

Abstract
Over the last several years, the sea lamprey (Petromyzon marinus) has grown substantially as a model for understanding the evolutionary fundaments and capacity of vertebrate developmental and genome biology. Recent work on the lamprey genome has resulted in a preliminary assembly of the lamprey genome and led to the realization that nearly all somatic cell lineages undergo extensive programmed rearrangements. Here we describe the development of a bacterial artificial chromosome (BAC) resource for lamprey germline DNA and use sequence information from this resource to probe the subchromosomal structure of the lamprey genome. The arrayed germline BAC library represents ~10× coverage of the lamprey genome. Analyses of BAC-end sequences reveal that the lamprey genome possesses a high content of repetitive sequences (relative to human), which show strong clustering at the subchromosomal level. This pattern is not unexpected given that the sea lamprey genome is dispersed across a large number of chromosomes (n~99) and suggests a low-copy DNA targeting strategy for efficiently generating informative paired-BAC-end linkages from highly repetitive genomes. This library therefore represents a new and biologically informed resource for understanding the structure of the lamprey genome and the biology of programmed genome rearrangement.
Introduction
Lampreys are a vestige of an ancient vertebrate group that branched from the majority of extant vertebrate lineages prior to the advent of jaws and paired appendages, approximately 500 million years ago (Janvier 2006). The lamprey is therefore positioned in the vertebrate tree of life wherein it can provide unique insight into the cellular and developmental processes that define the fundaments of vertebrate biology. For example, recent studies on lamprey have revealed fundamental features of the vertebrate immune system (Amemiya et al. 2007), neural crest regulatory network (Sauka-Spengler et al. 2007), and the diversification of mesodermal derivatives (Kusakabe and Kuratani 2007). The deep evolutionary history of lamprey also makes it an attractive system for understanding how basal cellular and developmental processes can be modified at the molecular level. This is because the extensive conservation of the basic vertebrate cellular and developmental mechanisms is seasoned by the evolution of novel genes and genetic pathways, which have been selected to regulate these processes over the last 500 MY of lamprey evolution. The lamprey genome thus represents a vast source of information regarding basal aspects of vertebrate cellular and developmental processes and novel genetic strategies for manipulating these deeply conserved processes. Consequently, the National Institutes of Health invested in the sequencing of the sea lamprey genome. Whole genome shotgun (WGS) sequencing was performed on liver DNA to approximately 7× genome coverage (Washington University Genome Sequencing Center 2007). Several attempts have been made to assemble this WGS dataset into a contiguous genome assembly; however, the current version remains highly fragmented (Washington University Genome Sequencing Center 2007; Rogozin et al. 2007; Libants et al. 2009). Elucidation of the broad-scale structure of the lamprey genome will presumably require the development of additional computational and genomic resources.
The biology of the lamprey genome differs significantly from that of other known vertebrate genomes. All vertebrate species undergo a small number of programmed local rearrangements during development (e.g., remodeling of immune receptors) (Dudley et al. 2005; Kapitonov and Jurka 2005; Kim et al. 2007; Rogozin et al. 2007), though a limited number of species are known to undergo much more extensive reorganizations (Kubota et al. 1997; Goto et al. 1998; Kubota et al. 2001; Smith et al. 2009). These changes mimic the dysregulated changes in genome architecture that give rise to cancers or other genomic disorders (Ye et al. 2007; Mitelman et al. 2007) but are presumably highly regulated and reproducible from generation to generation (Smith et al. 2009). We have recently reported the existence of widespread programmed genome rearrangements (PGRs) in the sea lamprey (Petromyzon marinus) (Smith et al. 2009). These rearrangements are tightly regulated, occur early in development, and result in the loss of transcribed genes. This discovery is significant with respect to existing lamprey genome resources because the large WGS dataset was derived from a somatic tissue (liver), which is missing approximately 20% of the DNA that is present in the germline progenitor lineages. This new understanding of the dynamic nature of the lamprey genome and the fragmentary status of the existing assembly argue strongly for the development of genomic resources that are targeted at the germline and effectively span assembly gaps (including gene-encoding regions that are discarded due to PGRs).
