

Abstract

C-O activation of mesylates by a palladium catalyst and subsequent cross-coupling with potassium cyclopropyltrifluoroborate have been achieved with high yield. Both electron-enriched and electron-deficient aryl mesylates are suitable electrophilic partners for the Suzuki-Miyaura reaction. The scope was successfully extended to heteroaryl mesylates with yields up to 94%.
Cyclopropanes are among the more useful subunits that can be incorporated within a target molecule to engender or improve biological activity. The number of newly discovered, biologically active natural products and pharmaceutical or crop protection compounds containing the cyclopropyl ring increases daily.1–3 Thus, it has become of interest to develop appropriate methods to install the cyclopropyl subunit within existing skeletons.
The cyclopropyl group, a small strained ring with unique hybridization, also exhibits a particular reactivity pattern in transition-metal catalyzed coupling reactions.4,5 Among these cross-coupling protocols, the Suzuki-Miyaura reaction is a method of choice because of its mild reaction conditions, excellent tolerance of a broad range of functional groups, and the use of environmentally sound, non-toxic boron species.6–9 Cyclopropylboronic acid10–26 has been extensively used, but because of its tendency to protodeboronate easily,27 recent work is currently more focused on various boronic acid derivatives. For example, Burke et al. have reported the use of cyclopropyl MIDA boronates in cross-coupling reactions with various aryl chlorides.27 The corresponding commercially available potassium cyclopropyltrifluoroborate, known to be air and moisture-stable and resistant to protodeboronation, has also been employed in various contexts.28–32 Stereodefined potassium cyclopropyltrifluoroborates were first engaged with aryl bromides to afford the cross-coupled products with retention of configuration.33,34 Our laboratory next developed an efficient method to cross-couple aryl and heteroaryl chloride electrophiles with potassium cyclopropyltrifluoroborate in high yields.35 In 2009, Hocek reported the synthesis of two purine derivatives from the corresponding bromide or chloride with potassium cyclopropyltrifluoroborate in moderate yields.36
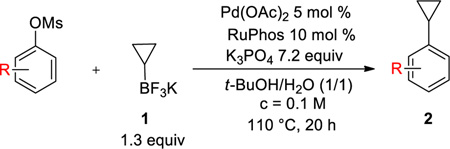
Although halides are usually employed as electrophilic partners, phenol derivatives bearing more environmentally sound, less expensive, and easier to handle nucleofuges offer an alternative of choice in terms of the electrophilic partner. Sulfonated phenol derivatives, especially, have emerged as very competitive cross-coupling substrates. Until now, aryl triflates have been successfully engaged in the Suzuki-Miyaura cross-coupling with cyclopropylboronic acid.13–23 However, triflating reagents such as Tf2O and PhNTf2 are relatively expensive, and some triflates are known to be unstable.37 When it comes to non-fluorinated sulfonated alcohols, only one example of the use of an aryl tosylate in the cross-coupling has been disclosed.24 This method requires the presence of a large excess of cyclopropylboronic acid (3 equiv) to afford the desired compound with a moderate yield. Moreover, to our knowledge, no example of mesylated counterparts has been reported to date. Even though these species are known to be among the least reactive sulfonated species, they display substantial advantages in that they are reasonably atom-economical, very stable, and have already been proven to be partners of choice for the Suzuki-Miyaura reaction.24,38–42 We disclose herein the first cross-coupling of both aryl and heteroaryl mesylates through C-O activation with potassium cyclopropyltrifluoroborate.
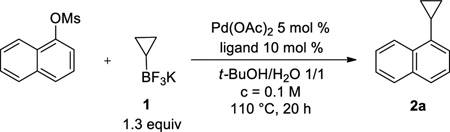
The catalytic system was first optimized on a model reaction between naphthalen-1-yl methanesulfonate and potassium cyclopropyltrifluoroborate 1. Our laboratory already reported that the use of a mixture of t-BuOH/H2O (1/1) and potassium phosphate as base were very efficient for the cross-coupling of mesylated counterparts.39,41,42 Based on these observations, we began our study by screening different ligands in combination with the air-stable Pd(OAc)2 catalyst (Table 1). Alkylphosphines and biarylphosphines, as well as monodentate or bidentate phosphines were tested (Figure 1), and 2-dicyclohexylphosphino-2',6'-di-isopropoxy-1,1'-biphenyl (RuPhos)43 appeared to be the most relevant ligand to obtain the desired cyclopropylnaphthalene 2a with total conversion and 87% isolated yield (Table 1, entry 4).
