with an external contact temperature probe. Thin-layer chromatography was performed with glass-backed 20 cm x 20 cm extra-hard layer 250 μm thickness 60 Å plates with F254 indicator cut down to 20 mm x 50 mm for analytical purposes. Compounds purified via preparative TLC were performed with glass-backed 20 cm x 20 cm extra-hard layer 1000 μm thickness 60 Å plates with F254 indicator.
(±) Seleno Acrylate 1 (R = cyclohexyl)
An oven-dried, single-necked 100 mL round-bottomed flask equipped with a magnetic stir bar was charged with dichloromethane (10 mL), DL-proline (0.07 g, 0.57 mmol, 0.2 equiv), 2-cyclohexylacetaldehyde (0.36 g, 2.84 mmol, 1.0 equiv), and was allowed to stir at room temperature under argon. After 10 min, N-(phenylseleno)phthalimide (1.00 g, 3.11 mmol, 1.1 equiv) was added and allowed to stir at room temperature under argon. After 2 h, hexanes (15 mL) were added and the slurry was gravity filtered and concentrated into a 50-mL round-bottomed flask on a rotary evaporator. The resultant seleno-aldehyde was dissolved in THF (10 mL) and methyl (triphenylphosphoranylidene)acetate (1.23 g, 3.68 mmol, 1.3 equiv) was added to the flask. The solution was allowed to stir at room temperature under argon. After 16 h, the reaction was quenched with NH4Cl (20 mL), extracted with diethyl ether (3 × 25 mL), dried over MgSO4, gravity filtered, and concentrated on a rotary evaporator. The yellow oil was purified via column chromatography (hexanes flush of initial yellow band followed by 50:1, hexanes: ethyl acetate) to afford the desired product (1) as a yellow oil (0.55 g, 1.70 mmol, 60%). IR (thin film) 3056, 2926, 2852, 1722, 1644, 1578, 1476, 1436, 1331, 1310, 1268, 1233, 1216, 1199, 1171, 1143, 1073, 1036, 1021, 978, 910, 739 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.47-7.44 (m, 2H), 7.30-7.21 (m, 3H), 6.91 (dd, J = 15.6, 11.2 Hz, 1H), 5.15 (d, J = 15.6 Hz, 1H), 3.65 (s, 3H), 3.54 (dd, J = 11.2, 7.6 Hz, 1H), 2.01 (d, J = 12.8 Hz, 1H), 1.79-1.69 (m, 3H), 1.60–1.69 (m, 2H), 1.28-1.05 (m, 5H). 13C NMR (100 MHz, CDCl3) δ 166.6, 146.7, 136.2, 128.9, 128.7, 128.2, 119.1, 54.4, 51.4, 41.5, 32.1, 31.4, 26.2, 26.2, 26.1. HRMS (ESI) m/z calcd for C17H23O2Se [M + H+] 339.0863, found 339.0852.
(±) α-Chloro Ester 2 (R = cyclohexyl)
An oven-dried, single-necked 50 mL round-bottomed flask equipped with a magnetic stir bar was charged with seleno-acrylate 1 (0.48 g, 1.42 mmol, 1.0 equiv), hexanes (3 mL), and ethyl vinyl ether (4.85 mL, 50.6 mmol, 6.0 equiv). Sulfuryl chloride (0.23 mL, 2.84 mmol, 2.0 equiv) was dropwise as a solution in hexanes (7 mL) over 30 min and the reaction was allowed to stir at room temperature under argon. After 30 min TLC analysis confirmed completion of reaction and the solution was concentrated on a rotary evaporator and the dark oil was purified immediately via column chromatography (1% ethyl acetate in hexanes) to afford the desired product (2) as a colorless oil (0.28 g, 1.30 mmol, 92%). IR (thin film) 2926, 2852, 2360, 1748, 1653, 1558, 1436, 1270, 1160, 967, 664 cm−1. 1H NMR (400 MHz, CDCl3) δ 5.79 (dd, J = 15.6, 6.4 Hz, 1H), 5.59 (ddd, J = 14.8, 8.4, 0.8 Hz, 1H), 4.71 (d, 8.8 Hz, 1H), 3.74 (s, 3H), 2.03-1.92 (m, 1H), 1.74-1.65 (m, 4H), 1.58–1.65 (m, 1H), 1.29-0.99 (m, 5H). 13C NMR (100 MHz, CDCl3) δ 169.0, 143.5, 122.2, 58.1, 52.9, 40.2, 32.2, 32.1, 25.9, 25.8. HRMS (ESI) m/z calcd for C11H18ClO2 [M + H+] 217.0995, found 217.0996.