The bacterial artificial chromosome (BAC) system can stably accommodate exogenous inserts that are very large (100–300 kilobases, kb), allowing entire eukaryotic genes (including flanking regulatory regions) to be encompassed in a single clone. The BAC system is based on plasmid vectors that are essentially composed of an F-factor origin of replication and an antibiotic resistance gene (Shizuya et al. 1992; Osoegawa et al. 1998; Amemiya et al. 1999). The F-factor replicon allows propagation of the bacterial plasmid as a single-copy entity in Escherichia coli, thus permitting stable propagation of cloned inserts greater than 100 kb pairs (kb). The ability to accommodate such large inserts is advantageous for many applications in genome biology, including positional cloning, targeted genomic sequencing, and as vehicles for generating transgenic animals. The entire procedure is conceptually simple although the actual generation and arraying of a library is technically challenging, highly empirical, and labor intensive (Osoegawa et al. 1998; Miyake and Amemiya 2004).
In this paper, we report the construction of a germline-specific BAC library from the lamprey. This represents the first lamprey genomic resource that is specifically targeted to the definitive germline genome and therefore provides representation of ~20% of single- and multi-copy genomic sequence that is not represented in any other existing genomic resource for this species. Indeed sequences from this library have proven valuable for identifying germline-specific DNAs for the sea lamprey and demonstrating that the species undergoes PGR on a global scale (Smith et al. 2009). The library contains 168,960 clones with an average insert size of ~140 kb, corresponding to ~10× coverage of the 2.31 gb sperm genome (Smith et al. 2009). Analysis of 3,072 clone-end reads from this library reveals that (1) relative to the human genome, the lamprey genome contains a large amount of long-repetitive DNA, (2) low-copy regions (e.g., containing single-copy genes) are strongly clustered and distributed non-randomly relative to high-copy regions, and (3) many repetitive sequences are unique to lamprey or are vestiges of repeats that were present in the early chordate lineage but lost in “higher” vertebrates. These observations are consistent with expectations given the lamprey’s evolutionary history and complex karyotype (n~99) and indicate that this BAC resource can provide critical long-distance linkages that will be necessary to improve the existing and highly fragmented lamprey genome assembly (Washington University Genome Sequencing Center 2007). Moreover, the resource provides access to long-insert clones that contain germline-specific sequences thereby filling a “biological” gap in the existing WGS dataset.
Materials and methods
BAC library construction
Preparation of high molecular weight (HMW) DNA
A BAC library was constructed from agarose-embedded sperm nuclei that were isolated from a single individual. Sperm was isolated from the testes of a single male adult lamprey. The specimen was first anesthetized in MS222 [1 g/l in 0.5× Marcs modified ringers solution] (Nikitina et al. 2009), and the testes were removed and immediately minced in 1× lamprey PBS (7.0 g/l NaCl, 0.2 g/l KCl, 0.29 g/l MgSO4 7H2O, 0.21 g/l MgCl2 6H2O, 0.46 g/l KH2PO4, 3.82 g/l Na2HPO4 7H2O, and 0.13 g/l CaCl2 2H20). Sperm cells were dispersed from minced testes by extensively triturating in fresh PBS. The sperm cell suspension was filtered through 20-μm mesh to remove connective tissue, and spermatozoa were pelleted by centrifugation at 1,000×g for 15 min at 4°C. The pellet of sperm was diluted in PBS to a concentration of 20 million cells per milliliter, equilibrated to 45°C for 5 min. and embedded in agarose. Preparation of DNA-embedded agarose plugs for library construction was performed using previously described methods (Amemiya et al. 1996).
Partial digestion of HMW DNA
Prior to partial digestion, plugs were equilibrated to 0.5× TE for 48 h at 4°C, then to 0.5× TBE overnight at 4°C. A pulsed field gel electrophoresis (PFGE) prerun was performed in order to remove unwanted, smaller-sized DNA molecules prior to restriction digestion (Osoegawa et al. 1998). Plugs were recovered from the wells of the gel then equilibrated to 0.5× TE overnight at 4°C. Pilot partial digestions using varying amounts of HindIII were carried out in order to optimize the digestion conditions prior to scale-up (Amemiya et al. 1996). DNA fragments were separated by PFGE on a 1% agarose gel (Pulse-Field Certified, Bio-Rad) using a CHEF XA Mapper (Bio-Rad) in 1/2× TBE buffer using previously described methods (Osoegawa et al. 1998). Gel slices were taken from the preparative lane that contained the HMW DNA fragments. A total of eight fractions ranging from 50–300 kb were excised from the gel. A sliver from each fraction was used in a step ladder gel to determine the size range of the DNA fragments. Fractions 3 (~110–140 kb) and 4 (~125–180 kb) were chosen for further processing and were equilibrated in 1/2× TBE. Electroelution, ligation into the pCC1BAC vector (Epicentre), and transformation were performed as previously described (Strong et al. 1997; Lang et al. 2006).