TABLE 1.
Optimization
 | |||
|---|---|---|---|
| entry | ligand | conv (%)a | yield(%)b |
| 1 | Cy3P•HBF4 | 32 | 10 |
| 2 | XPhos I | 100 | 68 |
| 3 | RuPhos II | 100 | 93 |
| 4b | RuPhos II | 100 | 93 (87)c |
| 5 | SPhos III | 100 | 79 |
| 6 | XantPhos IV | 12 | Traces |
| 7 | DPEPhos V | 29 | 7 |
| 8 | dippf VI | 63 | 36 |
Relative GC yield determined using dodecane as the internal standard.
2 mol % of Pd(OAc)2 and 4 mol % of RuPhos.
Isolated yield.
Figure 1.

Structure of ligands I to VI
The optimized conditions were next applied to a wide range of aryl mesylates bearing either electron-donating or electron-withdrawing groups (Table 2). For most of the functionalized mesylates, it was necessary to utilize a catalyst loading of 5 mol % for the reaction to go to completion. The reaction proceeded very well with almost all the deactivated electrophilic partners, and the desired compounds 2b–d, f, h were obtained with yields as high as 96%. It was more difficult to cross-couple electron-deficient activated mesylates owing to the competitive sulfonate hydrolysis reaction, which generally occurred faster and resulted in the corresponding alcohol as a major product. With a goal to circumvent this problem, another source of palladium [PdCl2(COD) instead of Pd(OAc)2] was tested in the reaction with two substrates: [1,1'-biphenyl]-4-yl methanesulfonate and 4-benzoylphenyl methanesulfonate. Unfortunately this effort was unsuccessful as similar cross-coupled yields were obtained (72% versus 78% for 2f and 59% versus 56% for 2g). However, we were pleased to observe that the reaction is compatible with diverse electron-withdrawing substituents such as nitrile, benzoyl, and ester groups to afford the cyclopropyl arene derivatives 2e, g, i, j with moderate yields. Importantly, by scaling up the reaction to 4.5 mmol of naphth-1-yl mesylate, we were able to reduce the amount of catalyst from 2 mol % to 0.5 mol %, obtaining the desired compound 2a with a yield of 91% (versus 87% at 0.25 mmol scale). Moreover, to avoid solvent waste on this larger scale, the reaction proved to be as efficient in a more concentrated media (91% yield when the reaction was performed at 0.25 M). Of particular note, most of these unsubstituted cyclopropyl arenes are volatile because of their relatively low molecular weight. Thus careful handling is required to isolate the product.
TABLE 2.
Scope of Functionalized Aryl Mesylates
 | |||
|---|---|---|---|
| entry | Ar-OMs | yield (%) | |
| 1 |  |
2a | 87a (91)b (91)c |
| 2 | 2b | 96 | |
| 3 |  |
2c | 91a |
| 4 | 2d | 91 | |
| 5 | 2e | 49 | |
| 6 | 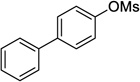 |
2f | 78 (72)d |
| 7 | 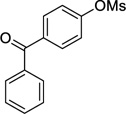 |
2g | 56a (59)a,d |
| 8 | 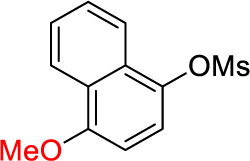 |
2h | 90e |
| 9 | 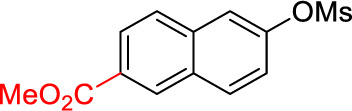 |
2i | 44 |
| 10 | 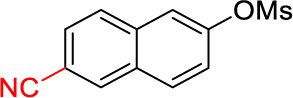 |
2j | 58a |
2 mol % of Pd(OAc)2 4 mol % of RuPhos,
0.5 mol % of Pd(OAc)2 and 1 mol % of RuPhos on a 4.5 mmol scale at a concentration of 0.1 M,
0.5 mol % of Pd(OAc)2 and 1 mol % of RuPhos on a 4.5 mmol scale at a concentration of 0.25 M,
PdCl2(COD)was used instead of Pd(OAc)2,
Using 10% of impurities that cannot be separated
The compatibility of heterocyclic substrates in the cross-coupling reaction with 1 was also examined. In this regard, the previously optimized conditions were initially applied to quinolin-6-yl methanesulfonate as a representative substrate: it transpired that the desired product 3a was obtained in only 50% yield, and the reaction did not proceed to complete conversion. Different palladium sources were thus screened, and in this way total conversion was achieved by using 2 mol % of PdCl2(COD) instead of palladium acetate. The desired product of this reaction was isolated in 94% yield. Using these new reaction conditions, the transformations proceeded very well with a variety of structurally diverse heterocycles. Mesylated quinoline, benzothiazole, dibenzofuran, benzothiophene and dibenzothiophene proved to be suitable partners, affording the corresponding cyclopropyl heteroarenes 3a–c, e, f with yields ranging between 72% and 94%. Only the quinolin-8-yl methanesulfonate afforded the cross-coupled compound 3d with a moderate yield, perhaps because of the coordination of the nitrogen to the palladium, which may partially inhibit the catalytic cycle.44,45,46
In conclusion, a convenient method to cross-couple a large array of aryl mesylates with potassium cyclopropyltrifluoroborate in high yields has been developed. The method is also efficient with diverse heterocyclic mesylates as electrophiles and provides cyclopropyl heteroarenes with very good yields. This environmentally sound new strategy based on C-O activation of mesylates affords a complementary way to obtain cyclopropyl functionalized molecules, known to be of interest for their biological properties.
