

Abstract
In more than 90 percent of cancers including glioma, telomere elongation reverse transcriptase (hTERT) is overexpressed. In the present study, we sought to explore whether matrix metalloproteinase-9 (MMP-9) shRNA could alter hTERT-mediated proliferation in glioma cells. MMP-9 shRNA induced senescence and apoptosis in glioma cells by inhibiting hTERT expression and telomere activity. MMP-9 silencing decreased oncogenic c-Myc expression (hTERT activator), whereas the expression of the c-Myc antagonist MAD increased drastically (hTERT repressor); both c-Myc and MAD are transcription factors for hTERT. In addition, MMP-9 suppression turns the switch from c-Myc/MAX to MAD/MAX heterodimer binding to the hTERT promoter as determined by chromatin immunoprecipitation assay. We also show that silencing MAD via siRNA restored hTERT expression and inhibited senescence in glioma cells. MMP-9 transcriptional suppression decreased the expression of FAK, phosphor FAK and β1 integrin in glioma xenograft cells. Further, MMP-9 suppression decreased the interaction of β1 integrin/FAK and also MMP-9/β1 integrin as confirmed by immunoprecipitation analysis. Studies with either function blocking β1 integrin or FAK shRNA indicate that suppression of MMP-9 decreased β1 integrin-mediated induction of FAK, which led to decreased hTERT expression. Moreover, 4910 and 5310 glioma xenograft tissue sections from mice treated with MMP-9 shRNA showed reduced expression of FAK/c-Myc and elevated MAD levels. Decreased co-localization of β1 integrin and MMP-9 was associated with MMP-9-suppressed tumor sections. Further, immunoprecipitation analysis showed decreased association of proteins involved in telomere end repair in MMP-9 shRNA-treated glioma cells. Elevated levels of p73 and TRAIL and the results of the FACS analysis show induction of apoptosis in MMP-9-silenced glioma cells. Taken together, these data provide new insights into the mechanisms underlying MMP-9-mediated hTERT expression in glioma proliferation.
Keywords: MMP-9, shRNA, hTERT, c-Myc/MAX/MAD, FAK, β1 integrin, senescence, apoptosis, glioma
1. Introduction
Tumor cell proliferation requires active telomerase activity with every cell division and to attain immortalization. Telomerase is a ribonucleic-acid protein involved in adding telomere repeats TTAGGG to linear chromosomes to maintain its stability. Telomerase is composed of an internal telomerase RNA template and the enzyme human telomerase elongation reverse transcriptase (hTERT). hTERT gene was overexpressed in more than 90 percent of cancer cells, thereby contributing to cancer cell proliferation.
The c-Myc/MAX/MAD network of transcription factors are implicated in cell growth, cell division and cell differentiation [1]. Expression of c-Myc proteins are involved in promoting cell proliferation, whereas MAD acts as a c-Myc antagonist by inhibiting proliferation [1,2]. It is also demonstrated that the high binding efficiency of c-Myc//MAX and MAD/MAX heterodimer to an E box element (CACGTG) acts as positive or negative switch for cell proliferation, senescence and apoptosis. hTERT and cyclin D2 are a few of the target genes regulated by this network [2]. Recent studies show that c-Myc/MAX or MAD/MAX binding to the hTERT promoter E-box regulates its transcriptional expression or repression [3]. Grade IV gliomas show close to 100% telomerase activity compared to other lower grades of gliomas, and normal brain cells show no activity.
Matrix metalloproteinase-9 (MMP-9) is a proteolytic enzyme that is overexpressed in most cancers as compared to normal tissue. It regulates the tumor microenvironment and has been shown to degrade structural components of the extracellular matrix such as integrins. MMP-9 regulates various cancer cell characteristics such as cell growth, differentiation, invasion, migration and apoptosis. Earlier studies and studies from our laboratory have shown that downregulation of MMP-9 induced apoptosis and inhibited cancer cell migration, invasion and proliferation by regulating the expression of MMP-9 and affecting the downstream cellular signaling of integrins [4,5]. In an earlier study, we demonstrated that MMP-9 shRNA in combination with uPAR/cathepsin B shRNA reduced proliferation by inducing apoptosis in glioma xenograft cells with accumulated DNA damage (Ponnala et al., under review).
hTERT is a catalytic subunit of telomerase and processes telomere ends, which are a form of DSBs [6,7]. Lack of or insufficient telomerase and its activity have been associated with an inability to process telomere ends, which contributes to genetic instability in the form of DNA damage and induces senescence [8,9]. Previously, we have shown that MMP-9 downregulation induced senescence and apoptosis in medulloblastoma [10]. Here, in the present study, we show that MMP-9 silencing induces senescence and apoptosis in glioma xenograft cells via β1 integrin-mediated, FAK-induced hTERT expression.
2. Materials and Methods
2.1 Cell culture conditions
Glioblastoma xenograft cells (4910 and 5310) were kindly provided by Dr. David James (University of California at San Francisco). Cells were generated and maintained in mice and were highly invasive in the mice brain [11]. At 3 to 4 passages of xenograft cells from mice, heterotrophic tumors were frozen. These frozen stocks were used for further experimental studies up to the 10th passage to obtain consistent results. Xenograft cells were maintained in RPMI 1640 buffer supplemented with 10% FBS (Invitrogen Corporation, Carlsbad, CA), 50 units/mL penicillin, and 50 μg/mL streptomycin (Life Technologies, Inc., Frederick, MD). Cell lines were maintained in a 37°C incubator with a 5% CO2 humidified atmosphere. We used the following antibodies from Santa Cruz Biotechnology (Santa Cruz, CA): hTERT, c-Myc, MAD, MAX, Ku80, ATM, POT1, EGFR, p73, p16, p18, FAK, phospho FAK (Tyr 397), IgG and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Antibody for TRAIL was purchased from Cell Signaling (Danvers, MA). We used species-specific secondary antibodies conjugated to HRP, Alexa Fluor 488, and Alexa Fluor 595 (Invitrogen, Carlsbad, CA) in this study. All transfections were carried out using FuGene HD transfection reagent according to the manufacturer’s instructions (Roche Applied Science, Madison, WI).
2.2 Construction of shRNA-expressing plasmids and transfection conditions
Plasmid constructs containing shRNA against MMP-9 (pMMP-9), FAK (pFAK) and scrambled vector (pSV) with an imperfect sequence were designed in our laboratory [12,13]. Xenograft cells at 70–80% confluence were transfected with plasmids expressing shRNA against MMP-9, FAK or SV for 72 hours. pDNR-CMV vector expressing full length MMP-9 (M-fl) plasmid was designed and constructed in our laboratory. MAD siRNA was purchased from Santa Cruz Biotechnology, and transfection was carried out according to the manufacturer’s instructions. For the inhibitor study, cells seeded in six-well plates were treated with MAD siRNA according to the manufacturer’s instructions (Santa Cruz Biotechnology, Santa Cruz, CA).