Insert size screening
An initial screening of clones was performed using Epilyse (Epicentre) on 52 random white colonies (26 per fraction), which determines the frequency of inserts and a rough estimate of size. For further analysis, DNA from 24 clones was isolated using a standard alkaline lysis miniprep procedure. Each clone was digested using NotI, and sizing was accomplished using PFGE (15 h and 1 s initial time, 20 s final time, 14°C, field angle 120°, and 6 V/cm) with the low-range PFG marker.
Arraying
Transformants were plated on Luria Bertani media (LB)/1.5% agar plates that were supplemented with 12.5 μg/ml chloramphenicol, 0.1 mM IPTG, and 120 μg/ml X-Gal. These were incubated overnight at 37°C and picked into 384-well microtiter plates (Genetix) containing LB supplemented with 12.5 μg/ml chloramphenicol and 5% v/v glycerol, using a colony-picking robot (Norgren Systems). A total of 440 plates were picked for this library. A Total Array System (BioRobotics) machine was used to spot high-density nylon filter sets (22 cm×22 cm) containing BAC DNA.
Sequencing and analysis
Four representative 384-well plates of BAC clones were sequenced by the Washington University Genome Sequencing Center. Base calls were generated, and sequences were quality-trimmed to Q20 using phred (Ewing and Green 1998; Ewing et al. 1998) and were vector-trimmed using phrap (Green 1994). Lamprey WGS reads were downloaded from the NCBI TraceArchives database (http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?), trimmed in the same manner, and formatted into a blast database consisting of 18,506,949 reads totaling 9,799,055,754 nucleotides. All BAC-end reads were aligned to all lamprey WGS reads using megablast (Zhang et al. 2000). Alignments were postprocessed to select long/high-identity alignments (≥400 bp in length, ≥95% nucleotide identity) that approximate a relevant range of lengths and sequence identities for whole genome assembly. Depth of WGS coverage was calculated for all end reads, and these coverage estimates were used to define sequences as low or high copy. Here, depth of coverage is defined as the average number of aligning reads per nucleotide unit length along the entire length of the query (BAC-end) sequence.
A second set of representative BAC and WGS datasets from a single diploid vertebrate was also selected for comparison with the lamprey genome. Paired-end reads from BAC and WGS libraries representing a single human genome were downloaded from the TraceArchives database and trimmed to remove vector and low-quality sequence (BAC dataset: n=229,578 reads; WGS dataset: n= 12,110,821 reads). Depth of coverage for human BAC ends was calculated using the same methods that were used for lamprey.
Lamprey repetitive reads that were identified in our ab initio screen were further characterized on the basis of sequence similarity to other sequences. Reads that corresponded to the Germ1 element were identified using blastn (Altschul et al. 1990), and the extended sequence of Germ1 was generated by assembling these sequences with the known fragment of Germ1, using ContigExpress (Vector NTI v11, Invitrogen). Other repeats were characterized by searching for similarity to a database of known repetitive elements (RepBase Update 20080801) (Jurka et al. 2005) using RepeatMasker (version open-3.2.5) (Smit et al. 2004).
Karyotypes
Chromosomes were prepared from lamprey testes and gill by first disaggregating the tissues in hypotonic KCl (75 mM) via gentle grinding in a Dounce homogenizer with a loose pestle. Single cells were allowed to swell in suspension for 1 h, prefixed by adding an equal volume of 3:1 methanol/glacial acetic acid (Farmer’s solution), then fixed through three changes of Farmer’s solution. Suspensions of fixed cells were dropped onto microscope slides and permitted to air dry at room temperature. Chromosome spreads were counterstained with DAPI (4′,6-diamidino-2-phenylindole).