Experimental Section
All of the mesylates were synthesized following a representative procedure.42
Procedure A
1-Cyclopropylnaphthalene (2a)
is used as an example. A Biotage microwave vial was charged with Pd(OAc)2 (1.1 mg, 5.0 µmol), RuPhos (4.7 mg, 10 µmol), naphthalen-1-yl methanesulfonate (55.5 mg, 0.25 mmol), cyclopropyltrifluoroborate (47.2 mg, 0.33 mmol) and K3PO4 (382 mg, 1.80 mmol). The test tube was sealed with a cap lined with a disposable Teflon septum and evacuated under vacuum and purged with argon three times. A mixture of t-BuOH/H2O (1.25 mL/1.25 mL) was added under argon. The reaction mixture was heated to 110 °C for 4 h before cooling to rt. The reaction mixture was extracted with EtOAc (3 × 2 mL) and then dried (MgSO4). The solvent was removed in vacuo, and the crude product was purified by preparative silica gel chromatography (elution with hexanes/ CH2Cl2 80:20) to yield 2a in 87% yield (36.6 mg) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 8.3 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.57-7.54 (m, 1H), 7.51-7.48 (m, 1H), 7.40-7.37 (m, 1H), 7.28-7.26 (m, 1H), 2.38-2.33 (m, 1H), 1.09-1.05 (m, 2H), 0.79-0.76 (m, 2H); 13C NMR (125 MHz, acetone-d6) δ 139.0, 133.6, 133.4, 128.3, 126.3, 125.6, 125.5, 125.4, 124.1, 123.2, 12.7, 6.0; FT-IR (neat) 1596, 1509 cm−1; HRMS (ESI) m/z calcd. for C13H13 (M+H)+ 169.1017, found 169.1015.
1-Cyclopropyl-4-methoxybenzene (2b)
Following procedure A, the reaction was carried out with 4-methoxyphenyl methanesulfonate (101 mg, 0.50 mmol), Pd(OAc)2 (5.6 mg, 25 µmol) and RuPhos (23.3 mg, 50.0 µmol) to obtain 2b (71.2 mg, 96%) as a yellow oil after silica gel chromatography (elution with hexanes/EtOAc 98:2). 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 8.6 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 3.78 (s, 3H), 1.88-1.82 (m, 1H), 0.90-0.87 (m, 2H), 0.62-0.60 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 157.5, 135.8, 126.8, 113.7, 55.3, 14.6, 8.5; FT-IR (neat) 1613, 1246, 1032 cm−1; HRMS (ESI) m/z calcd. for C10H13O (M+H)+ 149.0966, found 149.0963.
5-Cyclopropyl-1,2,3-trimethoxybenzene (2c)
Following procedure A, the reaction was carried out with 3,4,5-trimethoxyphenyl methanesulfonate (131 mg, 0.50 mmol) to obtain 2c (94.4 mg, 91%) as a yellow oil after silica gel chromatography (elution with hexanes/EtOAc 90:10). 1H NMR (500 MHz, CDCl3) δ 6.31 (s, 2H), 3.84 (s, 6H), 3.81 (s, 3H), 1.87-1.84 (m, 1H), 0.95-0.91 (m, 2H), 0.68-0.65 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 153.5, 140.0, 136.4, 103.2, 61.2, 56.4, 16.2, 9.2; FT-IR (neat) 1585, 1246, 1236, 1127 cm−1; HRMS (ESI) m/z calcd. for C12H17O3 (M+H)+ 209.1178, found 209.1171.
