

Abstract
The putative tumor metastasis suppressor nm23H1 was originally identified in murine melanomas by subtraction cloning. It displays nucleoside diphosphate kinase activity and regulates cellular events, including growth and development. Recently nm23H1 has been reported to also act as a GTPase-activating protein of the Ras-related GTPase Rad. We attempted to determine whether nm23H1 also regulates Rho-family GTPases. Although we were unable to detect a direct association between nm23H1 and Rho-family GTPases, nm23H1 was shown to be associated with a Rac1-specific nucleotide exchange factor, Tiam1, by interaction with its amino-terminal region in extracts from the cells expressing exogenous Tiam1 and from native tissue. Overexpression of nm23H1 inhibited the Tiam1-induced production of GTP-bound Rac1 and activation of c-Jun kinase. On the other hand, forced overexpression of the wild type, but not the kinase-inactivated mutant of nm23H1, converted the GDP-bound forms of Rac1, Cdc42, and RhoA to their GTP-bound forms in vitro by its nucleoside diphosphate kinase activity, but nm23H1 alone apparently did not produce the GTP-bound form of these GTPases in vivo. These results suggest that nm23H1 negatively regulates Tiam1 and inhibits Rac1 activation in vivo. Moreover, adhesion-stimulated membrane ruffles of Rat1 fibroblasts were reduced by overexpression of nm23H1. Based on these observations, we concluded that we had identified a function of nm23H1 as a regulator of Rac1 and that it may be related to the effect of nm23H1 as a tumor metastasis suppressor.
Protein nm23 belongs to a family of structurally conserved proteins of NDP kinases that are major suppliers of NTPs, such as GTP, where ATP is the phosphate donor (1). There are eight known nm23 genes in the human genome, and two of them, nm23H1 and nm23H2, have been the most widely studied. Although nm23H1 and nm23H2 have a highly homologous (88% identity) amino acid sequence, their function and cellular localization differ. Nm23H1 is a putative tumor metastasis suppressor and is expressed mainly in the brain and testis of the mouse, whereas nm23H2 is a DNA-binding protein, activates transcription of the c-myc gene, and is present in various organs (2). Both nm23H1 and -H2 proteins are found in the cytoplasm, but nm23H2 has also been detected in the nucleus through the use of an isoform-specific antibody (3, 4). A number of previous studies have reported a correlation between nm23H1 expression and poor prognosis for various human tumors, but the previous data are not always consistent (5, 6). Introduction of nm23H1 has reduced the metastatic potential and in vitro cell motility of tumor cells (7, 8), and Kantor et al. (8, 9) reported that murine melanoma cell lines and human breast carcinoma cells that were stably transfected with nm23H1 lost their ability to migrate in response to a variety of chemoattractants, including serum, platelet-derived growth factor, and insulin-like growth factor 1. Despite these previous studies, the basic mechanism of nm23H1 as a suppressor of tumor cell metastasis is still unclear. Zhu et al. (10) have reported that nm23H1 interacts with Rad, converts GDP-Rad to GTP-Rad, and acts as a GTPase-activating protein (GAP) for Rad, but as far as we have been able to determine, there is still little information concerning the presumed interactions between nm23 and the Ras superfamily GTPase in general.
The purpose of our study is to determine whether nm23H1 regulates Rho-family GTPases, which are closely related to cell migration. Furthermore, we pursue the possibility that this regulation may be one of the basic mechanisms of nm23H1 to modify the metastatic potential of tumor cells.
The Rho family of low-molecular-weight G proteins consists of the Rho, Rac, and Cdc42 subfamilies. The members of the Rho family have been recognized as key regulators of signal transduction pathways that mediate the distinct changes in the actin cytoskeleton required for transformation (11). Rho family GTPases are activated by guanine nucleotide exchange factors (GEFs) and negatively regulated by GAPs (12) and GDP dissociation inhibitors, which lock the GTPase in either the active or the inactive state (13). There are several GEFs for the Rho family GTPases, including Vav, PIX, and Tiam1, which possess a catalytic Dbl homology domain and convert GTPases from a GDP-bound state to the active GTP-bound state. Vav1 and PIX both are known to activate both Cdc42Hs and Rac1 (14, 15). Tiam1 is a specific GEF for Rac1, which originally was identified in T lymphoma cells as an invasion- and metastasis-inducing gene (16). Tiam1 protein is known to be highly expressed in the brain and testis of mice (17). Recent evidence has shown that Tiam1 stimulates the kinase activity of c-Jun kinase (JNK) via production of GTP-loaded Rac1 (18–20). Rac activation down-regulates Rho activity, which is required for the Tiam1-induced epithelial phenotype (21).
In this report we address the negative regulation of Tiam1 by nm23H1, which specifically inhibits Rac1 activation in Tiam1-overexpressing cells. This report demonstrates interaction between nm23H1 and Rho-family GTPases.
Materials and Methods
Plasmids and Constructs.