General Procedure for Synthesis of 5-(Z)-Alkylidine Rhodanines 3–5
An oven-dried, 2 dram vial, equipped with a magnetic stir bar, under a stream of Argon through a needle was charged with carbon disulfide (0.17 mmol, 1.5 equiv), amine (0.33 mmol, 3.0 equiv), 2-propanol (1.0 mL) and was stirred at room temperature under argon. After 6 h, α-chloro ester 2 (0.11 mmol, 1.0 equiv) was added as a solution in 2-propanol (0.5 mL), and the reaction was allowed to stir at room temperature under argon. After 24 h, the reaction was quenched with saturated ammonium chloride (1 mL) and extracted with diethyl ether (3 × 5 mL). The organic layers were combined, dried over MgSO4, gravity filtered, and concentrated on a rotary evaporator. The crude mixtures were purified via silica gel column chromatography (5% ethyl acetate in hexanes) or alternatively via preparative silica TLC (5% ethyl acetate in hexanes).
Rhodanine 3a
22.0 mg, 59% yield as a yellow oil. IR (thin film) 3062, 3029, 2926, 2857, 2368, 2345, 2092, 1717, 1628, 1603, 1495, 1454, 1425, 1375, 1348, 1197, 1080, 1030 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.43-7.41 (m, 2H), 7.33-7.17 (m, 8H), 6.97 (t, J = 7.6 Hz, 1H), 5.25 (s, 2H), 2.85 (t, 7.6 Hz, 2H), 2.55 (q, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 193.4, 166.1, 139.7, 136.9, 134.8, 128.9, 128.7, 128.5, 128.2, 128.0, 127.6, 126.6, 47.3, 33.8. 33.6. HRMS (ESI) m/z calcd for C19H17NOS2 339.0752, found 339.0758.
Rhodanine 3b
21.6 mg, 68% yield as a yellow oil. IR (thin film) 3061, 3026, 2924, 2853, 1720, 1626, 1495, 1453, 1417, 1365, 1337, 1213, 1136, 989, 934 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.33-7.13 (m, 5H), 6.97 (t, J = 7.2 Hz, 1H), 5.87-5.70 (m, 1H), 5.32-5.22 (m, 2H), 4.67 (d, J = 5.6 Hz, 2H), 2.86 (t, J = 7.6 Hz, 2H), 2.56 (q, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 193.2, 165.7, 139.7, 136.9, 129.6, 129.5, 128.7, 128.3, 127.7, 119.3, 46.2, 33.9, 33.7. HRMS (ESI) m/z calcd for C15H15NOS2 290.0668, found 290.0663.
Rhodanine 3c
21.3 mg, 64% yield as a yellow oil. IR (thin film) 3061, 3026, 2925, 2359, 2341, 1718, 1628, 1496, 1430, 1350, 1328, 1273, 1202, 1136 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.33-7.17 (m, 5H), 6.95 (t, J = 8.0 Hz, 1H), 5.83-5.72 (m, 1H), 5.11-5.07 (m, 2H), 4.15 (dd, J = 6.0, 2.8 Hz, 2H), 2.86 (t, J = 7.6 Hz, 2H), 2.46 (q, J = 7.2 Hz, 2H), 2.43-2.41 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 193.8, 166.2, 139.9, 136.8, 134.0, 128.9, 128.5, 127.8, 126.8, 117.9, 43.7, 34.1, 33.8, 31.4. HRMS (ESI) m/z calcd for C16H18NOS2 [M + H+] 304.0830, found 304.0829.