Results and discussion
Insert length and genome coverage
The arrayed P. marinus germline BAC library consists of 440 microtiter plates (384-well) with inserts ranging from 100 to 200 kb in length (~140 kb average). Electrophoresis of supercoiled DNAs from 76 clones from the library was performed in order to estimate the distribution of insert sizes for this library (Fig. 1). The insert distribution is highly skewed toward large insert sizes, and we observed very few clones with no or short inserts. With an average size of ~140 kb and a total of 168,960 arrayed clones, the library represents approximately 23 gb of germline sequence. Given a germline genome size of 2.3 gb, this resource provides ~10× coverage of the lamprey germline. A further 550,000 clones are retained as frozen unamplified pools and represent an additional ~30× coverage. Importantly, this library encompasses the ~20% of germline DNA that is lost during the establishment of somatic lineages, which is not represented by any other existing lamprey clone resource. Given the combination of long-insert size, high coverage, and germline source of genomic DNA, this library should provide excellent representation of nearly every clonable region of the lamprey genome.
Fig. 1.
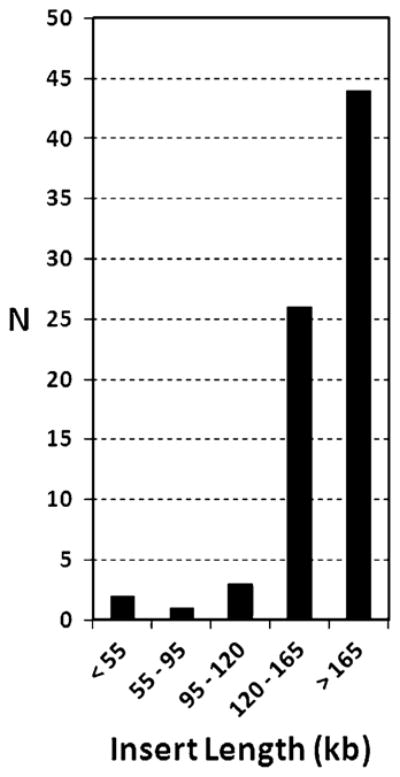
Frequency distribution of insert lengths for the sea lamprey (P. marinus) germline BAC library. A majority of inserts are longer than 120 kb
Sequencing analysis
Single-pass paired-end reads that were derived from a total of 1,536 clones yielded 1,846 sequences that were of sufficient quality/length (≥400 bp at Q20) to be included in subsequent analyses. A small proportion of these (n=11) had no/short inserts, consistent with our initial sizing estimates: these clones were not analyzed further. Paired-end analyses required that both end reads satisfied our quality thresholds, and thus, a total of 604 read pairs were used for analysis of the genome wide distribution of low-vs. high-copy DNAs. The distribution of coverage depths for lamprey and human BAC-end reads revealed that BAC-end coverage is generally bimodal, with one peak corresponding to roughly single-copy reads and the other corresponding to repetitive DNA (Fig. 2). The distributions of coverage depths imply that human and lamprey WGS datasets provide ~4× and ~7× coverage of single-copy DNAs, respectively. For both species, there are a non-trivial number of clones with a read depth of zero, which would be expected given the observed bell-shaped distributions with means that are not exceedingly far from zero. For lamprey, there is apparently an additional excess of zero-coverage reads. This is due, in part, to the fact that ~20% of germline sequences are absent from the somatic WGS dataset by virtue of the fact that they are removed during development via PGR (Smith et al. 2009).
Fig. 2.

Frequency distributions for alignment coverage depths of BAC-end sequences that were aligned to whole genome shotgun sequences datasets from the same species. The height of each bar represents the number of BAC-end reads that had a given range of coverage depths. The data that are presented here consider only those BACs that yielded >400 bp of Q20 sequence for both end reads. Low-copy (approximately single-copy) end reads fall within bell-shaped distributions with modal coverage depths <30
It is notable that lamprey appears to possess a much higher fraction of long/high-identity repetitive DNA (≤400 bp in length, ≤95% nucleotide identity) than does the human genome. The proportions of repetitive reads in the lamprey and human genomes are 0.581 and 0.045, respectively. Taken at face value, this extremely high repeat content might be interpreted as evidence that it will be extremely difficult to generate contigs from Sanger WGS sequencing data and existing automated assembly algorithms. However, it is also important to consider how these repetitive sequences are distributed throughout the genome. Essentially every vertebrate chromosome contains obligatory large stretches of highly repetitive DNA at the centromeres and near the telomeres, and the lamprey genome is no exception (Boan et al. 1996). Moreover, lamprey possess a karyotype (reported n~82–84) (Potter and Rothwell 1970) that is more complex than most vertebrates, including human (n=23).