Full-length nm23H1 tagged with a hemagglutinin (HA), myc, or flag epitope at the C terminus was cloned in pCS2 + (provided by D. Turner, Hutchinson Cancer Research Center, Seattle, WA) for transfection into 293T cells. To prepare a green fluorescent protein (GFP) fusion protein, nm23H1 was subcloned into pGFP-C3 (CLONTECH). Mutants of nm23H1, H118C and P96S, representing clones with a point mutation of His-118 to Cis-118 and Pro-96 to Ser-96, respectively, were generated by two-step PCR mutagenesis, as described (22). Full-length human Tiam1 cDNA was generated from the mRNA of HL60 cells by long and accurate PCR, as described (20). Deletion mutants of nm23H1 [ΔKpn; deletion of killer of prune (Kpn) loop of nm23H1; amino acids 96–116] and Tiam1 (C1199, C682, and N392; nomenclature refers to the number of COOH-terminal or NH2-terminal amino acids of the encoded Tiam1 proteins) also were generated by the PCR-based technique. Plasmids encoding full-length cDNAs of human βPIX and human Vav1 were gifts from T. Nagase (Kazusa DNA Research Institute, Kisarazu, Japan) and M. Shibuya (The Institute of Medical Science, University of Tokyo, Japan), respectively. Plasmids encoding human p21-activated kinase 1 (PAK1), JNK1, and wild-type or activated mutants of Rac1, Cdc42Hs, and RhoA were provided by B. J. Mayer, University of Connecticut Health Center, Farmington, CT (23). Plasmid encoding human Rho-GDP dissociation inhibitor was a gift from W. Lu, California Institute of Technology, Pasadena, CA (24). All constructs were cloned into pCS2+ or pGEX-2T (Amersham Pharmacia) to prepare glutathione S-transferase (GST) fusion proteins in bacteria. The coding regions of Rac1 and JNK1 were subcloned into pEBG for production of GST-tagged proteins in 293T cells. pGEX2T p21 binding domain (pGEX2T-PBD) was generated by cloning of a PCR-amplified fragment of putative p21 binding domain of human PAK1 (amino acids 70–133) into pGEX-2T for affinity precipitation as described (25).
Antibodies.
The mAbs for the HA epitope tag, myc epitope tag, and flag tag were from Berkeley Antibody (Richmond, CA), Santa Cruz Biotechnology, and Sigma, respectively. The polyclonal antibodies for the HA epitope tag, Tiam1, Rac1, nm23H1, and GST were from Santa Cruz Biotechnology. Anti-mouse or anti-rabbit Ig-conjugated or unconjugated to alkaline phosphatase and FITC-conjugated or tetramethylrhodamine B isothiocyanate-conjugated secondary antibodies were from Dako.
Cell Culture, Transfection, and Immunoprecipitation.
293T human embryonal kidney cells and Rat1 fibroblasts were cultured in DMEM (GIBCO/BRL and Life Technologies, Rockville, MD) supplemented with 10% FBS. For transient expression assays, 293T cells were transfected with a maximum of 12 μg of plasmid DNA per 10-cm-diameter dish or 2.0 μg of plasmid DNA for a 3.5-cm-diameter dish by a calcium phosphate coprecipitation method with concurrent treatment with 25 μM chloroquine, essentially as described (26). Rat1 cells were transfected with the use of Lipofectamine (GIBCO/BRL and Life Technologies) with a maximum of 1.0 μg of plasmid DNA per 3.5-cm-diameter dish. Cells were harvested 48 h after transfection, and cell lysates were prepared with protease inhibitors in TXB buffer (10 mM Tris, pH 7.6/150 mM NaCl/5 mM EDTA, pH 8.0/10% glycerol/1 mM Na3VO4/1% Triton X-100). Whole-brain tissue from 5-week-old Institute for Cancer Research (ICR) mice was also lysed in TXB buffer with a Dounce homogenizer. For peptide neutralization, anti-Tiam1c antibody was combined with a 5-fold (by weight) excess of blocking peptide in a small amount of PBS and incubated for 2 h at room temperature. Lysates were precleared by incubation with protein G agarose (Roche Molecular Biochemicals) for 3 h at 4°C. To purify the protein, 1 μg of monoclonal or affinity-purified polyclonal antibody was incubated with 500 μl of cell lysate for 2 h at 4°C and precipitated with protein G agarose for 4 h at 4°C. Immunoprecipitates were extensively washed with TXB buffer, separated by SDS/PAGE, and immunoblotted. Rat1 cell lines overexpressing GFP-tagged nm23H1 were established by transfection of pGFP-C3 encoding nm23H1 as described above. Cells were cultured in DMEM containing 0.4 μg/ml G418 (GIBCO/BRL) for 2–3 weeks. Well-isolated colonies were characterized further.
Determination of GTP Formation of Rho-Family GTPases.
Measurement of guanine nucleotides bound to Rho-family GTPases in vivo was carried out as described (27). Briefly, pEBG-Rac1, with or without plasmids encoding Tiam1 and nm23H1, was transfected into 2 × 105 293T cells in a 35-mm-diameter dish. Forty-eight hours after transfection, the cells were labeled with 0.05 mCi of 32Pi (NEN) in 0.5 ml of phosphate-free MEM (GIBCO/BRL) for 2 h and lysed in lysis buffer (20 mM Tris⋅HCl, pH 7.5/150 mM NaCl/20 mM MgCl2/1 mM Na3VO4/0.5% Triton X-100/5 μg/ml aprotinin/1 mM PMSF). GST-tagged Rac1 was purified by glutathione Sepharose, then eluted in buffer containing 20 mM Tris (pH 7.5), 20 mM EDTA, and 2% SDS. Eluted samples were resolved by TLC, and the results were visualized and quantitated with a Bio Imaging Analyzer (BAS1000; Fuji). To determine whether nm23H1 promotes the formation of GTP-Rac1 in situ, GDP was immobilized on GST-Rac1 by UV crosslinking with the use of Stratalinker (Stratagene). The samples were washed, incubated with [γ-32P]ATP (10 μCi) in the absence or presence of 0.1 μg of GST-nm23H1 at 25°C for 5 min, and resolved by SDS/PAGE and autoradiography as described (10).
Affinity Precipitation.