Rhodanine 3d
22.1 mg, 61% yield as a yellow oil. IR (thin film) 3734, 3609, 3583, 3026, 2924, 2854, 2360, 1721, 1626, 1496, 1453, 1413, 1338, 1243, 1180, 1077, 1010, 939, 743 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.33-7.28 (m, 3H), 7.22-7.15 (3H), 6.98 (t, J = 7.7 Hz, 1H), 6.38 (d, J = 3.6 Hz, 1H), 6.30-6.29 (m, 1H), 2.84 (t, J = 7.7 Hz, 2H), 2.54 (q, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 193.1, 165.8, 148.1, 142.8, 139.9, 137.3, 128.9, 128.5, 127.6, 126.8, 110.6, 110.5, 40.3, 34.1, 33.9. HRMS (ESI) m/z calcd for C17H15NO2S2 329.0544, found 329.0545.
Rhodanine 3e
18.4 mg, 57% yield as a yellow oil. IR (thin film) 3445, 3026, 2941, 1721, 1628, 1496, 1454, 1427, 1329, 1276, 1197, 1093, 729, 700 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.34-7.29 (m, 2H), 7.27-7.23 (m, 1H), 7.20-7.18 (m, 2H), 6.99 (t, 7.8 Hz, 1H), 4.30 (t, J = 5.4 Hz, 2H), 3.90 (t, J = 5.4 Hz, 2H), 2.86 (t, J = 7.2 Hz, 2H), 2.59 (q, J = 7.5 Hz, 2H), 1.93 (br s, 1H). 13C NMR (100 MHz, CDCl3) δ 194.4, 166.8, 139.7, 137.5, 128.7, 128.3, 127.4, 126.7, 60.1, 46.4, 33.9, 33.7. HRMS (ESI) m/z calcd for C14H16NO2S2 [M + H+] 294.06225, found 294.06231.
Rhodanine 3f
19.7 mg, 53% yield as a yellow oil. IR (thin film) 3025, 2935, 2835, 2090, 1723, 1628, 1496, 1454, 1419, 1359, 1324, 1219, 1187, 1127, 1102, 1066, 988, 822, 728 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.34-7.29 (m, 2H), 7.23-7.18 (m, 3H), 6.97 (t, J = 7.6 Hz, 1H), 4.87 (t, J = 5.6 Hz, 1H), 4.19 (d, J = 6.0 Hz, 2H), 3.37 (s, 6H), 2.86 (t, J = 7.2 Hz, 2H), 2.55 (q, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 194.0, 165.9, 139.7, 137.1, 128.7, 128.3, 127.4, 126.6, 99.3, 53.8, 44.9, 33.9. 33.7. HRMS (ESI) m/z calcd for C16H19NO3S2 337.08064, found 337.08056.
Rhodanine 3g
20.7 mg, 52% yield as a yellow oil. IR (thin film) 2925, 2854, 2181, 2101, 1726, 1575, 1495, 1454, 1437, 1347, 1266, 1230, 1174, 1137, 1065, 1020, 735 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.33-7.29 (m, 2H), 7.24-7.22 (m, 1H), 7.21-7.17 (m, 2H), 6.95 (t, J = 7.6 Hz, 1H), 4.04-4.01 (m, 2H), 2.86 (t, J = 7.6 Hz, 2H), 2.55 (q, J = 7.6 Hz, 2H), 1.68-1.55 (m, 2H), 1.33-1.23 (m, 10H), 0.89-0.85 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 193.7, 166.1, 139.8, 136.5, 128.7, 128.3, 127.8, 126.6, 44.6, 33.9, 33.7, 31.8, 29.1, 26.9, 26.7, 22.6, 14.1. HRMS (ESI) m/z calcd for C20H28NOS2 [M + H+] 362.1612, found 362.1614.