2.3 Cell proliferation and cell cycle analysis
BrdU incorporation assay was performed (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions. Clonogenic assay was carried out by seeding 500 cells in 100-mm plates after the transfection as described above, and survival fraction was calculated based on the colony forming ability of cells after two weeks. Fluorescence activate cell sorting (FACS) analysis was done 72 hours after transfection as described previously [14]. FACS analysis was carried out on at least 10,000 cells from each sample, and cell cycle data were analyzed using a FACS Calibur flow cytometer (BD BioSciences, San Jose, CA).
2.4 Immunoblotting and immunoprecipitation assay
For Western blot analysis, equal amounts of protein fraction were resolved over SDS-PAGE and immunoblotted with primary antibody followed by HRP-conjugated secondary antibodies. Signals were detected using the ECL Western blotting detection system (Pierce, Rockford, IL). Immunoprecipitation assays were carried out by incubating a minimum of 100–500 μg total cell lysate with antibody overnight at 4°C on a rotating shaker. Protein A/G agarose beads (Miltenyi Biotec, Auburn, CA) were added to the above complex and incubated on ice for 1 hour. Immunoprecipitates were eluted using μMac columns according to the manufacturer’s instructions and were immunoblotted with appropriate primary and secondary antibodies.
2.5 TRAP assay
Telomerase activity was measured in MMP-9-transfected and control glioma cells using the PCR-based telomere repeat amplification protocol according to the manufacturer’s instructions (TRAPeze kit, Chemicon, Temecula, CA). Briefly, the cell extracts of treated and untreated samples were washed in PBS. The cell pellet (105 cells) was collected and lyzed in 200 μL CHAPS lysis buffer. The suspension was mixed and incubated on ice for 30 min and then centrifuged at 14,000 rpm for 30 min. The resulting supernatant was aliquoted and stored at −80°C. The TS primer in the kit served as a telomerase substrate. The cell extract from an individual sample was incubated at 37°C with TRAP reaction mixture for 30 min and followed by a three-step PCR (94°C for 30 sec, 50°C for 30 sec, and 72°C for 45 sec; total of 35 cycles). The PCR products were analyzed by 15% non-denaturing PAGE. The gels were stained with ethidium bromide and visualized with a UV trans-illuminator. Telomerase activities were quantified by comparing the mean band intensity of each lane and with the control. The control mean band intensity was defined as 100% telomerase positive. Appropriate positive and negative heat-inactivated cell extracts were set up with test samples. Each experiment was carried out in triplicate.
2.6 SA-β-gal staining
Cells were seeded in six-well plates, transfected with pMMP-9, and SA-β-gal staining was performed according to the manufacturer’s instructions (Cell Signaling, Danvers, MA). Control and transfected cells were washed twice with PBS and fixed for 10 min at room temperature in 37% formaldehyde. After washing, the cells were incubated at 37°C (without CO2) overnight with freshly prepared 1 mg/mL concentration of β-gal stain. For each sample, at least 200 cells were counted under the microscope. Each experiment was performed in triplicate.
2.7 Reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from transfected cells using TRIZOL reagent (Invitrogen, CA) following standard protocol. RNA (1 μg) was used as a template for reverse transcription reaction (Roche Applied Science, Indianapolis, IN) and followed by PCR analysis. We used the following sequences for the forward and reverse primers:
| Gene product (F: forward) | Primer sequence 5′ to 3′ | Gene product (R: reverse) | Primer sequence 5′ to 3′ |
| hTERT F | gtgaccgtggtttctgtgtg | hTERT R | tcgcctgaggagtagaggaa |
| Max F | gaacgaaaacgtagggacca | Max R | cttgctggtgtgtgtggttt |
| Madl1 F | tgtcagcagaacttggatgc | Madl1 R | tttttgtgttgcaggtccag |
| c-MYC F | cagatcagcaacaaccgaaa | c-MYC R | ggccttttcattgttttcca |
| TINF2 F | ctgagcccatggaacagaat | TINF2 R | tccttatggcctcccctagt |
| RAP1 F | gtctcactgcaccttcaatggca | RAP1 R | tggccctgctctttgccaact |
| TNKS2 F | caaatgggctttcacacctt | TNKS2 R | gcttctcacaccattgagca |
| POT1 F | ccttacgtgtttgggcatct | POT1 R | tttgtagccgatggatgtga |
| STAU1 F | tcctctcagccacctctgat | STAU1 R | ctcccacacacagacattgg |
| TEP1 F | cccaagtccctgaactgtgt | TEP1 R | acattgaaggccaaggtacg |
| DKC1 F | atgtgcttgatgctcagtgg | DKC1 R | gcagattgcttctcctttgg |
| TRF1 F | ggcagcggcaaaagtagtag | TRF1 R | gtcttgttgctgggttccat |
| TRF2 F | gtacccaaaggcaagtggaa | TRF2 R | tgacccactcgctttcttct |
| Ku80 F | tgacttcctggatgcactaatcgt | Ku80 R | ttggagccaatggtcagtcg |
| GAPDH F | agccacatcgctcagacacc | GAPDH R | gtactcagcggccagcatcg |
Reverse transcriptase PCR was set up using the following PCR cycle: 95°C for 5 min, (95°C for 30 sec, 55–60°C for 30 sec, and 72°C for 30 sec) × 30 cycles, and 72°C for 10 min. PCR products were resolved on a 1.6% agarose gel, visualized, and photographed under UV light. All reactions were performed in triplicate.
2.8 Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed according to the manufacturer’s instructions (Roche Applied Science, Madison, WI). In brief, cells (~2×106 cells/100-mm dish) were fixed by adding 20 mL of minimal essential medium with 0.54 μL of 37% formaldehyde prepared in 15% methanol. Then, cells were incubated for 10 min at room temperature (RT) on an end-to-end rotor. The cells were washed once with ice-cold PBS. After preparing the lysates, 4 μg of specific antibodies (c-Myc and MAD; Santa Cruz Biotechnology, Santa Cruz, CA) were used to immunoprecipitate the protein-DNA complexes. Antibody controls were also included for each ChIP assay; no precipitation was observed. The antibody/protein complexes were collected using magnetic beads and processed further according to the manufacturer’s instructions. The samples were treated with proteinase K for 1 hour, and the DNA was purified by phenol/chloroform extraction and precipitated using ethanol. The recovered DNA was resuspended in elution buffer and used as templates for PCR of TERT or β-actin promoters. The following primers were used for PCR: hTERT promoter-sense, 5′-AGCCCCTCCCCTTCCTTT-3′, and hTERT promoter-antisense, 5′-AGCACCTCGCGGTAGTGG-3′; β-actin promoter-sense, 5′-CCAACGCCAAAACTCTCCC-3′, and β-actin promoter-antisense, 5′-AGCCATAAAAGGCAACTTTCG-3′. Initially PCR was performed with different numbers of cycles or dilutions of input DNA to determine the linear range of the amplification; all results shown fall within this range. Following 30 cycles of amplification, PCR products were run on 2% agarose gels and analyzed by ethidium bromide staining.