Given our recent discovery that the lamprey somatic genome is actually a rearranged variant(s) of the lamprey germline genome (Smith et al. 2009), we sought to characterize the germline karyotype in order to better understand the expected distribution and abundance of centromeric and telomeric repeat clusters within the germline genome. Analysis of eight testes and eight gill karyotypes gave modal counts of n=99 and n=82, respectively (Table 1), revealing that the lamprey germline possesses an even greater degree of karyotypic complexity than the somatic genome. This variation in chromosome number presumably reflects fusion or deletion events that differentiate somatic lineages from germline. It is also possible that chromosomes counts may vary among somatic tissues, which could explain the high variation in previously reported chromosome counts (Potter and Rothwell 1970). Alternately, variation in somatic counts may reflect preparative or counting errors. More extensive surveys of somatic tissues will be necessary to fully address the issue of somatic karyotype variation in lamprey.
Table 1.
Chromosome counts (1N) for eight metaphase spreads from gill (mitotic) and eight metaphase spreads from testes (meiotic metaphase 1)
| Tissue | Replicate count
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Gill | 77 (154) | ~82 (163) | 82 (164) | 82 (164) | 82 (164) | ~82 (165) | 85 (170) | ~89 (177) |
| Testes | 97 | 97 | 98 | 98 | 99 | 99 | 99 | 101 |
Gill and testes chromosomes were prepared from the same animal. Raw chromosome counts are given in parentheses for gill; note that sister chromatids are paired at meiotic metaphase 1. Modal values are presented in italics
In light of lamprey’s karyotypic complexity, it seems well within reason that the lamprey should carry an additional burden of repetitive DNA. This is because of the simple fact that the lamprey genome contains several times the number of centromeres and telomeres than are present in the typical vertebrate genome. Moreover, these chromosomes are parsed from a genome that is only two thirds the size of the human genome. Importantly though, this architecturally obligatory repetitive DNA is expected to cluster distinctly from the majority of (assembly-relevant) low-copy DNA and should therefore prove much less disruptive to assembly of genic regions than if it were randomly distributed throughout the genome.
To better characterize the subchromosomal distribution (i.e., 140-kb spacing) of low- vs. high-copy DNA in the lamprey and human genomes, we tabulated the number of BACs that fell into each of three categories: LL, both ends are low copy (i.e., represented by ≤30 reads); HH, both ends are high copy (i.e., represented by >30 reads); and HL, one high-copy end and one low-copy end. For both species, the distributions of HH and LL classes are strongly enriched relative to expectations of random sampling from the observed numbers of high- and low-copy ends (lamprey: χ2=214.0, df=2, N=604 pairs, P=3.4E−47; human: χ2= 6.1E4, df=2, N=11,490 pairs, P≤1E−50) (Table 2). In total, 30% of lamprey BACs possess paired ends representing low-copy DNA; this represents approximately twice the number that would be expected given random sampling from the genome. The overabundance of HH and LL classes provides direct evidence that repetitive sequences are strongly clustered within the lamprey genome. This inference is further supported by restriction fingerprints of BACs from this library, which frequently yield low-complexity fingerprints (Fig. 3), indicating that these long (>100 kb) inserts are composed almost entirely of tandemly repeated sequences. This new knowledge of the repetitive structure of the lamprey genome indicates that it should be possible to assemble long contiguous sequences for genic regions of the lamprey genome. Moreover, these results imply that single-copy targeting strategies may represent a more efficient approach toward generation of BAC-based linkage information for lamprey and other similarly structured genomes. For lamprey, sequencing efforts could be reduced by ~30% if one end of each BAC was sequenced and the paired read only generated on the contingency that the first is non-repetitive.
Table 2.