Rhodanine 4a
16.9 mg, 48% yield as a yellow oil. IR (thin film) 3032, 2953, 2926, 2855, 2359, 1718, 1627, 1604, 1494, 1455, 1425, 1375, 1348, 1319, 1300, 1194, 1108, 1080, 949, 821, 730 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.45-7.42 (m, 2H), 7.32-7.28 (m, 3H), 6.97 (t, J = 7.6 Hz, 1H), 5.25 (s, 2H), 2.23 (q, J = 7.2 Hz, 2H), 1.57-1.52 (m, 4H), 1.28-1.32 (m, 6H), 0.91-0.88 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 193.7, 166.2, 138.8, 134.9, 128.9, 128.6, 128.1, 126.9, 47.4, 32.1, 31.5, 28.9, 27.9, 22.5, 14.0. HRMS (ESI) m/z calcd for C17H21NOS2 319.1065, found 319.1068.
Rhodanine 5a
26.9 mg, 74% yield as a yellow oil. IR (thin film) 2921, 2850, 2362, 1718, 1626, 1494, 1447, 1424, 1375, 1347, 1318, 1298, 1193, 1079, 1029, 939, 729 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.46-7.42 (m, 2H), 7.32-7.27 (m, 3H), 7.0 (t, 8.0 Hz, 1H), 5.26 (s, 2H), 2.12 (dd, J = 8.0, 6.8 Hz, 2H), 1.76-1.68 (m, 5H), 1.26-1.14 (m, 4H), 1.12-0.97 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 193.8, 166.1, 137.8, 134.9, 129.0, 128.5, 128.1, 127.6, 47.3, 39.9, 37.7, 33.2, 26.1. HRMS (ESI) m/z calcd for C18H22NOS2 [M + H+] 332.1143, found 332.1135.
Rhodanine 5f
19.9 mg, 55% yield as a yellow oil. IR (thin film) 2924, 2851, 2360, 2341, 1723, 1628, 1557, 1540, 1506, 1447, 1419, 1359, 1323, 1217, 1187, 1127, 1103, 1010, 985, 890, 750, 723 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.00 (t, 8 Hz, 1H), 4.87 (t, 5.2 Hz, 1H), 4.19 (d, 4.4 Hz, 2H), 3.38 (s, 6H), 2.12 (dd, J = 14.8, 7.6 Hz, 2H), 1.75-1.63 (m, 5H), 1.28-1.18 (m, 4H), 1.12-0.97 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 194.5, 166.1, 138.1, 127.5, 99.5, 53.9, 45.0, 40.1, 37.9, 33.4, 26.3, 26.2. HRMS (ESI) m/z calcd for C15H23NO3S2 329.1119, found 329.1105.
Epalrestat Analogue 8
An oven-dried, 2 dram vial, equipped with a magnetic stir bar under argon was charged via syringe with carbon disulfide (25.0 mg, 0.33 mmol, 1.5 equiv), amine orthoester 6 (105.0 mg, 0.66 mmol, 3.0 equiv), 2-propanol (2.0 mL) and was stirred at room temperature, under argon. After 6 h, α-chloro ester 2 (R = PhCH2) (50.0 mg, 0.22 mmol, 1.0 equiv) was added as a solution in 2-propanol (1.0 mL), and the reaction was allowed to stir at room temperature, under argon. After 24 h, the reaction was quenched with saturated ammonium chloride (1 mL) and extracted with diethyl ether (3 × 5 mL). The organic layers were combined, dried over MgSO4, gravity filtered, and concentrated on a rotary evaporator at room temperature. The crude oil was dissolved in methanol (1.5 mL), transferred to a microwave vial, charged with p-toluenesulfonic acid (3.8 mg, 0.02 mmol, 0.1 equiv), sealed and heated at 90 °C for 10 h. Dichloromethane (5 mL) was added, washed with water (3 × 5 mL), dried over MgSO4, and concentrated. The residue was purified via silica gel column chromatography (10% ethyl acetate in hexanes) to afford 33.9 mg of 8 as a yellow oil in 48% yield. IR (thin film) 3024, 2924, 2851, 2362, 1750, 1718, 1625, 1578, 1495, 1454, 1435, 1400, 1364, 1321, 1265, 1198, 1054, 1008, 954, 734 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.32-7.29 (m, 2H), 7.22-7.17 (m, 3H), 7.02 (t, J = 7.6 Hz, 1H), 4.79 (s, 2H), 3.76 (s, 3H), 2.87 (t, J = 7.6 Hz, 2H), 2.58 (q, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 193.2, 166.4, 166.3, 139.6, 137.8, 128.8, 128.3, 127.4, 126.7, 52.8, 44.5, 33.9, 33.8. HRMS (ESI) m/z calcd for C15H16NO3S2 [M + H+] 322.0572, found 322.0571.