2.9 Immunofluorescence
Cells grown in two-well chamber slides were washed with PBS, fixed with ice-cold methanol, permeabilized with 0.3% triton X, and blocked with 2% BSA in PBS. Cells were incubated with primary antibodies for either 2 hours at room temperature or overnight at 4°C, washed with PBS, and incubated with Alexa Fluor® conjugated secondary antibodies for 1 hour at room temperature. Nuclei were counterstained with DAPI and analyzed under a microscope (Olympus BX61 Fluoview, Minneapolis, MN).
2.10 Gelatin zymography
MMP-9 activity in the conditioned medium was determined by gelatin zymography. 4910 and 5310 human glioma xenograft cells were transfected with pSV, pMMP-9 and pFAK for 72 hours. Cells were washed and incubated in serum-free medium overnight. Equal amounts of protein were electrophoresed in 10% SDS-polyacrylamide gels containing 1.5 mg/mL gelatin. The gels were washed and gently shaken in three consecutive washings in 2.5% Triton X-100 solution to remove SDS. The gels were then incubated at 37°C overnight in incubation buffer [50 mmol/L Tris-HCl (pH 7.5), 0.05% NaN3, 5 mmol/L CaCl2 and 1 μmol/L ZnCl2]. Next, the gels were stained with 0.1% amido black in 10% acetic acid and 10% isopropanol and subsequently destained for 1 hour. Gelatinolytic activities were identified as clear zones of lysis against a dark background.
2.11 DNA extraction and DNA fragmentation assay
DNA was extracted from all the treated and untreated glioma cells as described earlier [15]. 1 μg of genomic DNA was loaded onto 0.8% agarose gel for analysis.
2.12 Immunohistochemistry
The 4910 and 5310 glioma xenograft cells were injected intracerebrally into nude mice with a 10 μl aliquot (0.2×105 cells/μL) under isofluorane anesthesia with the aid of a stereotactic frame. Tumors were allowed to grow for 10 to 12 days, and the animals were separated into groups (six animals per group). Alzet mini pumps were implanted for pMMP-9 delivery at the rate of 0.2 μL/h. The concentration of the plasmid solution was 2 μg/μL (100 μl per mouse, six mice in each group). After five weeks or when the control animals started showing symptoms the mice were sacrificed by intracardiac perfusion, first with PBS and then with 4% paraformaldehyde in normal saline. The brains were collected, stored in 4% paraformaldehyde, processed, embedded in paraffin, and sectioned (5 μm thick) using a microtome. Paraffin embedded sections were stained with hematoxylin and eosin to visualize tumor cells and to examine tumor volume.
Paraffin-embedded brain sections (5 μm thick) from control and treatment groups were deparaffinized following standard protocol. Antigen retrieval was performed by treating the section with citrate buffer at 95° for 15–20 minutes followed by hydrogen peroxide treatment for 30 min. The sections were rinsed with PBS and blocked with 1% BSA in PBS to prevent non-specific staining and further incubated overnight with primary antibodies (1:50 dilution) at 4°C. The sections were then incubated with HRP-conjugated secondary antibodies for 1 hour at room temperature followed by incubation with DAB for 30 min. The sections were counterstained with hematoxylin to visualize the nucleus, mounted and observed under a light microscope. Negative controls were maintained using IgG.
2.13 Densitometry
ImageJ software (National Institutes of Health) was used to quantify the band intensity. Data represent intensities relative to the indicated loading control.
2.14 Statistical analysis
All data are presented as mean ± standard deviation (SD) of at least three independent experiments. Statistical comparisons were performed using Graph Pad Prism software (version 3.02). Bonferroni’s post hoc test (multiple comparison tests) was used to compare any statistical significance between groups. Differences in the values were considered significant at p<0.05.
3. Results
3.1 MMP-9 silencing regulates hTERT expression and activity
MMP-9 is known to play a role in senescence induction [10]. GBM often express 100% of telomerase activity, a key factor in cancer cell immortalization. Telomerase function prevents progressive loss of chromosomal ends with every cell division. We examined the effect of MMP-9 shRNA on glioma cell-induced telomerase expression and activity. Figure 1 shows that downregulation of MMP-9 in 4910 and 5310 glioma xenograft cells significantly lowered the expression of hTERT at both the mRNA and protein levels when compared to control cells (Figures 1A and 1B). Telomerase activity as assessed by TRAPeze kit showed markedly decreased telomere repeat amplification in pMMP-9-transfected 4910 and 5310 glioma cells as compared to pSV and controls (Figures 1C and 1D). SA-β-gal stain indicated that number of senescent cells was high in pMMP-9-treated 4910 (43%) and 5310 (45%) cells as compared to untreated cells (<5%) (p<0.05) (Figure 1E and 1F).
Figure 1. Effect of MMP-9 on the expression of hTERT expression and its activity.

A. RT-PCR and Western blot analysis shows effect of pMMP-9 transfection on hTERT expression in 4910 and 5310 glioma xenograft cells. B. Further, quantification of the Western blots using software to determine levels of protein after pMMP-9 treatments is shown. Values shown are the mean (±SD). *p<0.05 vs. control. (n = 3). C and D. TRAP assay was performed to evaluate the effect of pMMP-9 treatment on the activity of telomerase in both glioma cell types. 36bp band shows internal control. Bar diagram showing densitometry quantified data of TRAP product in single lane/positive control (PC) of TRAP reaction ratios from three independent experiments. IC - Internal control for TRAP reaction. Each bar represents triplicate analyses of mean ± SD. *p<0.05 vs. control. E. Senescence associated beta gal (SAβgal) staining shows effect of pMMP-9 transfection on 4910 and 5310 glioma cells in induction of senescence. Percent SAβgal positivity was calculated by scoring 200 cells from three different fields from single treatment. Mean was obtained from three independent experiments and values shown are the mean (±SD). *p<0.05 vs. control. (n=3).
3.2 pMMP-9 transfection retards glioma proliferation
In the present study, BrdU incorporation assay and survival fraction showed significant decreases in the proliferation of 4910 and 5310 glioma cells treated with pMMP-9 as compared to control and pSV cells. Cell proliferation by BrdU assay showed 35–45% reduced proliferation in pMMP-9-treated 4910 and 5310 glioma cells as compared to control and pSV (Supplementary Figure 1A). Survival fraction from a clonogenic assay showed a 70–80% decrease in colony forming units two weeks after pMMP-9 treatment as compared to controls and pSV treatment (Supplementary Figure 1B).