1-Cyclopropyl-2-methoxybenzene (2d)
Following procedure A, the reaction was carried out with 2-methoxyphenyl methanesulfonate (115 mg, 0.57 mmol), Pd(OAc)2 (6.3 mg, 28 µmol) and RuPhos (26.6 mg, 57.0 µmol) to obtain 2d (77.3 mg, 91%) as a yellow oil after silica gel chromatography (elution with hexanes/EtOAc 98:2). 1H NMR (500 MHz, CDCl3) δ 7.15-7.12 (m, 1H), 6.89-6.83 (m, 3H), 3.87 (s, 3H), 2.20-2.15 (m, 1H), 0.94-0.90 (m, 2H), 0.67-0.63 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 158.1, 131.9, 126.1, 124.7, 120.4, 110.1, 55.5, 9.2, 7.6; FT-IR (neat) 1244, 1029 cm−1; HRMS (ESI) m/z calcd. for C10H13O (M+H)+ 149.0966, found 149.0966.
4-Cyclopropyl-3-methoxybenzonitrile (2e)
Following procedure A, the reaction was carried out with 4-cyano-2-methoxyphenyl methanesulfonate (56.8 mg, 0.25 mmol), Pd(OAc)2 (2.8 mg, 12 µmol) and RuPhos (11.7 mg, 25.0 µmol) to obtain 2e (28.1 mg, 91%) as a white solid after preparative silica gel chromatography (elution with hexanes/EtOAc 90:10). mp: 82–83 °C; 1H NMR (500 MHz, CDCl3) δ 7.16 (d, J = 7.9 Hz, 1H), 7.03 (s, 1H), 6.83 (d, J = 7.9 Hz, 1H), 3.88 (s, 3H), 2.25-2.20 (m, 1H), 1.04-1.00 (m, 2H), 0.71-0.68 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 158.3, 138.9, 125.1, 125.0, 119.4, 112.9, 109.4, 55.9, 9.7, 9.1; FT-IR (neat) 2224, 1508, 1266, 1036 cm−1; HRMS (ESI) m/z calcd. for C11H11NO (M)+ 173.0841, found 173.0843.
4-Cyclopropyl-1,1'-biphenyl (2f)
Following procedure A, the reaction was carried out with [1,1'-biphenyl]-4-yl methanesulfonate (124 mg, 0.50 mmol), Pd(OAc)2 (5.6 mg, 25 µmol) and RuPhos (23.3 mg, 50.0 µmol) to obtain 2f (40.4 mg, 42%) as a white solid after silica gel chromatography (elution with petroleum ether). mp: 68–71 °C; 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J = 7.3 Hz, 2H), 7.52 (d, J = 8.3 Hz, 2H), 7.47-7.45 (m, 2H), 7.44-7.34 (m, 1H), 7.18 (d, J = 8.3 Hz, 2H), 2.00-1.94 (m, 1H), 1.05-1.01 (m, 2H), 0.79-0.77 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 143.4, 141.3, 138.5, 128.9, 127.2, 127.1, 126.2, 15.3, 9.5; FT-IR (neat) 1488 cm−1; HRMS (ESI) m/z calcd. for C15H15 (M+H)+ 195.1174, found 195.1176.
4-Cyclopropylphenylmethanone (2g)
Following procedure A, the reaction was carried out with 4-benzoylphenyl methanesulfonate (69.0 mg, 0.25 mmol) to obtain 2g (31.1 mg, 56%) as an off-white solid after preparative silica gel chromatography (elution with hexanes/EtOAc 95:5). mp: 60–62 °C; 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 7.7 Hz, 2H), 7.71 (d, J = 7.9 Hz, 2H), 7.58-7.55 (m, 1H), 7.48-7.45 (m, 2H), 7.14 (d, J = 7.9 Hz, 2H), 1.99-1.94 (m, 1H), 1.08-1.06 (m, 2H), 0.80-0.79 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 196.5, 149.9, 138.2, 134.8, 132.3, 130.6, 130.0, 128.3, 125.4, 15.9, 10.5; FT-IR (neat) 1648, 1605 cm−1; HRMS (ESI) m/z calcd. for C16H15O (M+H)+ 223.1123, found 223.1129.