Analysis of paired-end depths for human and lamprey BACs
| Copy # | Species | Count | Prop (obs) | Prop (exp) | % Enrichment | χ2 (DF) | P |
|---|---|---|---|---|---|---|---|
| LL | Lamprey | 181 | 0.30 | 0.16 | 91 | 77.9 (1) | 1.1E−18 |
| Human | 103,198 | 0.93 | 0.88 | 5 | |||
| HL | Lamprey | 117 | 0.19 | 0.48 | −60 | 102.4 (1) | 4.5E−24 |
| Human | 3,235 | 0.03 | 0.11 | −74 | |||
| HH | Lamprey | 306 | 0.51 | 0.36 | 39 | 33.6 (1) | 6.6E−09 |
| Human | 5,057 | 4.5E−3 | 0.00 | 1,166 | |||
| Total | Lamprey | 604 | 214.0 (2) | 3.4E−47 | |||
| Human | 111,490 |
BACs were assigned to one of three categories (LL, HL, and HH—see manuscript text for explanation). Proportions of LL and HH BACs were significantly greater than expected given random sampling and the observed frequencies of low- and high-copy BAC ends in each species
% Enrichment the percentage of the expected values by which the observed exceed expectation [(100×(obs−exp))−100], obs observed, exp expected, DF degrees of freedom
Fig. 3.
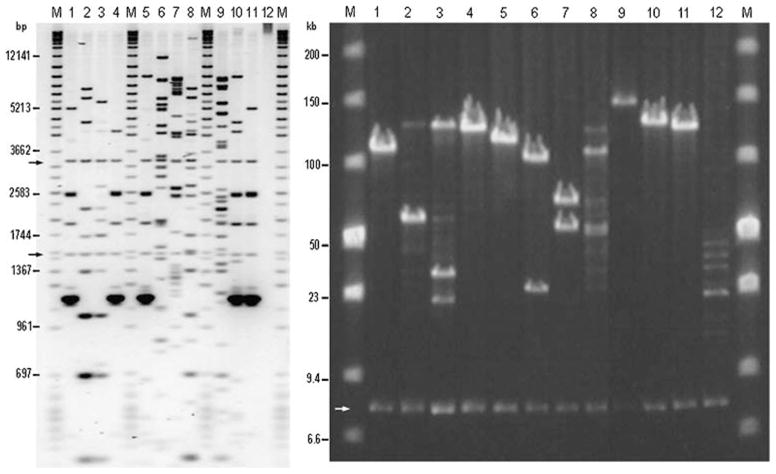
Left panel: Pst I digest of 12 BACs from the lamprey germline BAC library. Clones 6 and 7 show complex banding patterns that are consistent with single-copy/high-complexity DNA. Clones 1–5, 10, and 11 show simple banding patterns and exceptionally dark bands, indicating that they do not contain high-complexity DNA but are rather composed of a small number of repeated sequences. Clones 8 and 9 show moderate complexity, but also contain banding patterns that are seen in repetitive clones. These clones were selected on the basis of hybridization to a degenerate probe for homeobox genes. The repetitive sequence class that is present in this sample strongly cross-hybridizes with the probe. Right panel: NotI digest of these same clones. M DNA size marker: left panel—analytical marker DNA Wide Range (Promega) and MarkerV (Roche); right panel—low-range PFG Marker (New England Biolabs). Arrows mark bands that correspond to the BAC vector sequence
Content of the repetitive fraction
As the repetitive sequences represent a major component of the lamprey genome, we sought to further classify these sequences. It is known that sequence element Germ1 is enriched in germline, relative to soma, and represents a substantial fraction of the germline genome (Smith et al. 2009). As our BAC resource provides the most extensive sequence survey of lamprey germline to date, we reasoned that it might be possible to extend the known sequence of Germ1. By aligning the known 10,120-bp sequence of Germ1 to our BAC-end sequences, we were able to assign 357 BAC-end sequences to this repeat. Assembling these with the known sequence allowed us to extend Germ1 an additional 1,194-bp 5′ and 576-bp 3′ for a final length of 11,900 bp.