3.3 MMP-9 regulates hTERT expression via the c-Myc/MAX/MAD network of transcription factors
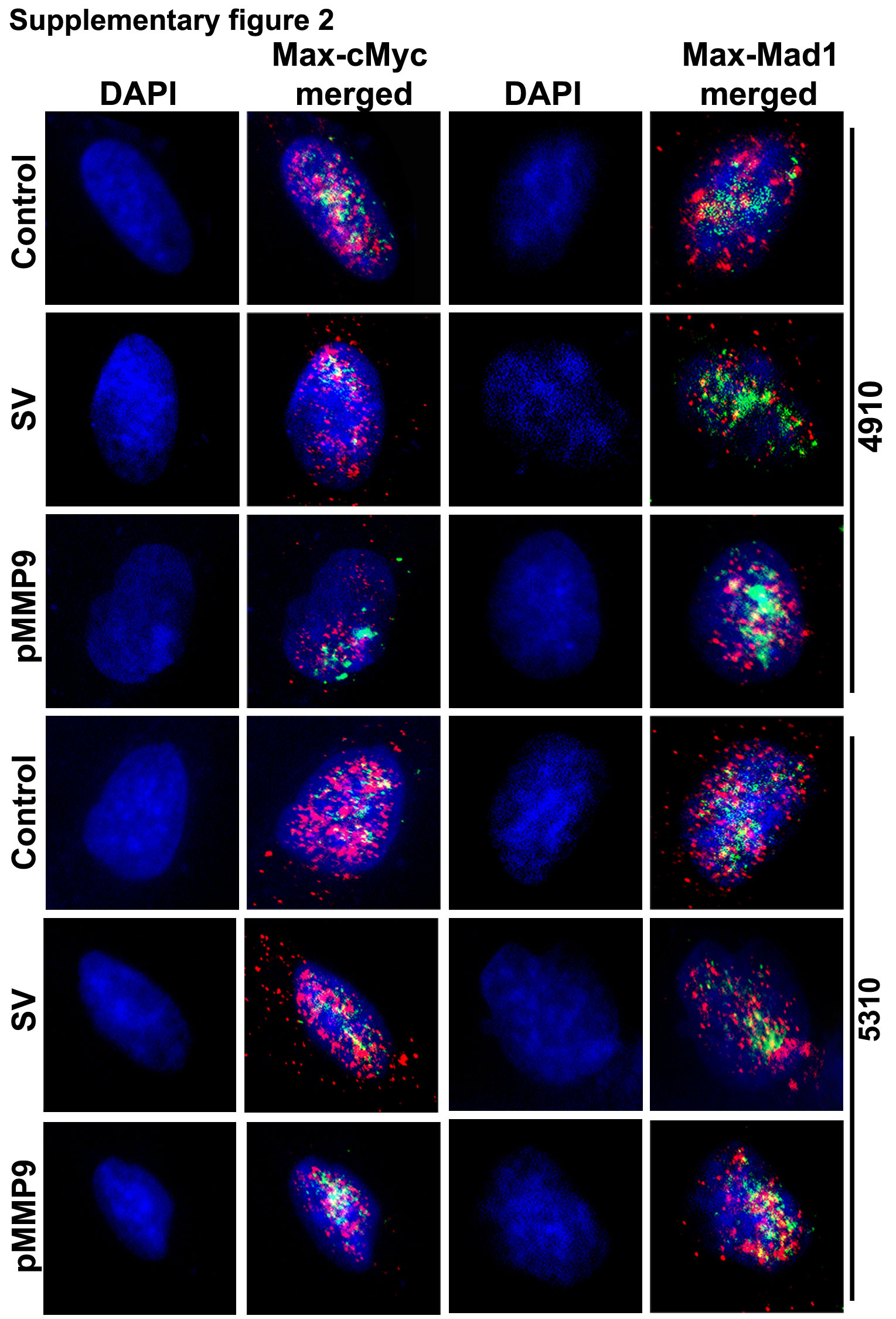
The c-Myc/MAX/MAD network has been shown to regulate the expression of hTERT. c-Myc/MAX and MAD/MAX heterodimers bind to E-boxes of the hTERT promoter to either activate or repress its transcription, respectively. In the present study, pMMP-9-transfected glioma cells altered the expression of c-Myc and MAD at both the mRNA and protein levels as compared to control and pSV (Figure 2A). 4910 and 5310 glioma cells treated with pMMP-9 showed significant decreases in oncogenic c-Myc levels and increases in the expression of MAD protein levels as compared to control and pSV-treated cells (Figure 2B). However, no noticeable difference in the expression of MAX was observed in both glioma cell types after pMMP-9 treatment as compared to control and pSV-treated cells (Figure 2A). Immunoprecipitation (IP) analysis of 4910 and 5310 glioma cells after pMMP-9 transfection was carried out to analyze the interaction of c-Myc/MAX and MAD/MAX complex interaction. Figure 2C clearly shows that IP with MAX followed by immunoblot with c-Myc and MAD antibodies showed reduced c-Myc/MAX and increased MAD/MAX heterodimer in pMMP-9-treated 4910 and 5310 glioma xenograft cells as compared to control and pSV (Figure 2C). pMMP-9 lowered the co-localization of c-Myc/MAX heterodimer whose binding to E-box of promoter acts as a gene transcription activator and increased the interaction of MAD/MAX heterodimer, which acts as a transcriptional repressor, in both 4910 and 5310 glioma cells as assessed by immunofluorescence (Supplementary Figure 2). We have analyzed the interaction between c-Myc and MAD proteins at the hTERT proximal promoter E-box with the use of the chromatin immunoprecipitation assay. The results showed lowered association of c-Myc with hTERT E-box compared to MAD binding in pMMP-9-treated glioma cells as compared to control and pSV-treated cells (Figure 2D). The ratio of c-Myc and MAD binding to hTERT promoter in pMMP-9-transfected cells compared to control and pSV showed significant differences in both 4910 and 5310 glioma cells (Figure 2E)
Figure 2. MMP-9 regulates expression of c-Myc/MAD/MAX and switch c-Myc/MAX and MAD/MAX interaction.

A. RT-PCR and Western blot analysis shows effect of pMMP-9 transfection on c-Myc, MAD and MAX expression in 4910 and 5310 glioma xenograft cells compared to control and pSV. B. Further, quantification of the Western blots using software to determine levels of protein after pMMP-9 treatments is shown. (n = 3). Values shown are the mean (±SD). *p<0.05 vs. control. C. Immunoprecipitation (IP) analysis. IP of MAX was immunoprobed with c-Myc and MAD in both 4910 and 5310 glioma xenograft cells transfected with pMMP-9 along with control and pSV cells. D. ChIP assay was performed on pMMP-9-treated 4910 and 5310 glioma cells. The formaldehyde cross linked chromatin was IP with antibodies specific for c-Myc and MAD. Purified DNA was amplified for the hTERT promoter associated proximal E-Box by using specific primers. Controls show input genomic DNA before the addition of antibody and eluants from no antibody immunoprecipitations. E. Quantification of the c-Myc and MAD ChIP fractions on the hTERT promoter in both 4910 and 5310 glioma cells. Each bar represents triplicate analyses of mean ± SD. Significant changes are represented by an asterisk (*) (P <0.05). (n=3).