1-Cyclopropyl-4-methoxynaphthalene (2h)
Following procedure A, the reaction was carried out with 4-methoxynaphthalen-1-yl methanesulfonate (63.0 mg, 0.25 mmol), Pd(OAc)2 (2.8 mg, 12 µmol) and RuPhos (11.7 mg, 25.0 µmol) to obtain 2h (44.5 mg, 90%) as a colorless oil after preparative silica gel chromatography (elution with petroleum ether/hexanes/EtOAc 68:30:2). 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 8.3 Hz, 1H), 8.34 (d, J = 8.3 Hz, 1H), 7.63-7.60 (m, 1H), 7.56-7.53 (m, 1H), 7.23 (d, J = 7.9 Hz, 1H), 6.74 (d, J = 7.9 Hz, 1H), 4.00 (s, 3H), 2.30-2.25 (m, 1H), 1.06-1.04 (m, 2H), 0.76-0.74 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 154.4, 134.5, 131.3, 126.4, 125.8, 125.2, 124.5, 124.3, 122.5, 103.3, 55.6, 13.1, 6.2; FT-IR (neat) 1588, 1271, 1098 cm−1; HRMS (ESI) m/z calcd. for C14H14O (M)+. 198.1045, found 198.1047.
Methyl 6-cyclopropyl-2-naphthoate (2i)
Following procedure A, the reaction was carried out with methyl 6-((methylsulfonyl)oxy)-2-naphthoate (70.0 mg, 0.25 mmol), Pd(OAc)2 (2.8 mg, 12 µmol) and RuPhos (11.7 mg, 25.0 µmol) to obtain 2i (24.7 mg, 44%) as a colorless oil after preparative silica gel chromatography (elution with hexanes/EtOAc 98:2). 1H NMR (500 MHz, CDCl3) δ 8.55 (s, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.76 (d, J = 8.5 Hz, 1H), 7.55 (s, 1H), 7.23 (d, J = 8.5 Hz, 1H), 3.97 (s, 3H), 2.10-2.05 (m, 1H), 1.10-1.06 (m, 2H), 0.86-0.83 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 167.5, 144.7, 135.9, 131.0, 131.0, 129.5, 127.5, 126.5, 125.6, 125.4, 123.7, 52.3, 16.0, 9.9; FT-IR (neat) 1706, 1292, 1209 cm−1; HRMS (ESI) m/z calcd. for C15H14O2 (M+H)+ 227.1072, found 227.1075.
6-Cyclopropyl-2-naphthonitrile (2j)
Following procedure A, the reaction was carried out with 6-cyanonaphthalen-2-yl methanesulfonate (61.8 mg, 0.25 mmol), Pd(OAc)2 (2.8 mg, 12 µmol) and RuPhos (11.7 mg, 25.0 µmol) to obtain 2j (28.0 mg, 58%) as a white solid after preparative silica gel chromatography (elution with hexanes/EtOAc 90:10). mp: 103–104 °C; 1H NMR (500 MHz, CDCl3) δ 8.15 (s, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.76 (d, J = 8.6 Hz, 1H), 7.56-7.54 (m, 2H), 7.28 (dd, J = 8.6, 1.7 Hz, 1H), 2.11-2.05 (m, 1H), 1.12-1.10 (m, 2H), 0.86-0.85 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 145.6, 134.8, 133.8, 130.5, 128.3, 128.3, 126.5, 126.2, 123.7, 119.4, 108.0, 15.8, 9.9; FT-IR (neat) 2226, 1626 cm−1; HRMS (ESI) m/z calcd. for C14H12N (M+H)+ 194.0970, found 194.0973.
Procedure B
6-Cyclopropylquinoline (3a)
is used as an example. A Biotage microwave vial was charged with PdCl2(COD) (1.4 mg, 5.0 µmol), RuPhos (4.7 mg, 10 µmol), quinolin-6-yl methanesulfonate (55.8 mg, 0.25 mmol), cyclopropyltrifluoroborate (47.2 mg, 0.33 mmol) and K3PO4 (382 mg, 1.80 mmol). The test tube was sealed with a cap lined with a disposable Teflon septum and evacuated under vacuum and purged with argon three times. A mixture of t-BuOH/H2O (1.25 mL/1.25 mL) was added under argon. The reaction mixture was heated to 110 °C for 16 h before cooling to rt. The reaction mixture was extracted with EtOAc (3 × 2 mL) and then dried (MgSO4). The solvent was removed in vacuo, and the crude product was purified by preparative silica gel chromatography (elution with hexanes/ EtOAc 80:20) to yield 3a in 94% yield (39.6 mg) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.81 (s, 1H), 8.03 (d, J = 8.1 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.46 (s, 1H), 7.40 (d, J = 8.8 Hz, 1H), 7.33-7.31 (m, 1H), 2.08-2.05 (m, 1H), 1.06-1.04 (m, 2H), 0.81-0.80 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 149.3, 147.0, 142.3, 135.2, 129.2, 128.2, 128.1, 123.3, 121.0, 15.4, 9.4; FT-IR (neat) 1592, 1499 cm−1; HRMS (ESI) m/z calcd. for C12H12N (M+H)+ 170.0970, found 170.0975.