Affinity precipitation with the GST-tagged p21 binding domain of PAK1 (GST-PBD) was performed as described (25). Briefly, 293T cells were lysed in the lysis buffer (20 mM Tris⋅HCl, pH 7.5/150 mM NaCl/20 mM MgCl2/1 mM Na3VO4/5% Triton X-100/5 μg/ml aprotinin/1 mM PMSF), and incubated with GST-PBD on Sepharose for 1 h at 4°C. Precipitants were washed three times in the same buffer, and endogenous Rac1 was detected by immunoblot.
In Vitro Kinase Assay of JNK1.
GST-JNK1 was purified by glutathione-Sepharose from 293T cells transfected with pEBG-JNK1 together with the plasmids cited. The in vitro kinase assay was carried out as described with GST-cJun as substrate (26). The results were visualized and quantitated with a Bio Imaging Analyzer (BAS1000; Fuji).
Cell Staining.
For immunohistochemical analysis, cells were plated on fibronectin-coated slides (Roche Molecular Biochemicals) for 15 min or 120 min, fixed with PBS containing 4% paraformaldehyde for 20 min at 25°C, and then permeabilized with PBS containing 0.2% Triton X-100 for 5 min. The cells were preincubated in 1% BSA for 10 min and incubated with the primary antibody for 1 h at 25°C. The cells then were rinsed in PBS containing 1% BSA and incubated with anti-mouse or anti-rabbit Ig for 1 h at 25°C to enhance the signal. After the cells were rinsed, they were further incubated with FITC- or tetramethylrhodamine B isothiocyanate-conjugated secondary antibody for 1 h at 25°C, and filamentous F-actin was stained with tetramethylrhodamine B isothiocyanate-conjugated phalloidin (Sigma). Cells were examined with an Axiophot microscope (Zeiss).
Results and Discussion
Nm23H1 Specifically Associates with Guanine Nucleotide Exchanging Factor Tiam1.
We attempted to determine, by coprecipitation analysis, whether nm23H1 interacts with regulators of Rac1, including Tiam1, Vav1, and PIX. Fig. 2a shows that nm23H1 associated with Tiam1 in vivo (lanes 1 and 6).
Figure 2.

Nm23H1 specifically associates with the guanine nucleotide exchanging factor Tiam1. (a) 293T cells were transiently transfected with plasmid encoding myc or HA-tagged nm23H1 together with the Tiam1 constructs indicated above the lanes. Cells were lysed and immunoprecipitated (IP) with anti-myc (lanes 1–4) or anti-Tiam1-c (lanes 5–7), which reacts with epitopes located at the carboxyl terminus of Tiam1. Bound proteins were immunoblotted (IB) with the antibodies indicated. Expression of Tiam1 and nm23H1 in individual cell lysates (total lysate) was confirmed by immunoblotting (Bottom). (b) 293T cells were transiently cotransfected with flag-tagged wild type or ΔKpn nm23H1 together with Tiam1 constructs as indicated. Cell lysates were immunoprecipitated with anti-flag and immunoblotted with the antibodies indicated (lanes 1–3). HA-tagged wild-type or ΔKpn nm23H1 constructs were transiently transfected with flag-tagged wild-type or ΔKpn nm23H1 constructs (lanes 4–6). Immunoprecipitation and immunoblotting were performed as indicated. Expression of nm23H1 and Tiam1 is shown at the bottom. (c) 293T cells were transiently transfected with plasmid encoding myc-tagged nm23H1 together with Tiam1, HA-tagged βPIX, or Vav1 as indicated. Cell lysates were immunoprecipitated with anti-myc and immunoblotted sequentially with anti-Tiam1-c and anti-HA antibodies. (d) Whole brains of adult Institute for Cancer Research (ICR) mice were lysed and immunoprecipitated with anti-Tiam1-c antibody, normal rabbit Ig fraction, or peptide-neutralized anti-Tiam1-c antibody. Precipitants were subjected to immunoblot with the indicated antibodies. These experiments were performed at least three times.
To determine the location of the nm23H1 binding site in the Tiam1 protein, truncated Tiam1 cDNA constructs were used (Fig. 1). Coprecipitation analysis revealed that a truncated Tiam1 encoding 392 amino-terminal aa (N392) tightly bound to nm23H1 in vivo (Fig. 2a, lane 4, and Fig. 2c, lane 2). N-terminal deleted Tiam1 constructs (C1199 and C682), on the other hand, did not associate with nm23H1 (Fig. 2a, lanes 2, 3, 5, and 7, and Fig. 2c, lane 1). Previous studies demonstrated that His-118 corresponds to a phosphorylated region in nm23H1 and is essential for NDP kinase activity (28). Substitution of His-118 by Cys in nm23H1 (H118C) abolishes the histidine kinase activity of nm23H1. The NDP kinase activity of nm23 was not involved in the association with Tiam1, because a mutation in nm23H1 (H118C) did not affect the results of Tiam1 interaction (data not shown). Another mutation, Pro-96 to Ser (P96S), which is known as a killer of the prune mutation in the Drosophila nm23 homolog that causes developmental defects, also was examined for ability to bind to Tiam1. Association of nm23H1 (P96S) with Tiam1 also was detected, at almost the same level as the wild-type nm23H1 association with Tiam1 (data not shown). X-ray crystallography studies have shown that nm23H1 forms hexamers, and an essential role of the Kpn loop in nm23H1 in oligomerization has been proposed (29). We prepared a Kpn loop deletion mutant (ΔKpn) of nm23H1. This mutant could not oligomerize with wild-type nm23H1 or itself under the conditions that enabled oligomerization of nm23H1(WT)s in vivo (Fig. 2b, lanes 4–6). Nor could this mutant bind to Tiam11–392 in vivo (Fig. 2b, lane 3). Considering that H118C and P96S mutants are the most essential for known biological functions of nm23 (in particular, mutation of Pro-96 abrogates its motility inhibitory activity upon transfection into human breast carcinoma cells) (9), our observation is striking in terms of the independence of previously known mechanisms. Nm23H1s (wild-type, histidine-mutated, and P96S-mutated forms) inhibited the Rac1 activation via negative regulation of Tiam1's GEF activity in the environment of Tiam1-activated cells in the same way. The effect of nm23H1 through protein–protein interaction (actually, interaction of Tiam1 and nm23H1 shown here was quite an unexpected observation) may drastically change the view of the metastasis suppressor function of nm23H1 and will provide an alternative interpretation of the commitment of this molecule to the biological phenomena underlying a more complex situation, i.e., tumor metastasis.