3.4 MAD silencing activates telomerase activity
To further confirm that the MAD/MAX heterodimer was responsible for silencing hTERT, the expression of MAD was silenced using specific MAD siRNA capable of degrading mRNA transcripts in a target-specific manner. MAD siRNA significantly reduced the expression of MAD at both the mRNA and protein levels (Figure 3A). The TRAP assay results demonstrated stimulation in telomerase activity in MAD siRNA-transfected cells as compared with pSV-transfected and control glioma cells (Figure 3B and 3C). Next, we analyzed the SA-β-gal staining in MAD siRNA-treated cells. As expected, siRNA-treated glioma cells revealed that MAD siRNA treatment resulted in a decrease in the induction of senescence in glioma cells. A similar trend was observed in 5310 glioma cells treated with MAD siRNA (data not shown). Overall, our results suggest that MAD inhibition upregulated hTERT expression, which led to a lowered percentage of senescence-positive cells.
Figure 3. Effect of MAD silencing on hTERT expression in glioma cells.

A. Western blot analysis shows MAD protein expression levels after MAD siRNA treatment in 4910 and 5310 glioma cells. B. RT-PCR analysis shows expression of hTERT after MAD silencing in 4910 and 5310 glioma cells compared to pSV and control cells. C. TRAP assay was performed to evaluate the effect of MAD treatment on the activity of telomerase in 5310 glioma cells. 36bp band shows internal control.
3.5 pMMP-9 seeks focal adhesion kinase (FAK)-mediated signaling in altering the c-Myc/MAD expression in glioma cells
ECM interaction with FAK via integrins is known to initiate a number of intracellular signaling pathways in cancer [16,17]. Therefore, we determined the effect of MMP-9 suppression on the expression of β1 integrin and FAK. As shown in Figure 4A, transfection with pMMP-9 inhibited the expression of β1 integrin, FAK and phosphor FAK (Tyr 397) at both the mRNA and protein levels in glioma cells. Co-localization of β1 integrin and FAK in pMMP-9-transfected glioma cells was markedly reduced as compared to control cells (Figure 4B). To determine the contribution of FAK signaling in pMMP-9-mediated c-Myc family of protein expression, we transfected the glioma cells with FAK shRNA. As shown in Figure 4A, transient transfection of glioma cells with FAK shRNA lowered the expression and activity of MMP-9. Further, β1 integrin expression was also reduced in FAK-silenced glioma cells (Figure 4A). Immunoprecipitation analysis showed lowered signaling of FAK mediated by p130CAS and Src in pMMP-9- and FAKsh-treated glioma cells compared to control and pSV cells (Figure 4C). We next sought to find out if FAK signaling was involved in regulating the c-Myc family of protein expression. As shown in Figure 4D, expression of c-Myc was markedly reduced whereas the expression of the c-Myc antagonist MAD was increased drastically. At least in part, these results show that the deregulation of c-Myc/MAD expression in MMP-9-suppressed glioma cells is mediated through FAK signaling.
Figure 4. MMP-9 regulates hTERT expression via FAK signaling.

A. Western blot, RT-PCR and gelatin zymography analyses show effect of FAK silencing on MMP-9 expression in 4910 and 5310 glioma xenograft cells compared to control and pSV. Western blot analysis shows MMP-9 and FAK silencing on β1 integrin. B. Immunocytochemistry analysis was carried out for co-localization of FAK and β1 integrin in pMMP-9-transfected 4910 and 5310 glioma xenograft cells along with control and pSV cells. C. Immunoprecipitation (IP) analysis. IP of FAK was immunoprobed with p130CAS and Src in 5310 glioma xenograft cells transfected with pMMP-9 and FAKsh along with control and pSV cells. D. Western blot analysis shows effect of FAKsh transfection on c-Myc, MAD and MAX expression in 4910 and 5310 glioma xenograft cells compared to control and pSV (n=3).
3.6 Transcriptional silencing of MMP-9 inhibits hTERT expression via β1 integrin in glioma cells
hTERT expression is regulated by various growth factors and the expression of transcription factors. The interaction of β1 integrin and FAK has been well established, and the signal transduction mediated by coordinated interplay between these molecules in proliferation, cell migration, invasion and apoptosis has been documented [16,17]. In the present study, we determined the interaction of MMP-9 and β1 integrin in FAK-regulated hTERT expression in glioma cells. Immunocytochemical analysis showed that the co-localization of MMP-9 and β1 integrin decreased in pMMP-9-transfected glioma cells as compared to control and pSV-transfected cells (Figure 5A). Immunoprecipitation analysis revealed significantly reduced interaction of MMP-9 and β1 integrin in both glioma cell types with pMMP-9 treatment as compared to control and pSV treatment (Figure 5B). To further evaluate the effect of MMP-9 and β1 integrin interaction on hTERT expression, we transfected glioma cells with FL-MMP-9 and carried out co-immunoprecipitation using an antibody for MMP-9. As expected, interaction of MMP-9 with β1 integrin was significantly increased with MMP-9 overexpression in control compared to pMMP-9-transfected cells (Figure 5B). FL-MMP-9 expression noticeably increased the expression of FAK, pFAK, c-Myc and hTERT in control and pMMP-9-treated cells (Figure 5C) as compared to glioma cells transfected with pMMP-9 only. Similarly, incubation of glioma cells with β1 integrin for protein neutralization prevented the FL-MMP-9-induced levels of FAK, pFAK, c-Myc and hTERT (Figure 5C).
Figure 5. MMP-9 silencing inhibits β1-integrin-mediated hTERT expression in glioma cells.

A. pMMP-9-transfected glioma cells were analyzed by immunocytochemistry for co-localization (yellow color) of MMP-9 (green color) and β1-integrin (red color) using specific antibodies. B. After 24 hours of pMMP-9 transfection, 4910 and 5310 glioma cells were double transfected with FL-MMP-9 and incubated for 24 hours. Western blot analysis shows effect of FL-MMP-9 transfection on MMP-9 expression in 4910 and 5310 glioma xenograft cells compared to control and pSV. Immnunoprecipitation was carried out by anti-MMP-9 antibody and immuprobed with β1-integrin. C. After 24 hours of pMMP-9 transfection, 4910 and 5310 glioma cells were double transfected with FL-MMP-9, non-specific IgG and/or anti-β1-integrin antibodies for another 24 hours. Western blot analysis was performed for FAK, phospho FAK (Tyr 397), hTERT and c-Myc using total cell lysates. GAPDH served as loading control. Protein band intensities were quantified by densitometric analysis using ImageJ software (National Institutes of Health). Each bar represents triplicate analyses of mean ± SD. Significant changes are represented by an asterisk (*) (P <0.05). (n=3).