Supplementary Material
Acknowledgments
We thank the NIH (AI 34885) for financial support.
Footnotes
1H and 13C NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Magedov IV, Frolova L, Manpadi M, Bhoga U, Tang H, Evdokimov NM, George O, Georgiou KM, Renner S, Getlik M, Kinnibrugh TL, Fernandes MA, Van Slambrouck S, Steelant FA, Shuster CB, Rogelj S, van Otterlo WAL, Kornienko A. J Med Chem. 2011;54:4234–4246. doi: 10.1021/jm200410r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tietze LF, Brasche G, Gericke KM. Angew Chem Int Ed. 2007;46:2977. [Google Scholar]
- 3.Ramón DJ, Yus M. Angew Chem Int Ed. 2005;44:1602–1634. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]
- 4.Posner GH. Chem Rev. 1986;86:831–844. [Google Scholar]
- 5.Ugi I. Angew Chem Int Ed. 1982;21:810–819. [Google Scholar]
- 6.Tomašić T, Mašić LP. Curr Med Chem. 2009;16:1596–1629. doi: 10.2174/092986709788186200. [DOI] [PubMed] [Google Scholar]
- 7.Opletalova V, Dolezel J, Kralova K, Pesko M, Kunes J, Jampilek J. Molecules. 2011;16:5207–5227. doi: 10.3390/molecules16065207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernardo PH, Sivaraman T, Wan KF, Xu J, Krishnamoorthy J, Song CM, Tian L, Chin JSF, Lim DSW, Mok HYK, Yu VC, Tong JC, Chai CLL. J Med Chem. 2010;53:2314–2318. doi: 10.1021/jm901469p. [DOI] [PubMed] [Google Scholar]
- 9.Chen ZH, Zheng CJ, Sun LP, Piao HR. Eur J Med Chem. 2010;45:5739–5743. doi: 10.1016/j.ejmech.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 10.Dolezel J, Hirsova P, Opletalova V, Dohnal J, Marcela V, Kunes J, Jampilek J. Molecules. 2009;14:4197–4212. doi: 10.3390/molecules14104197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sing WT, Lee CL, Yeo SL, Lim SP, Sim MM. Bioorg Med Chem Lett. 2001;11:91–94. doi: 10.1016/s0960-894x(00)00610-7. [DOI] [PubMed] [Google Scholar]
- 12.Lee CL, Sim MM. Tetrahedron Lett. 2010;41:5729–5732. [Google Scholar]
- 13.Sortino M, Delgado P, Juárez S, Quiroga J, Abonía R, Insuasty B, Nogueras M, Rodero L, Garibotto FM, Enriz RD, Zacchino SA. Bioorg Med Chem. 2007;15:484–494. doi: 10.1016/j.bmc.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 14.Bourahla K, Derdour A, Rahmouni M, Carreaux F, Bazureau JP. Tetrahedron Lett. 2007;48:5785–5789. [Google Scholar]
- 15.Ohishi Y, Mukai T, Nagahara M, Yajima M, Kajikawa N, Miyahara K, Takano T. Chem Pharm Bull. 1990;38:1911–1919. doi: 10.1248/cpb.38.1911. [DOI] [PubMed] [Google Scholar]
- 16.Grant EB, Guiadeen D, Baum EZ, Foleno BD, Jin H, Montenegro DA, Nelson EA, Bush K, Hlasta DJ. Bioorg Med Chem Lett. 2000;10:2179–2182. doi: 10.1016/s0960-894x(00)00444-3. [DOI] [PubMed] [Google Scholar]