With a more complete knowledge of the Germ1 sequence in hand, we sought to further characterize the remaining 557 repetitive BAC ends that were identified in our ab initio survey. Ninety-two of these represented low-complexity repeats and were not analyzed further. Comparison to known vertebrate repeat classes resulted in matches for 75 of the remaining 465 sequences (Table 3). The majority of these are similar to known vertebrate repeats, largely classes of DNA transposons (n=38) and LINE retroelements (n=22). Comparison to known repetitive elements from invertebrates identified matches for an additional 62 reads. Many of these “invertebrate” repeats (n=33) are most similar to sequences from ascidians: Ciona intestinalis and Ciona savignyi. This observation is consistent with the fact that the ascidians are one of the closest outgroups to the vertebrate clade, and suggests that some of the elements that constitute the repetitive fraction of the lamprey genome may be vestiges of sequences that were present in the vertebrate ancestor and subsequently lost within the jawed vertebrate lineage. The additional 231 of the repetitive reads that were identified in this study (41% of non-Germ1 repeats) are not similar to any known repeat class. These may represent elements that evolved independently in the lamprey lineage or elements that were present in the vertebrate ancestor but not retained in jawed vertebrates. Notably, this diversity of repeats implies that repeat content of BACs is not strongly correlated with extensive cloning bias, in which case most clone ends should represent one or few repeat classes. The abundance and diversity of novel repeat sequences in the lamprey genome, in conjunction with the taxon’s intermediate phylogenetic position between the invertebrates and the bony vertebrates, underscores the importance of ongoing efforts to characterize the repeat content and large-scale structure of the lamprey genome.
Table 3.
Classification of repetitive reads that were identified among lamprey germline BAC-end sequences
| Repeat element | Class | Subclass | Class | Subclass |
|---|---|---|---|---|
| Germ1 | 357 | |||
| Lamprey specific | 231 | |||
| Identifiable | 226 | |||
| Vertebrate | Invertebrate | |||
| DNA transposon | 38 | 6 | ||
| hAT | 29 | 5 (1:Ci) | ||
| Helotron | 5 | – | ||
| TC1 | 3 | – | ||
| Other | 1 | 1 | ||
| LINE (retro) | 22 | 46 | ||
| CIR2 | – | 30 (Ci) | ||
| Poseidon | 12 | – | ||
| Expander | 4 | – | ||
| CR1 | 3 | 4 | ||
| Other | 3 | 12 (1:Ci) | ||
| LTR (retro) | 1 | 1 | ||
| SINE (retro) | 8 | – | ||
| 5S RNA | 6 | – | ||
| U2-Satellite | – | 2 (Ci) | ||
| tRNA | – | 6 | ||
| Low complexity | 92 | – | ||
Non-redundant counts of end reads are reported for each repeat class. Subclasses of vertebrate repeats immediately below their parent class and are indented
Retro retrotransposable element, (Ci) all hits to a repeat subclass are from Ciona intestinalis or C. savignyi, (n:Ci) no hits to the subclass are from Ciona intestinalis or C. savignyi, (–) no hits identified within a class or subclass
Conclusions
Here we describe a BAC resource for the lamprey germline genome. This resource provides access to long-insert clones spanning the majority of the lamprey genome, and represents the only existing clone/sequence resource that provides representation of the ~20% of the lamprey genome that is lost during early development, including transcribed genes (Smith et al. 2009). Analysis of end reads from this library reveals that the lamprey genome possesses an exceptionally large fraction of repetitive DNA and that this repetitive DNA is strongly clustered at the subchromosomal level. This library represents a new and biologically informed resource for dissecting the structure of the lamprey genome and the biology of programmed genome rearrangement.
Acknowledgments
This work was supported by the National Institutes of Health [grant number GM079492] and the National Science Foundation [grant number MCB-0719558] to CTA. This work was supported by the National Institutes of Health [grant number T32-HG00035, F32-GM087919] to JJS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Contributor Information
Jeramiah J. Smith, Email: jsmith@benaroyaresearch.org, Benaroya Research Institute at Virginia Mason, 1201 Ninth Avenue, Seattle, WA 98101-2795, USA
Andrew B. Stuart, Benaroya Research Institute at Virginia Mason, 1201 Ninth Avenue, Seattle, WA 98101-2795, USA
Tatjana Sauka-Spengler, California Institute of Technology, Pasadena, CA 92125, USA.