3.7 MMP-9 depletion retards telomere end processing and induces apoptosis in glioma cells
Telomerase complex-associated proteins play important roles in regulating telomerase activity. In the present study, we investigated the expression of various proteins involved in telomere end processing using semi-quantitative RT-PCR. Our results clearly show that pMMP-9 altered the expression of the telomere end processing complex (Figure 6A). We further sought to determine the interaction of these proteins in repairing telomere ends. Our results demonstrate that pMMP-9-transfected cells have decreased interaction of ATM, Ku80 and POT1 as compared to control and pSV-transfected cells (Figure 6B). Moreover, downregulation of MMP-9 in 4910 and 5310 glioma cells resulted in increased expression of TRAIL and p73 proteins associated with apoptosis, and p16 and p18, which are associated with cell cycle arrest (Figure 6C [I], [II]). EGFR, a key survival signaling growth receptor, was downregulated in pMMP-9-treated 4910 and 5310 glioma cells as compared to control and pSV-treated cells (Figure 6C [III]). Induction of apoptosis was confirmed by FACS analysis. A characteristic ladder pattern indicative of DNA degradation due to apoptotic cell death was observed in MMP-9 silenced glioma cells compared to intact DNA of control and pSV-treated cells (data not shown). Figure 6D demonstrates accumulation of pMMP-9-transfected cells in the sub-G0–G1 phase (apoptotic cells) as compared to control and pSV-treated cells. pMMP-9-treated glioma cells showed 35–40% apoptosis in both 4910 and 5310 glioma cells as compared to control and pSV-treated cells (Figure 6D).
Figure 6. pMMP-9 lowers telomere end processivity and effects expression of apoptotic, cell cycle and growth signaling proteins induces apoptosis in glioma.

A. RT-PCR and Western blot analyses show effect of MMP-9 transfection on telomere end processing proteins in 4910 and 5310 glioma xenograft cells. B. Immunoprecipitation (IP) analysis. IP of hTERT was immunoprobed with Ku80 and POT1 in both 4910 and 5310 glioma xenograft cells transfected with pMMP-9 along with control and pSV samples. IgG shows equal loading. C. Western blot analysis shows effect of pMMP-9 treatment on apoptotic [I], cell cycle [II] and growth signaling [III] related protein expression in both glioma cell types compared to control and pSV cells. D. FACS analysis shows cell cycle distribution after pMMP-9 transfection along with control and pSV in 4910 glioma cells. 10,000 cells were scored for the analysis (n=3).
3.8 pMMP-9 treatment decrease MMP-9, β1 integrin interaction, reduces expression of FAK, c-Myc and increases MAD expression in in vivo nude mice
We next extended our work to an in vivo model to determine the effect of pMMP-9 transfection on c-Myc/MAD transcription factors that regulate hTERT expression in vivo. Our previous study demonstrated that pMMP-9 treatment decreased tumor growth and completely prevented tumor growth [18]. Aggressive tumor formation was noticed in xenograft-injected mouse brains as compared to pMMP-9-treated group. Co-localization of MMP-9 and β1 integrin in tumors from mice treated with pMMP-9 was markedly reduced when compared to untreated tumor tissue sections from control mice (Figure 7A). Figure 7B shows immunohistochemical analysis of paraffin-embedded brain tissue sections of nude mice pre-injected with 4910 and 5310 glioma cells revealing a decrease in the expression of c-Myc (hTERT transcriptional activator) and an increase in MAD expression (hTERT transcriptional repressor) after pMMP-9 treatment as compared to controls. We observed a drastic reduction in the expression of FAK in tissues treated with pMMP-9 as compared to untreated tumor tissue sections (Figure 7B).
Figure 7. MMP-9 silencing decreased FAK, c-Myc/MAD expression associated with hTERT transcription and co-localization of MMP-9 and β1 integrin in 4910 and 5310 glioma xenograft cells in vivo.

A. Immuohistochemistry was performed for the co-localization (yellow) of MMP-9 (green) and β1 integrin (red) using specific antibodies. B. IHC analysis shows expression of FAK, c-Myc and MAD in pre-established 4910 and 5310 glioma tumors treated with pMMP-9 construct. Inset show negative control (IgG). Magnification 60X. Bar = 20 μm. (n=6).
4. Discussion
More than 90 percent of cancers show activated telomerase [19,20]. Telomerase activity permits cancer cell immortalization and promotes tumorigenesis. Based on expression of telomerase and its genetic variation, it has been correlated with malignant glioma progression [21–25]. Loss of telomere length below the critical threshold induces growth arrest associated with cellular senescence and apoptosis [26,27]. Even though telomerase inhibitors are considered potential anti-tumor agents, the effect of these inhibitors on normal stem cells limits its application, and the potential use of our constructs may have the added advantage over these inhibitors. In the present study, we have demonstrated that MMP-9 inhibition using shRNA induced senescence and apoptosis in glioma xenograft cells both in vitro and in vivo. Further, our results indicated that MMP-9 inhibition altered the expression of hTERT via deregulation of the Myc family of transcription factors involved in hTERT transcription. hTERT is one of the target genes of the c-Myc/MAX/MAD network of transcription factors and alteration of these transcription factors regulates hTERT gene transcription [3,28–34]. However, it still remains unclear how hTERT activation or repression takes place in cancer and normal cells.
The c-Myc/MAX/MAD network of transcription factors are involved in the control of cell growth, proliferation, differentiation and apoptosis, and their expression is altered in a large number of human tumors [35]. c-Myc dimerization with MAX binds E-box (5′-CACGTG-3′). Recruitment of the c-Myc/MAX dimer acts as a repressor of the target gene. Like c-Myc, MAD also interacts with MAX. However, MAD/MAX dimer binding to E-box activates the target gene expression [35,36]. In this study, immunoprecipitation (IP) and chromatin IP assays showed reduced interaction of c-Myc/MAX and increased MAD/MAX association in cell lysates and also at hTERT promoter-associated E-box. This retards the hTERT expression and lowers its activity in glioma cells, thereby inducing senescence. Recent studies suggest that switching of c-Myc/MAX to MAD/MAX dimer binding to hTERT promoter decreases its promoter activity [3,37–39]. Inhibition of MAD1 has shown to reverse the hTERT expression and reduced the percentage of senescence of glioma cells. This observation is consistent with earlier findings [3,37].
To further establish signaling mediated by MMP-9 inhibition in the regulation of the Myc family of proteins, we anticipated early signaling events. Integrins form the basic cellular structures in attaching cells to the extracellular matrix and form the basis to activate various intracellular signaling events. Integrins engagement onto ECM is associated with phosphorylation of focal adhesion kinase (FAK), which triggers various intracellular signal transduction pathways and promotes cell survival [16,17]. In the present study, we noticed markedly lowered expression of FAK in pMMP-9-treated glioma cells. Further, to delineate the role of FAK in c-Myc/MAD signaling, we transfected the glioma cells with FAK shRNA. Western blot analysis showed lowered expression of c-Myc and an increase in MAD expression. This clearly shows that MMP-9 inhibition altered the expression of the Myc family of proteins and that this is mediated by FAK signaling. Moreover, inhibition of β1 integrin by antibody neutralization studies showed a decrease in the phosphorylation of FAK. Association of β1 integrin and FAK-mediated signaling has been described in earlier studies [40,41]. In corroboration with these findings, our studies with FL-MMP-9 overexpression showed increased expression of hTERT, β1 integrin and FAK, which was downregulated markedly by MMP-9 and FAK shRNA. The cumulative effect of FL-MMP-9 expression provides further evidence of MMP-9-induced downregulation of hTERT mediated by β1 integrin and FAK intracellular signaling via deregulation of c-Myc and MAD expression.