5-Cyclopropyl-2-methylbenzo[d]thiazole (3b)
Following procedure B, the reaction was carried out with 2-methylbenzo[d]thiazol-5-yl methanesulfonate (60.8 mg, 0.25 mmol) to obtain 3b (40.3 mg, 85%) as a yellow oil after silica gel chromatography (elution with hexanes/EtOAc 95:5). 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 1.5 Hz, 1H), 7.09 (dd, J = 8.3, 1.5 Hz, 1H), 2.80 (s, 3H), 2.04-1.99 (m, 1H), 1.02-0.98 (m, 2H), 0.77-0.73 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 167.0, 153.8, 142.3, 132.4, 123.3, 120.8, 118.9, 20.0, 15.3, 9.4; FT-IR (neat) 1525 cm−1; HRMS (ESI) m/z calcd. for C11H12NS (M+H)+ 190.0690, found 190.0695.
4-Cyclopropyldibenzo[b,d]thiophene (3c)
Following procedure B, the reaction was carried out with dibenzo[b,d]furan-4-yl methanesulfonate (69.5 mg, 0.25 mmol) to obtain 3c (49.7 mg, 89%) as a colorless oil after silica gel chromatography (elution with hexanes/EtOAc 95:5). 1H NMR (500 MHz, CDCl3) δ 8.17-8.14 (m, 1H), 8.00 (d, J = 7.7 Hz, 1H), 7.91-7.89 (m, 1H), 7.48-7.45 (m, 2H), 7.42-7.39 (m, 1H), 7.13 (d, J = 7.3 Hz, 1H), 2.18-2.12 (m, 1H), 1.12-1.08 (m, 2H), 0.89-0.86 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 140.8, 139.6, 137.7, 136.3, 135.5, 126.7, 124.9, 124.4, 123.2, 122.9, 121.9, 119.3, 15.0, 7.3; FT-IR (neat) 1442 cm−1; HRMS (ESI) m/z calcd. for C15H12S (M)+ 224.0660, found 224.0662.
8-Cyclopropylquinoline (3d)
Following procedure B, the reaction was carried out with quinolin-8-yl methanesulfonate (55.8 mg, 0.25 mmol) to obtain 3d (19.3 mg, 46%) as a red oil after preparative silica gel chromatography (elution with hexanes/EtOAc 80:20). 1H NMR (500 MHz, CDCl3) δ 8.99 (dd, J = 3.9, 1.4 Hz, 1H), 8.15 (dd, J = 8.2, 1.4 Hz, 1H), 7.63 (d, J = 8.2 Hz, 1H), 7.47-7.40 (m, 2H), 7.20 (d, J = 7.0 Hz, 1H), 3.22-3.16 (m, 1H), 1.20-1.18 (m, 2H), 0.87-0.85 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 149.5, 147.7, 142.7, 136.6, 128.4, 126.5, 125.2, 123.2, 121.1, 10.7, 9.6; FT-IR (neat) 1498 cm−1; HRMS (ESI) m/z calcd. for C12H12N (M+H)+ 170.0970, found 170.0977.
4-Cyclopropyldibenzo[b,d]furan (3e)
Following procedure B, the reaction was carried out with dibenzo[b,d]furan-4-yl methanesulfonate (65.5 mg, 0.25 mmol), PdCl2(COD) (3.6 mg, 12 µmol) and RuPhos (11.8 mg, 25.0 µmol) to obtain 3e (47.0 mg, 72%) as a colorless oil after preparative silica gel chromatography (elution with pentane). 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 7.7 Hz, 1H), 7.76 (dd, J = 7.7, 0.9 Hz, 1H), 7.62 (d, J = 8.3 Hz, 1H), 7.49-7.47 (m, 1H), 7.37-7.35 (m, 1H), 7.28-7.25 (m, 1H), 7.04 (d, J = 7.7 Hz, 1H), 2.48-2.42 (m, 1H), 1.15-1.11 (m, 2H), 1.01-0.98 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 156.2, 155.3, 128.2, 127.1, 124.7, 123.8, 123.1, 123.0, 122.7, 120.9, 117.6, 111.8, 10.3, 8.2; FT-IR (neat) 1450, 1186, 1068 cm−1; HRMS (ESI) m/z calcd. for C15H12O (M)+ 208.0888, found 208.0886.