- 17.Russell AJ, Westwood IM, Crawford MHJ, Robinson J, Kawamura A, Redfield C, Laurieri N, Lowe ED, Davies SG, Sim E. Bioorg Med Chem. 2009;17:905–918. doi: 10.1016/j.bmc.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 18.Gabillet S, Lecerclé D, Loreau O, Carboni M, Dézard S, Gomis JM, Taran F. Org Lett. 2007;9:3925–3927. doi: 10.1021/ol701563e. [DOI] [PubMed] [Google Scholar]
- 19.Attanasi OA, Crescentini LD, Gavi G, Filippone P, Giorgi G, Mantellini F, Moscatelli G, Behalo MS. Org Lett. 2009;11:2265–2268. doi: 10.1021/ol900545v. [DOI] [PubMed] [Google Scholar]
- 20.Yavari I, Hajinasiri R, Sayyed-Alangi SZ, Iravani N. Monatsh Chem. 2008;139:1029–1031. [Google Scholar]
- 21.Alizadeh A, Zohreh N. Synlett. 2009;13:2146–2148. [Google Scholar]
- 22.Anderluh M, Jukič M, Petrič R. Tetrahedron. 2009;65:344–350. [Google Scholar]
- 23.Radi M, Botta L, Casaluce G, Bernardini M, Botta M. J Comb Chem. 2010;12:200–205. doi: 10.1021/cc9001789. [DOI] [PubMed] [Google Scholar]
- 24.Baldwin JE. J C S Chem Comm. 1976:734–736. [Google Scholar]
- 25.Hess LC, Posner GH. Org Lett. 2010;12:2120–2122. doi: 10.1021/ol100615j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genna DT, Hencken CP, Siegler M, Posner GH. Org Lett. 2010;12:4694–4697. doi: 10.1021/ol102142a. [DOI] [PubMed] [Google Scholar]
- 27.Hencken CP, Genna DT, Siegler M, Posner GH. J Org Chem. 2011;76:5149–5155. doi: 10.1021/jo200795f. [DOI] [PubMed] [Google Scholar]
- 28.Kandeel KA. Chem Pap. 2004;58:334–340. [Google Scholar]
- 29.Ottaná R, Maccari R, Barreca ML, Bruno G, Rotondo A, Rossi A, Chiricosta G, Di Paola R, Sautebin L, Cuzzocrea S, Vigorita MG. Bioorg Med Chem. 2005;13:4243–4252. doi: 10.1016/j.bmc.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 30.Vicini P, Geronikaki A, Anastasia K, Incerti M, Zani F. Bioorg Med Chem. 2006;14:3859–3864. doi: 10.1016/j.bmc.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 31.Kasmi-Mir S, Djarfri A, Paquin L, Hamelin J, Rahmouni M. Molecules. 2006;11:597–602. doi: 10.3390/11080597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamila S, Ankati H, Biehl ER. Tetrahedron Lett. 2011;52:4375–4377. doi: 10.1016/j.tetlet.2011.05.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corey EJ, Raju N. Tetrahedron Lett. 1983;24:5571–5574. [Google Scholar]
- 34.Miyamoto S. Chem-Bio Informatics Journal. 2002;2:74–85. [Google Scholar]
- 35.Demopoulos VJ, Nicolaou I, Alexiou P, Zika C, Sturm K, Kristl A. In: Brilliant Light in Life and Material Sciences. Tsakanov V, Wiedemann H, editors. Springer; 2007. p. 241. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.