Figure 1.
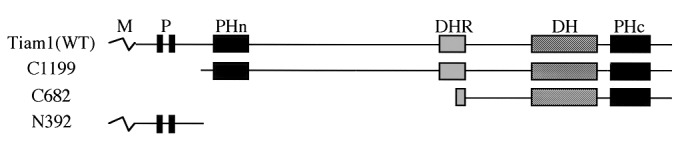
Schematic representation of wild-type and truncated Tiam1 cDNA constructs used in this study. Proteins are depicted to scale. M, myristoylation signal; P, region rich in proline, glutamate, serine, and threonine; PHn and PHc, NH2-terminal and COOH-terminal pleckstrin homology domains; DHR, Discus-large homology region; DH, Dbl homology domain; WT, wild type.
There are numerous possible mechanistic bases for this phenomenon. The results obtained with Kpn loop deletion mutants indicate that the oligomerization of nm23H1 is probably important for in vivo binding to Tiam1. Alternatively, it is also possible that the Kpn loop of nm23H1 could be bound to Tiam1. Because we could not demonstrate any direct interaction between bacterially expressed nm23H1 protein and Tiam1 in vitro (data not shown), the preferential association of nm23H1 with Tiam1 (Tiam11–392) may require some other linking proteins. Although it has been determined that Tiam11–392 contains two domains rich in proline, glutamate, serine, and threonine (amino acids 58–92 and 100–132), which are considered to be predictors of protein instability (16, 30), the presence of an nm23-binding motif in Tiam11–392 should be elucidated. On the other hand, as shown in Fig. 2c, neither PIX nor Vav1 was coprecipitated with nm23H1, thereby indicating that the association of nm23H1 with Tiam1 is rather specific among the GEFs. Among native mouse tissues, Tiam1 has been reported to be highly expressed in the brain and the testis (16). Nm23H1 was coprecipitated with Tiam1 from mouse brain extract by specific Tiam1 antibody. However, nm23H1 was not detected against mouse brain extract immunoprecipitated by normal rabbit Ig fraction or peptide-neutralized Tiam1 antibody (Fig. 2d). This finding indicates that physiological interaction between nm23H1 and Tiam1 occurs in the brain, although the biological function of Tiam1 in the brain is unclear. Further analysis, including analysis of the histological distribution of these proteins in the brain, will be necessary.
Nm23H1 Down-Regulates Rac1 by Association with Tiam1.
Next we attempted to determine whether nm23H1 modified the GEF activity of Tiam1 by labeling cells with 32Pi in vivo. The amount of GTP-bound Rac1 was markedly increased by overexpression of wild-type Tiam1 (Fig. 3a, lane 3). Coexpression of wild-type nm23H1 inhibited the increase in GTP-bound Rac1 induced by Tiam1 in a dose-dependent manner (Fig. 3a, lanes 4 and 5), and Tiam1-induced Rac1 activation also was inhibited by coexpression of kinase-inactivated nm23H1 (H118C) (Fig. 3b, lane 4). The modification of Tiam1-induced Rac1 activation by nm23H1 also was examined by affinity precipitation of GTP-bound Rac1 with the GST-PBD, as described (25). The PAK family of serine/threonine kinases is activated by binding to the GTP-bound form of Rac1 or Cdc42, which leads to the activation of mitogen-activated protein kinases (31, 32). Therefore the PBD of PAK1 was used to pull down GTP-bound Rac1 in this experiment. GTP-bound Rac1 precipitated with GST-PBD was reduced by coexpression of wild-type or H118C mutant nm23H1 (Fig. 4a, lanes 3–6). As in other proteins containing a Dbl homology domain, N-terminal truncation of the Tiam1 protein is known to cause it to activate itself into an oncogenic form, establishing Tiam1 as a protooncogene (33, 34). GTP-bound Rac1 was markedly increased by N-terminal truncated Tiam1 (C1199), and the increase was not reduced by cotransfection with the wild-type or H118C mutant of nm23H1 under conditions in which wild-type Tiam1-induced Rac1 activation was significantly inhibited (Fig. 3c). This finding is also compatible with the prediction that nm23H1 associates with N-terminal Tiam11–392. In conclusion, nm23H1 negatively regulates Tiam1, independently of its NDP kinase activity, and interaction between Tiam1 and nm23H1 is necessary for its negative regulation. Recent evidence has shown that Tiam1 stimulates the JNK pathway through activation of Rac1 (18–20). Observation of the interaction of nm23H1 with Tiam1 led us to investigate possible nm23H1 modulation of Tiam1-induced JNK activation. As shown in Fig. 4b, the expression of wild-type Tiam1 induced JNK activation (lane 2), but the activation was abrogated by cotransfection with the wild-type or H118C mutant of nm23H1 (lanes 3 and 4). In contrast, coexpression of nm23H1 did not affect the JNK activation induced by Tiam1 (C1199) (Fig. 4b, lanes 5–7). Although Tiam1 (N392) did not stimulate JNK activation as expected (Fig. 4c, lane 2), coexpression of an excess of Tiam1 (N392) partially rescued the nm23H1-blocked JNK activation (Fig. 4c, lane 5). These results further support an inhibitory role for nm23H1 in the Tiam1-Rac1-induced JNK activation based on the association with the N-terminal region of Tiam1. Although the mechanism of down-regulation of Tiam1 by nm23H1 is unclear, we found that nm23H1 inhibited the GEF activity of Tiam1. A structural change in Tiam1 protein as a result of the association with nm23H1 may induce inhibition of the GEF activity of Tiam1.