In cancer cells, stabilization of telomeres by reactivation of telomerase is suggested to be a crucial step during cellular immortalization and tumorigenesis. Moreover, its inhibition is associated with induction of apoptosis and senescence [42–44]. Earlier studies have shown that selective silencing of hTERT using hTERT siRNA and oligonucleotides targeting the RNA component of telomerase induced both apoptosis and senescence [27,45]. Accumulating evidence suggests that unstable telomere ends increase the chances of chromosomal rearrangements and DNA fusions, thereby elevating genetic instability in the cells. This is further supported by the expression of the HR 23B gene, an early DNA damage marker in adenocarcinoma SEG 1 cells with reduced telomerase activity [46]. In the present study, we anticipate that loss of telomerase activity mediated by MMP-9 silencing contributes to an increase in DNA damage, wherein cells reached a point of crisis to initiate signaling for programmed cell death by activation of p73 and TRAIL expression. FACS analysis showed an increase in apoptotic cell population in MMP-9-inhibited glioma cells. These results are in accord with our earlier findings, which showed that silencing of MMP-9 in combination with uPAR/cathepsin B induced apoptotic cell death in DNA damage accumulated glioma cells (Ponnala et al, under review). Altered telomerase activity mimics cellular DNA damage inducing senescence, and continued replication of cells leads to induction of cell death [47]. Telomere shortening mediated by ATM has been associated with senescence induction and telomerase inhibition induces apoptosis in human tumor cells [44,48]. Reduced expression of ATM, Ku80 and POT1 at both the mRNA and protein levels (along with other proteins involved in telomere end processivity) was observed in pMMP-9-transfected glioma cells. In addition, the interaction of these proteins was drastically decreased in MMP-9-silenced glioma cells. The telomere end processivity involving ATM, Ku80 and POT1 further strengthen our hypothesis regarding the induction of apoptosis in cells with accumulated DNA damage. Taken together, these results show that DNA repair proteins like ATM and Ku80, which are involved in telomere length maintenance and telomere protection, are downregulated in MMP-9-silenced glioma cells. The reduced activity of these repair proteins has a direct role in regulating telomere length and protection [49].
Progressive loss of telomere length has been shown to regress the growth of various cancers [43,50–52]. In the present study, intracranial implantation of glioma xenograft cells in nude mice followed by treatment with pMMP-9 resulted in significantly reduced tumor growth. In vivo expression of Myc/MAD transcription factors was consistent with our in vitro data. In conclusion, silencing MMP-9 acts as telomerase antagonist and induces apoptosis and senescence in glioma cells.
Supplementary Material
A. pMMP-9-transfected glioma cells were assessed for cell proliferation using BrdU incorporation assay. B. Survival fraction of glioma cells treated with pMMP-9 was evaluated by counting number of colony forming units. 200 cells were plated in 6-well plates. (n=3). Each bar represents triplicate analyses of mean ± SD where significant difference from cells treated with pMMP-9 compared to pSV and control is represented by an asterisk * (P <0.05).
{kind=link}
Immunocytochemistry analysis was carried out for co-localization of c-Myc/MAX and MAD/MAX heterodimers in pMMP-9-transfected 4910 and 5310 glioma xenograft cells along with control and pSV cells.
{kind=link}
Highlights.
MMP-9 induced senescence and apoptosis in glioma by silencing telomerase.
MMP-9 regulates hTERT levels via c-Myc/MAX/MAD network of transcription factors.
MMP-9 seeks FAK-mediated signaling in altering c-Myc/MAD expression in glioma.
MMP-9 retard hTERT expression via β1 integrin both in vitro and in vivo in glioma.
MMP-9 depletion retards telomere end processing and induced apoptosis in glioma.
Acknowledgments
The authors wish to thank Shellee Abraham for manuscript preparation, and Diana Meister and Sushma Jasti for manuscript review.
Funding
This research was supported by a grant from National Institute of Neurological Disorders and Stroke (N.I.N.D.S), NS047699 (to JSR). The contents are solely the responsibility of the authors and do not necessarily represent the official views of National Institute of Health (NIH).
Abbreviations
- MMP-9
Matrix metalloprotease 9
- shRNA
small hairpin RNA
- FAK
Focal adhesion kinase
- hTERT
human telomeasre revesre transcriptase
- FACS
Fluorescence activated cell sorting
- ATM
ataxia telangiectasia mutated
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Grandori C, Cowley SM, James LP, Eisenman RN. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 2.Luscher B. Gene. 2001;277:1–14. doi: 10.1016/s0378-1119(01)00697-7. [DOI] [PubMed] [Google Scholar]
- 3.Gunes C, Lichtsteiner S, Vasserot AP, Englert C. Cancer Res. 2000;60:2116–2121. [PubMed] [Google Scholar]
- 4.Iyer V, Pumiglia K, DiPersio CM. J Cell Sci. 2005;118:1185–1195. doi: 10.1242/jcs.01708. [DOI] [PubMed] [Google Scholar]
- 5.Veeravalli KK, Chetty C, Ponnala S, Gondi CS, Lakka SS, Fassett D, Klopfenstein JD, Dinh DH, Gujrati M, Rao JS. PLoS One. 2010;5:e11583. doi: 10.1371/journal.pone.0011583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelleher C, Teixeira MT, Forstemann K, Lingner J. Trends Biochem Sci. 2002;27:572–579. doi: 10.1016/s0968-0004(02)02206-5. [DOI] [PubMed] [Google Scholar]
- 7.Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V, Iliakis G, Shay JW, Young CS, Pandita TK. Oncogene. 2003;22:131–146. doi: 10.1038/sj.onc.1206063. [DOI] [PubMed] [Google Scholar]
- 8.Cerni C. Mutat Res. 2000;462:31–47. doi: 10.1016/s1383-5742(99)00091-5. [DOI] [PubMed] [Google Scholar]
- 9.Mathon NF, Lloyd AC. Nat Rev Cancer. 2001;1:203–213. doi: 10.1038/35106045. [DOI] [PubMed] [Google Scholar]