4-Cyclopropylbenzo[b]thiophene (3f)
Following procedure B, the reaction was carried out with benzo[b]thiophen-4-yl methanesulfonate (57.0 mg, 0.25 mmol) to obtain 3f (40.3 mg, 93%) as a yellow oil after silica gel chromatography (elution with pentane). 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 8.1 Hz, 1H), 7.67 (dd, J = 5.6, 0.9 Hz, 1H), 7.47 (d, J = 5.6 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 7.01 (d, J = 7.5 Hz, 1H), 2.35-2.29 (m, 1H), 1.07-1.04 (m, 2H), 0.83-0.80 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 140.0, 139.7, 138.6, 125.9, 124.5, 122.3, 120.5, 120.1, 13.8, 7.5; FT-IR (neat) 1449, 1408 cm−1; HRMS (ESI) m/z calcd. for C11H11S (M+H)+ 175.0581, found 175.0579.
Supplementary Material
TABLE 3.
Scope of Heteroaryl Mesylates
 | |||
|---|---|---|---|
| entry | HetAr-OMs | yield (%) | |
| 1 | 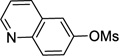 |
3a | 94 |
| 2 | 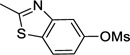 |
3b | 85 |
| 3 | 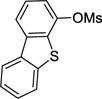 |
3c | 89 |
| 4 | 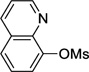 |
3d | 46 |
| 5 | 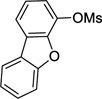 |
3e | 72a |
| 6 | 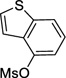 |
3f | 93 |
5 mol % of PdCl2(COD) 10 mol % of RuPhos
Acknowledgments
This research was supported by a National Priorities Research Program (NPRP) grant from the Qatar National Research Fund (Grant No. 08-035-1-008) and the NIGMS (R01GM-035249). We acknowledge Johnson Matthey for its donation of Pd(OAc)2 and PdCl2(COD), and AllyChem USA, Inc. and BASF for gifts of cyclopropylboronic acid. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data.
Footnotes
Supporting Information: 1H NMR and 13C NMR spectral data for compounds 2a–j and 3a–f. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Patai S, Rappoport Z, editors. The Chemistry of the Cyclopropyl Group. New York: John Wiley & Sons; 1987. [Google Scholar]
- 2.Wessjohann LA, Brandt W, Thiemann T. Chem. Rev. 2003;103:1625–1647. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- 3.de Meijere A, Kozhushkov SI. Mendeleev Commun. 2010;20:301–311. [Google Scholar]
- 4.de Meijere A. Angew. Chem. Int. Ed. 1979;18:809–826. [Google Scholar]
- 5.Rubin M, Rubina M, Gevorgyan V. Chem. Rev. 2007;107:3117–3179. doi: 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]
- 6.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483. [Google Scholar]
- 7.Suzuki A. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Weinheim: Wiley-VCH; 1998. [Google Scholar]
- 8.Suzuki A. J. Organomet. Chem. 1999;576:147–168. [Google Scholar]
- 9.Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou SM, Deng MZ, Xia LJ, Tang MH. Angew. Chem. Int. Ed. 1998;37:2845–2847. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2845::AID-ANIE2845>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 11.Wallace DJ, Chen CY. Tetrahedron Lett. 2002;43:6987–6990. [Google Scholar]
- 12.Lemhadri M, Doucet H, Santelli M. Synth. Commun. 2006;36:121–128. [Google Scholar]
- 13.Yao ML, Deng MZ. Synthesis. 2000:1095–1100. [Google Scholar]
- 14.Yao ML, Deng MZ. New J. Chem. 2000;24:425–428. [Google Scholar]
- 15.Imbriglio JE, Chang S, Liang R, Raghavan S, Schmidt D, Smenton A, Tria S, Schrader TO, Jung JK, Esser C, Taggart AKP, Cheng K, Carballo-Jane E, Waters MG, Tata JR, Colletti SL. Bioorg. Med. Chem. Lett. 2009;19:2121–2124. doi: 10.1016/j.bmcl.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Whelligan DK, Solanki S, Taylor D, Thomson DW, Cheung KMJ, Boxall K, Mas-Droux C, Barillari C, Burns S, Grummitt CG, Collins I, van Montfort RLM, Aherne GW, Bayliss R, Hoelder S. J. Med. Chem. 2010;53:7682–7698. doi: 10.1021/jm1008727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Xi, Dragoli DR, Fan P, Gleason MM, Jaen JC, Li L, Mcmahon JP, Powers J, Zeng Y, Zhang P, Fan J. 2010 US100311712. [Google Scholar]