Figure 3.

Nm23H1 down-regulates Rac1 by association with Tiam1. (a–c) 293T cells transfected with plasmids indicated at the top were labeled with 32Pi for 2 h. Lanes 4 and 5 (a): transfected with 0.5 μg and 0.2 μg, respectively, of plasmid encoding nm23H1 tagged with myc. The total amount of transfected plasmid DNA was equalized by adding empty vector pCS2+. The labeled guanine nucleotides that bound GST-tagged Rac1 were separated by TLC and visualized and quantitated with imaging analyzer (BAS 1000; Fuji). Comparable expression of Tiam1, GST-Rac1, and nm23H1 was confirmed by immunoblotting (Bottom). ori, origin of the TLC plate.
Figure 4.

Nm23H1 down-regulates Rac1 by association with Tiam1. (a) 293T cells were transfected with the plasmids indicated at the top. A total of 0.25 μg (lanes 3 and 5) and 0.5 μg (lanes 4 and 6) of HA-tagged plasmids encoding wild-type or H118C mutant nm23H1 (HC), respectively, was transfected. Cells were lysed for affinity precipitation (AP) with immobilized GST-PBD as described in Materials and Methods. Precipitated endogenous GTP-bound Rac1 was detected by immunoblotting with anti-Rac1 antibody. Expression of Tiam1 and nm23H1 was detected by immunoblotting (Bottom). (b and c) 293T cells were transfected with the plasmids at the Top. GST-tagged JNK1 was purified from cell lysates and subjected to in vitro kinase assay with GST-c-jun as substrate. Comparable expression of Tiam1, GST-JNK1, and nm23H1 in individual lysates was confirmed by immunoblotting (Bottom). (c) The relative activity of JNK1 against the reference (Lane 2) was calculated by adjusting the densitometric value and standardization. Similar results were obtained in three independent experiments.
Nm23H1 Transphosphorylates to Rho-Family GTPases in Vitro.
NDP kinase family genes modulate GTP-binding proteins by direct phosphorylation or indirectly by modulation of nucleotide pool sizes, resulting in regulation of G-protein activity (35–38). Nm23H1, but not nm23H1 (H118C), phosphorylated the Rho-family GTPases Rac1, Cdc42Hs, and RhoA in the presence of ATP in vitro (data not shown). Zhu et al. (10) showed that Rad-bound GDP was directly converted to GTP in situ as a result of phosphorylation by nm23H1. We therefore attempted to determine whether nm23H1 can convert GDP to GTP in situ by fixing GDP on GST-Rac1 by UV irradiation as described (10). As shown in Fig. 5a, the wild-type nm23H1 was able to convert GDP-Rac1 to GTP-Rac1 without release of GDP from Rac1, whereas nm23H1 (H118C) and the control GST could not.
Figure 5.

Nm23H1 transphosphorylates to Rac1 in vitro. (a) GDP-loaded GST-Rac1 on glutathione Sepharose (1 μg) was UV-irradiated as described in Materials and Methods, followed by incubation with 10 μCi [γ-32P]ATP in the presence of 0.1 μg GST-nm23H1, GST-nm23H1 (H118C), or control GST, as indicated above the lanes. The reactants were washed and resolved by 12% SDS/PAGE. Precipitated GST-tagged Rac1 was detected by Coomassie stain (Bottom). (b) 293T cells were transfected with pEBG-Rac1 in combination with either mock vector, wild type (WT), H118C mutant of nm23H1 or wild-type Tiam1, as indicated above. Cells were labeled with 32Pi for 2 h. The labeled guanine nucleotides bound to GST-tagged GTPases were separated by TLC. Expression of GST-tagged Rac1 in individual lysates was confirmed by immunoblotting (Bottom). Similar results were obtained in three independent experiments.
We next attempted to determine whether nm23H1 had functional GEF activity for Rac1 in vivo, which seemed to be the opposite of nm23H1's function as the negative regulator of Tiam1-Rac1 signaling described above. Although nm23H1 induced a significant increase in the GTP-bound form of GTPases in vitro, the amount of GTP-loaded Rac1 was very small in nm23H1-transfected cells (Fig. 5b, lane 2), which was determined by labeling cells with 32Pi as described in Fig. 3. This finding is consistent with the observation that overexpression of wild-type nm23H1 or the kinase-inactivated mutant of nm23H1 (H118C) markedly reduced Rac1 activation induced by Tiam1. There are several possible explanations for why nm23H1 can directly phosphorylate Rac in vitro but not in vivo. The sensitivity of our in vivo assay for GTP-bound Rac1 may be insufficient, and other proteins also may contribute to regulation of Rac1 activation in vivo. Further inspection of Fig. 4b, lanes 3 and 4, revealed that nm23H1 (H118C) induced slightly greater reduction than did wild-type nm23H1 in JNK activation, probably reflecting the persistence of barely detectable functional GEF activity for Rac1 of nm23H1.