- 10.Bhoopathi P, Chetty C, Kunigal S, Vanamala SK, Rao JS, Lakka SS. J Biol Chem. 2008;283:1545–1552. doi: 10.1074/jbc.M707931200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, James CD. Neuro-oncol. 2005;7:164–176. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakka SS, Gondi CS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Rao JS. Oncogene. 2004;23:4681–4689. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- 13.Gogineni VR, Nalla AK, Gupta R, Gujrati M, Klopfenstein JD, Mohanam S, Rao JS. Int J Oncol. 2011;38:1615–1624. doi: 10.3892/ijo.2011.987. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Gopinath S, Malla RR, Gondi CS, Alapati K, Fassett D, Klopfenstein JD, Dinh DH, Gujrati M, Rao JS. PLoS One. 2010;5:e11668. doi: 10.1371/journal.pone.0011668. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Ponnala S, Rao KP, Chaudhury JR, Ahmed J, Rama RB, Kanjilal S, Hasan Q, Das UN. Prostaglandins Leukot Essent Fatty Acids. 2009;80:43–50. doi: 10.1016/j.plefa.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 16.Schlaepfer DD, Hauck CR, Sieg DJ. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 17.Burridge K, Turner CE, Romer LH. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chetty C, Lakka SS, Bhoopathi P, Gondi CS, Veeravalli KK, Fassett D, Klopfenstein JD, Dinh DH, Gujrati M, Rao JS. Mol Cancer Ther. 2010;9:2605–2617. doi: 10.1158/1535-7163.MCT-10-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins K, Mitchell JR. Oncogene. 2002;21:564–579. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 20.de LT. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 21.Harada K, Kurisu K, Tahara H, Tahara E, Ide T, Tahara E. J Neurosurg. 2000;93:618–625. doi: 10.3171/jns.2000.93.4.0618. [DOI] [PubMed] [Google Scholar]
- 22.Hiraga S, Ohnishi T, Izumoto S, Miyahara E, Kanemura Y, Matsumura H, Arita N. Cancer Res. 1998;58:2117–2125. [PubMed] [Google Scholar]
- 23.Mergny JL, Riou JF, Mailliet P, Teulade-Fichou MP, Gilson E. Nucleic Acids Res. 2002;30:839–865. doi: 10.1093/nar/30.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon M, Hosking FJ, Marie Y, Gousias K, Boisselier B, Carpentier C, Schramm J, Mokhtari K, Hoang-Xuan K, Idbaih A, Delattre JY, Lathrop M, et al. Clin Cancer Res. 2010;16:5252–5259. doi: 10.1158/1078-0432.CCR-10-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le S, Zhu JJ, Anthony DC, Greider CW, Black PM. Neurosurgery. 1998;42:1120–1124. doi: 10.1097/00006123-199805000-00099. [DOI] [PubMed] [Google Scholar]
- 26.Harley CB, Futcher AB, Greider CW. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 27.Shammas MA, Koley H, Batchu RB, Bertheau RC, Protopopov A, Munshi NC, Goyal RK. Mol Cancer. 2005;4:24. doi: 10.1186/1476-4598-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerezo A, Kalthoff H, Schuermann M, Schafer B, Boukamp P. J Cell Sci. 2002;115:1305–1312. doi: 10.1242/jcs.115.6.1305. [DOI] [PubMed] [Google Scholar]
- 29.Cong YS, Wright WE, Shay JW. Microbiol Mol Biol Rev. 2002;66:407–25. doi: 10.1128/MMBR.66.3.407-425.2002. table. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horikawa I, Barrett JC. Carcinogenesis. 2003;24:1167–1176. doi: 10.1093/carcin/bgg085. [DOI] [PubMed] [Google Scholar]
- 31.Lin SY, Elledge SJ. Cell. 2003;113:881–889. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- 32.Liu JP. FASEB J. 1999;13:2091–2104. doi: 10.1096/fasebj.13.15.2091. [DOI] [PubMed] [Google Scholar]
- 33.Ogretmen B, Kraveka JM, Schady D, Usta J, Hannun YA, Obeid LM. J Biol Chem. 2001;276:32506–32514. doi: 10.1074/jbc.M101350200. [DOI] [PubMed] [Google Scholar]
- 34.Xu D, Dwyer J, Li H, Duan W, Liu JP. J Biol Chem. 2008;283:23567–23580. doi: 10.1074/jbc.M800790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole MD, Henriksson M. Semin Cancer Biol. 2006;16:241. doi: 10.1016/j.semcancer.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Ayer DE, Lawrence QA, Eisenman RN. Cell. 1995;80:767–776. doi: 10.1016/0092-8674(95)90355-0. [DOI] [PubMed] [Google Scholar]
- 37.Oh S, Song YH, Yim J, Kim TK. Oncogene. 2000;19:1485–1490. doi: 10.1038/sj.onc.1203439. [DOI] [PubMed] [Google Scholar]
- 38.Kyo S, Takakura M, Taira T, Kanaya T, Itoh H, Yutsudo M, Ariga H, Inoue M. Nucleic Acids Res. 2000;28:669–677. doi: 10.1093/nar/28.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu D, Popov N, Hou M, Wang Q, Bjorkholm M, Gruber A, Menkel AR, Henriksson M. Proc Natl Acad Sci U S A. 2001;98:3826–3831. doi: 10.1073/pnas.071043198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geiger B, Bershadsky A, Pankov R, Yamada KM. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 41.Shibue T, Weinberg RA. Proc Natl Acad Sci USA. 2009;106:10290–10295. doi: 10.1073/pnas.0904227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cassar L, Nicholls C, Pinto AR, Li H, Liu JP. FASEB J. 2009;23:1880–1892. doi: 10.1096/fj.08-119529. [DOI] [PubMed] [Google Scholar]
- 43.Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. Nat Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Mar V, Zhou W, Harrington L, Robinson MO. Genes Dev. 1999;13:2388–2399. doi: 10.1101/gad.13.18.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herbert BS, Pongracz K, Shay JW, Gryaznov SM. Oncogene. 2002;21:638–642. doi: 10.1038/sj.onc.1205064. [DOI] [PubMed] [Google Scholar]
- 46.Volker M, Mone MJ, Karmakar P, van HA, Schul W, Vermeulen W, Hoeijmakers JH, van DR, van Zeeland AA, Mullenders LH. Mol Cell. 2001;8:213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 47.Stewart SA, Weinberg RA. Annu Rev Cell Dev Biol. 2006;22:531–557. doi: 10.1146/annurev.cellbio.22.010305.104518. [DOI] [PubMed] [Google Scholar]
- 48.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 49.Blasco MA. Nat Rev Genet. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 50.Dikmen ZG, Gellert GC, Jackson S, Gryaznov S, Tressler R, Dogan P, Wright WE, Shay JW. Cancer Res. 2005;65:7866–7873. doi: 10.1158/0008-5472.CAN-05-1215. [DOI] [PubMed] [Google Scholar]
- 51.Djojosubroto MW, Chin AC, Go N, Schaetzlein S, Manns MP, Gryaznov S, Harley CB, Rudolph KL. Hepatology. 2005;42:1127–1136. doi: 10.1002/hep.20822. [DOI] [PubMed] [Google Scholar]
- 52.Hochreiter AE, Xiao H, Goldblatt EM, Gryaznov SM, Miller KD, Badve S, Sledge GW, Herbert BS. Clin Cancer Res. 2006;12:3184–3192. doi: 10.1158/1078-0432.CCR-05-2760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. pMMP-9-transfected glioma cells were assessed for cell proliferation using BrdU incorporation assay. B. Survival fraction of glioma cells treated with pMMP-9 was evaluated by counting number of colony forming units. 200 cells were plated in 6-well plates. (n=3). Each bar represents triplicate analyses of mean ± SD where significant difference from cells treated with pMMP-9 compared to pSV and control is represented by an asterisk * (P <0.05).
Immunocytochemistry analysis was carried out for co-localization of c-Myc/MAX and MAD/MAX heterodimers in pMMP-9-transfected 4910 and 5310 glioma xenograft cells along with control and pSV cells.