- 18.Heald R, Jackson P, Lyssikatos JP, Price S, Savy PP. 2010 WO2010003025. [Google Scholar]
- 19.Schmitz FU, Rai R, Roberts CD, Kazmierski W, Grimes R. 2010 WO2010062821. [Google Scholar]
- 20.Gibbons P, Hanan E, Liu W, Lyssikatos JP, Magnuson SR, Mendonca R, Pastor R, Rawson TE, Siu M, Zak ME, Zhou A, Zhu B-Y. 2011 WO2011003065. [Google Scholar]
- 21.Taniguchi T, Kawada A, Kondo M, Quinn JF, Kunitomo J, Yoshikawa M, Fushimi M. 2010 US100197651. [Google Scholar]
- 22.Pracitto R, Kadow JF, Bender JA, Beno BR, Grant-young KA, Han Y, Hewawasam P, Nickel A, Parcella KE, Yeung K-S, Chupak LS. 2010 US100184800. [Google Scholar]
- 23.Pracitto R, Kadow JF, Bender JA, Beno BR, Grant-young KA, Han Y, Hewawasam P, Nickel A, Parcella KE, Yeung K-S, Chupak LS. 2010 US100063068. [Google Scholar]
- 24.Bhayana B, Fors BP, Buchwald SL. Org. Lett. 2009;11:3954–3957. doi: 10.1021/ol9015892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng MZ, Yao M-L. Tetrahedron Lett. 2000;41:9083–9087. [Google Scholar]
- 26.Charette AB, De Freitas-Gil RP. Tetrahedron Lett. 1997;38:2809–2812. [Google Scholar]
- 27.Knapp DM, Gillis EP, Burke MD. J. Am. Chem. Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49–56. [Google Scholar]
- 29.Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 30.Stefani HA, Cella R, Adriano S. Tetrahedron. 2007;63:3623–3658. [Google Scholar]
- 31.Darses S, Genet J-P. Chem. Rev. 2008;108:288–325. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- 32.Butters M, Harvey JN, Jover J, Lennox AJJ, Lloyd-Jones GC, Murray PM. Angew. Chem., Int. Ed. 2010;49:5156–5160. doi: 10.1002/anie.201001522. [DOI] [PubMed] [Google Scholar]
- 33.Fang GH, Yan ZJ, Deng MZ. Org. Lett. 2004;6:357–360. doi: 10.1021/ol036184e. [DOI] [PubMed] [Google Scholar]
- 34.Charette AB, Mathieu S, Fournier JF. Synlett. 2005:1779–1782. [Google Scholar]
- 35.Molander GA, Gormisky PE. J. Org. Chem. 2008;73:7481–7485. doi: 10.1021/jo801269m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasnik Z, Pohl R, Hocek M. Synthesis. 2009:1309–1317. [Google Scholar]
- 37.Kuroda JI, Inamoto K, Hiroya K, Doi T. Eur. J. Org. Chem. 2009:2251–2261. [Google Scholar]
- 38.Chow WK, So CM, Lau CP, Kwong FY. J. Org.Chem. 2010;75:5109–5112. doi: 10.1021/jo100846t. [DOI] [PubMed] [Google Scholar]
- 39.Molander GA, Beaumard F. Org. Lett. 2010;12:4022–4025. doi: 10.1021/ol101592r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita A-M, Garg NK, Percec V. Chem. Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molander GA, Beaumard F. Org. Lett. 2011;13:1242–1245. doi: 10.1021/ol200128y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molander GA, Beaumard F. Org. Lett. 2011;13:3948–3951. doi: 10.1021/ol201469r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:6338–6361. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ariafard A, Hyland CJT, Canty AJ, Sharma M, Brookes NJ, Yates BF. Inorg. Chem. 2010;49:11249–11253. doi: 10.1021/ic1020912. [DOI] [PubMed] [Google Scholar]
- 45.Heydenrych G, von Hopffgarten M, Stander E, Schuster O, Raubenheimer HG, Frenking G. Eur. J. Inorg. Chem. 2009:1892–1904. [Google Scholar]
- 46.Fairlamb IJS. Chem. Soc. Rev. 2007;36:1036–1045. doi: 10.1039/b611177g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.