Zhu et al. (10) showed that nm23H1 associates with Rad and regulates Rad in two ways: it converts GDP-Rad to GTP-Rad in vitro, depending on its NDP kinase activity, and it acts as a GAP for Rad. Although the possibility that nm23H1 may also have GAP activity for Rac1 was examined by measurement of the release of [γ-32P]GTP from Rac1 in vitro, no GAP activity of nm23H1 for Rac1 was detected in our study (data not shown). Furthermore, no direct association of nm23H1 with Rho-family GTPases was detected in vitro in our experiments. Neither the wild-type nor the activated form of Rac1 or Cdc42Hs was coprecipitated with nm23H1 when Rac1 and Cdc42Hs were associated with the PBD of PAK1, which was used as a control. Moreover, nm23H1 was not coprecipitated with either GTP-loaded or GDP-loaded GTPases under conditions in which PAK1 and Rho-GDP dissociation inhibitor, which were used as controls, associated strongly with these GTPases (data not shown). The absence of a direct association of nm23H1 with GTPases seems to be compatible with the modest effect of nm23H1 on the activation of Rac1 in vivo.
Overexpression of nm23H1 Reduces Membrane Ruffles.
Rac1 and Tiam1 both have been shown to be involved in the formation of lamellipodia and the establishment of cadherin-mediated cell–cell adhesion (21, 39). When Rat1 fibroblasts adhered to the extracellular matrix protein fibronectin, F-actin-containing membrane ruffling was stimulated as the initial response (compare Fig. 6 b and d), as described by others (40). Although endogenous Tiam1 is expressed homogeneously in the cytoplasm of Rat1 cells under usual conditions (Fig. 6a), it had localized to the lamellipodia by 15 min after cells were placed on fibronectin (Fig. 6c). The membrane ruffles were prominent in the control GFP-overexpressed cells (Fig. 6e), but overexpression of GFP-tagged nm23H1 reduced the number of F-actin-containing ruffles formed (Fig. 6f). Consistent with this finding, the control GFP did not affect the peripheral localization of Tiam1, but Tiam1 was not expressed on the periphery of the cell membrane in nm23H1-overexpressed cells (Fig. 6 g and h). On the other hand, expression of nm23H1 did not affect the membrane ruffle formation induced by transfection of Tiam1 (C1199) mutant or the peripheral localization of Tiam1 (C1199) (Fig. 6 l–o). This observation is compatible with the result that nm23H1 did not inhibit Tiam1 (C1199)-induced Rac1/JNK activation as shown in Figs. 3c and 4b. The fact that the level of expression of Tiam1 protein was not significantly different in nm23H1-overexpressed Rat1 cells and control GFP-overexpressed cells indicates that nm23H1 did not down-regulate expression of the Tiam1 gene (Fig. 6k). Inhibition of membrane ruffles by nm23H1, demonstrated here, is compatible with the previous finding that nm23H1 prevents cell migration in response to a variety of chemoattractants, including whole serum, platelet-derived growth factor, and insulin-like growth factor 1 (8, 9). The mutations in nm23H1 that abrogate NDP kinase activity have not been reported to be correlated with the motility-suppressive phenotype of nm23H1 (9). Nor did we detect any significant difference between the wild type and H118C mutant of nm23H1 in the inhibitory effect on ruffle and lamellipodium formation in Rat1 cells (data not shown). Obviously questions remain to be elucidated. It is especially necessary to determine whether the abrogation of ruffling formation in nm23H1-overexpressed cells depends on inactivation of endogenous Tiam1 or modification of other pathways. It also should be elucidated whether nm23H1 inhibits Rac1 activity by changing the subcellular localization of Tiam1. Wild-type Tiam1 has its own myristoylation signal in the amino terminus, but this signal is not sufficient for membrane localization (34). The additional myristoylation signal of the N-terminal v-Src sequence stabilizes Tiam1 on the cell membrane (34). It therefore may be interesting to investigate the possibility that nm23H1 also effectively inhibits the Rac1 activation induced by a Tiam1 mutant that is forcibly localized on the membrane by the addition of the myristoylation signal of v-Src.
Figure 6.
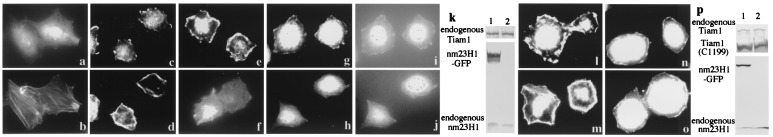
Overexpression of nm23H1 reduces the adhesion-stimulated membrane ruffles of Rat1 cells. (a–d) Rat1 cells were stained for Tiam1 (a and c) and for F-actin (b and d) on uncoated slides (a and b) or replated on fibronectin-coated slides for 15 min (c and d). (e–j and l–o) Rat1 cells stably expressing GFP (e, g, i, l, and n) or GFP-tagged nm23H1 (f, h, j, m, and o) were either untransfected (e–j) or were transfected with Tiam1 C1199 (myc tagged at the C terminus) (l–o), then replated on fibronectin-coated slides for 15 min and stained for F-actin (e, f, l, and m), endogenous Tiam1 (g and h), or Tiam1 C1199 (anti-myc) (n and o). The same cells are shown in a–d, g, i, and h, j. (i and j) Stable expression of GFP or GFP-nm23H1. Expression of endogenous or transfected Tiam1 and nm23H1 in Rat1 cells is shown by immunoblot (k and p). Three independent clones expressing GFP-tagged nm23H1 or GFP were examined, and similar results were obtained.
As shown in other reports, constitutively activated Tiam1 (C1199) induces epithelium-like morphology in fibroblasts when cells are plated at high density, and Tiam1 is dominantly expressed at sites of the cell–cell adhesion (21). No such accumulation of Tiam1 protein at cell junctions was observed in wild-type Tiam1-transfected Rat1 cells or NIH 3T3 cells in our experiments (data not shown). This lack of accumulation of Tiam1 protein may indicate that fully activated Tiam1 may be necessary for the induction of cadherin-mediated cell–cell contacts. Therefore the inhibitory effect of nm23H1 on Tiam1 activity may lead to different phenotypes of tumor cells: decreased cell motility, as suggested by our results, or decreased intercellular adhesion. Although nm23H1 is a putative tumor metastasis suppressor, expression of nm23H1 has not always been correlated with lower metastatic potential in some human tumors (5, 41). Total analyses, including the level of expression of Tiam1, nm23H1, and cadherin and the activity of Tiam1, may provide a more useful indicator of the metastatic potential of tumor cells.
On the other hand, the proteins that interact with nm23H1 are not fully understood. Members of the ROR/RZR nuclear orphan receptor subfamily are reported to interact with nm23 in vitro and regulate the transcription of genes, including N-myc (42). Moreover, an isoform of mouse nm23, nm23 M1, forms molecular complexes with β-tubulin in myogenic cells during the process of differentiation, although its biological significance is not well known (43).
We observed that nm23H1 associates with Tiam1 and down-regulates Tiam1-Rac1 signaling. Because nm23H1 did not associate with Rho-family GTPases and was not a functional GEF for GTPases in vivo, we concluded that the major role of nm23H1 in Tiam1-activated cells is to act as a negative regulator of Rac1. Although the molecules that stimulate Tiam1 activity are not well understood, Bourguignon et al. (44) recently reported a direct interaction between CD44v3 [the hyaluronic acid-binding receptor] and Tiam1 and that binding of hyaluronic acid to CD44v3 stimulated Tiam1-catalyzed Rac1 signaling and tumor cell migration. It would be interesting to investigate the possible interactions of Tiam1 and nm23H1 with other molecules such as CD44 in physiological and pathological conditions. Our findings reported here suggest a function of nm23H1 as a regulator of Rac1 GTPase that may lead to modification of the cell motility and metastatic potential of tumor cells. Furthermore, as both Tiam1 and nm23H1 are predominantly expressed in brain and testis, the physiological association of these two proteins and the functional significance of this association in these organs should be examined.
Acknowledgments
This work is supported by the Smoking Research Foundation, and a grant-in-aid by The Ministry of Education, Culture, Science, Sports, and Technology of Japan.
Abbreviations
- GFP
green fluorescent protein
- PAK
p21-activated kinase
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- JNK
c-Jun kinase
- HA
hemagglutinin
- PBD
p21 binding domain
- Kpn
killer of prune
- GST
glutathione S-transferase
- GST-PBD
GST-tagged p21 binding domain of PAK1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wagner P D, Vu N D. J Biol Chem. 1995;270:21758–21764. doi: 10.1074/jbc.270.37.21758. [DOI] [PubMed] [Google Scholar]
- 2.Steeg P S, Bevilacqua G, Kopper L, Thorgeirsson U P, Talmadge J E, Liotta L A, Sobel M E. J Natl Cancer Inst. 1988;80:200–204. doi: 10.1093/jnci/80.3.200. [DOI] [PubMed] [Google Scholar]
- 3.Pinon V P, Millot G, Munier A, Vassy J, Linares-Cruz G, Capeau J, Calvo F, Lacombe M L. Exp Cell Res. 1999;246:355–367. doi: 10.1006/excr.1998.4318. [DOI] [PubMed] [Google Scholar]
- 4.Kraeft S K, Traincart F, Mesnildrey S, Bourdais J, Veron M, Chen L B. Exp Cell Res. 1996;227:63–69. doi: 10.1006/excr.1996.0250. [DOI] [PubMed] [Google Scholar]
- 5.Hailat N, Keim D R, Melhem R F, Zhu X X, Eckerskorn C, Brodeur G M, Reynolds C P, Seeger R C, Lottspeich F, Strahler J R, et al. J Clin Invest. 1991;88:341–345. doi: 10.1172/JCI115299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacDonald N J, De la Rosa A, Benedict M A, Freije J M, Krutsch H, Steeg P S. J Biol Chem. 1993;268:25780–25789. [PubMed] [Google Scholar]
- 7.Leone A, Flatow U, King C R, Sandeen M A, Margulies I M, Liotta L A, Steeg P S. Cell. 1991;65:25–35. doi: 10.1016/0092-8674(91)90404-m. [DOI] [PubMed] [Google Scholar]
- 8.Kantor J D, McCormick B, Steeg P S, Zetter B R. Cancer Res. 1993;53:1971–1973. [PubMed] [Google Scholar]
- 9.MacDonald N J, Freije J M P, Stracke M L, Manrow R E, Steeg P S. J Biol Chem. 1996;271:25107–25116. doi: 10.1074/jbc.271.41.25107. [DOI] [PubMed] [Google Scholar]
- 10.Zhu J, Tseng Y H, Kantor J D, Rhodes C J, Zetter B R, Moyers J S, Kahn C R. Proc Natl Acad Sci USA. 1999;96:14911–14918. doi: 10.1073/pnas.96.26.14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 12.Scheffzek K, Ahmadian M R, Wittinghofer A. Trends Biochem Sci. 1998;23:257–262. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- 13.Boguski M S, McCormick F. Nature (London) 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 14.Crespo P, Schuebel K E, Ostrom A A, Gutkind J S, Bustelo X R. Nature (London) 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- 15.Manser E, Loo T H, Koh C G, Zhao Z S, Chen X Q, Tan L, Tan I, Leung T, Lim L. Mol Cell. 1998;1:183–192. doi: 10.1016/s1097-2765(00)80019-2. [DOI] [PubMed] [Google Scholar]
- 16.Habets G G, Scholtes E H, Zuydgeest D, van der Kammen R A, Stam J C, Berns A, Collard J G. Cell. 1994;77:537–49. doi: 10.1016/0092-8674(94)90216-x. [DOI] [PubMed] [Google Scholar]
- 17.Hart M J, Eva A, Zangrilli D, Aaronson S A, Evans T, Cerione R A, Zheng Y. J Biol Chem. 1994;269:62–65. [PubMed] [Google Scholar]
- 18.Coso O A, Chiariello M, Yu J C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind J S. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 19.Minden A, Lin A, Claret F X, Abo A, Karin M. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 20.Kawazoe N, Watabe M, Masuda Y, Nakajo S, Nakaya K. Oncogene. 1999;18:2413–2421. doi: 10.1038/sj.onc.1202555. [DOI] [PubMed] [Google Scholar]
- 21.Sander E E, ten Klooster J P, van Delft S, van der Kammen R A, Collard J G. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka M, Lu W, Gupta R, Mayer B J. Proc Natl Acad Sci USA. 1997;94:4493–4498. doi: 10.1073/pnas.94.9.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu W, Katz S, Gupta R, Mayer B J. Curr Biol. 1997;7:85–94. doi: 10.1016/s0960-9822(06)00052-2. [DOI] [PubMed] [Google Scholar]
- 24.Lu W, Mayer B J. Oncogene. 1999;18:797–806. doi: 10.1038/sj.onc.1202361. [DOI] [PubMed] [Google Scholar]
- 25.Yoshii S, Tanaka M, Otsuki Y, Wang D Y, Guo R J, Zhu Y, Takeda R, Hanai H, Kaneko E, Sugimura H. Oncogene. 1999;18:5680–5690. doi: 10.1038/sj.onc.1202936. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka M, Gupta R, Mayer B J. Mol Cell Biol. 1995;15:6829–6837. doi: 10.1128/mcb.15.12.6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M. Genes Dev. 1998;12:3331–3336. doi: 10.1101/gad.12.21.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okabe-Kado J, Kasukabe T, Hozumi M, Honma Y, Kimura N, Baba H, Urano T, Shiku H. FEBS Lett. 1995;363:311–315. doi: 10.1016/0014-5793(95)00338-a. [DOI] [PubMed] [Google Scholar]
- 29.Chiadmi M, Morera S, Lascu I, Dumas C, Le Bras G, Veron M, Janin J. Structure (London) 1993;1:283–293. doi: 10.1016/0969-2126(93)90016-a. [DOI] [PubMed] [Google Scholar]
- 30.Rogers S, Wells R, Rechsteiner M. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 31.Brown J L, Stowers L, Baer M, Trejo J, Coughlin S, Chant J. Curr Biol. 1996;6:598–605. doi: 10.1016/s0960-9822(02)00546-8. [DOI] [PubMed] [Google Scholar]
- 32.Zhang S, Han J, Sells M A, Chernoff J, Knaus U G, Ulevitch R J, Bokoch G M. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- 33.van Leeuwen F N, van der Kammen R A, Habets G G, Collard J G. Oncogene. 1995;11:2215–2221. [PubMed] [Google Scholar]
- 34.Michiels F, Stam J C, Hordijk P L, van der Kammen R A, Ruuls-Van Stalle L, Feltkamp C A, Collard J G. J Cell Biol. 1997;137:387–398. doi: 10.1083/jcb.137.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kikkawa S, Takahashi K, Shimada N, Ui M, Kimura N, Katada T. J Biol Chem. 1990;265:21536–21540. [PubMed] [Google Scholar]
- 36.Randazzo P A, Northup J K, Kahn R A. Science. 1991;254:850–853. doi: 10.1126/science.1658935. [DOI] [PubMed] [Google Scholar]
- 37.Randazzo P A, Northup J K, Kahn R A. J Biol Chem. 1992;267:18182–18189. [PubMed] [Google Scholar]
- 38.Freije J M, Blay P, MacDonald N J, Manrow R E, Steeg P S. J Biol Chem. 1997;272:5525–5532. doi: 10.1074/jbc.272.9.5525. [DOI] [PubMed] [Google Scholar]
- 39.Sander E E, van Delft S, ten Klooster J P, Reid T, van der Kammen R A, Michiels F, Collard J G. J Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark E A, King W G, Brugge J S, Symons M, Hynes R O. J Cell Biol. 1998;142:573–586. doi: 10.1083/jcb.142.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Postel E H. Int J Biochem Cell Biol. 1998;30:1291–1295. doi: 10.1016/s1357-2725(98)00087-9. [DOI] [PubMed] [Google Scholar]
- 42.Paravicini G, Steinmayr M, Andre E, Becker-Andre M. Biochem Biophys Res Commun. 1996;227:82–87. doi: 10.1006/bbrc.1996.1471. [DOI] [PubMed] [Google Scholar]
- 43.Lombardi D, Sacchi A, D'Agostino G, Tibursi G. Exp Cell Res. 1995;217:267–271. doi: 10.1006/excr.1995.1086. [DOI] [PubMed] [Google Scholar]
- 44.Bourguignon L Y, Zhu H, Shao L, Chen Y W. J Biol Chem. 2000;275:1829–1838. doi: 10.1074/jbc.275.3.1829. [DOI] [PubMed] [Google Scholar]