Sandra W. Clifton, Washington University School of Medicine Genome Center, St. Louis, MO 63108, USA
Chris T. Amemiya, Benaroya Research Institute at Virginia Mason, 1201 Ninth Avenue, Seattle, WA 98101-2795, USA
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amemiya CT, Ota T, Litman GW. Construction of P1 artificial chromosome (PAC) libraries from lower vertebrates. In: Lai E, Birren B, editors. Analysis of nonmammalian genomes. Academic; San Diego: 1996. pp. 223–256. [Google Scholar]
- Amemiya CT, Zhong TP, Silverman GA, Fishman MC, Zon LI. Zebrafish YAC, BAC, and PAC genomic libraries. Methods Cell Biol. 1999;60:235–258. doi: 10.1016/s0091-679x(08)61904-4. [DOI] [PubMed] [Google Scholar]
- Amemiya CT, Saha NR, Zapata A. Evolution and development of immunological structures in the lamprey. Curr Opin Immunol. 2007;19:535–541. doi: 10.1016/j.coi.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boan F, Vinas A, Rodriguez JM, Sanchez L, Gomez-Marquez J. A new EcoRI family of satellite DNA in lampreys. FEBS Lett. 1996;394:187–190. doi: 10.1016/0014-5793(96)00947-7. [DOI] [PubMed] [Google Scholar]
- Dudley DD, Chaudhuri J, Bassing CH, Alt FW. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol. 2005;86:43–112. doi: 10.1016/S0065-2776(04)86002-4. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Goto Y, Kubota S, Kohno S. Highly repetitive DNA sequences that are restricted to the germ line in the hagfish Eptatretus cirrhatus: a mosaic of eliminated elements. Chromosoma. 1998;107:17–32. doi: 10.1007/s004120050278. [DOI] [PubMed] [Google Scholar]
- Green P. Phrap. University of Washington, Department of Genome Sciences; 1994. http://www.phrap.org/phredphrapconsed.html. [Google Scholar]
- Janvier P. Palaeontology: modern look for ancient lamprey. Nature. 2006;443:921–924. doi: 10.1038/443921a. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005;3:e181. doi: 10.1371/journal.pbio.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Oh SC, Lim KJ, Kasamatsu J, Heo JY, Park BS, Lee H, Yoo OJ, Kasahara M, Lee JO. Structural diversity of the hagfish variable lymphocyte receptors. J Biol Chem. 2007;282:6726–6732. doi: 10.1074/jbc.M608471200. [DOI] [PubMed] [Google Scholar]
- Kubota S, Ishibashi T, Kohno S. A germline restricted, highly repetitive DNA sequence in Paramyxine atami: an interspecifically conserved, but somatically eliminated, element. Mol Gen Genet. 1997;256:252–256. doi: 10.1007/s004380050567. [DOI] [PubMed] [Google Scholar]
- Kubota S, Takano J, Tsuneishi R, Kobayakawa S, Fujikawa N, Nabeyama M, Kohno S. Highly repetitive DNA families restricted to germ cells in a Japanese hagfish (Eptatretus burgeri): a hierarchical and mosaic structure in eliminated chromosomes. Genetica. 2001;111:319–328. doi: 10.1023/a:1013751600787. [DOI] [PubMed] [Google Scholar]
- Kusakabe R, Kuratani S. Evolutionary perspectives from development of mesodermal components in the lamprey. Dev Dyn. 2007;236:2410–2420. doi: 10.1002/dvdy.21177. [DOI] [PubMed] [Google Scholar]
- Lang M, Miyake T, Braasch I, Tinnemore D, Siegel N, Salzburger W, Amemiya CT, Meyer A. A BAC library of the East African haplochromine cichlid fish Astatotilapia burtoni. J Exp Zoolog B Mol Dev Evol. 2006;306:35–44. doi: 10.1002/jez.b.21068. [DOI] [PubMed] [Google Scholar]
- Libants S, Carr K, Wu H, Teeter JH, Chung-Davidson YW, Zhang Z, Wilkerson C, Li W. The sea lamprey Petromyzon marinus genome reveals the early origin of several chemosensory receptor families in the vertebrate lineage. BMC Evol Biol. 2009;9:180. doi: 10.1186/1471-2148-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- Miyake T, Amemiya CT. BAC libraries and comparative genomics of aquatic chordate species. Comp Biochem Physiol C Toxicol Pharmacol. 2004;138:233–244. doi: 10.1016/j.cca.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Nikitina N, Bronner-Fraser M, Sauka-Spengler T. Emerging model organisms: a laboratory manual. CSHL; Cold Spring Harbor: 2009. The sea lamprey Petromyzon marinus: a model for evolutionary and developmental biology; pp. 405–429. [DOI] [PubMed] [Google Scholar]
- Osoegawa K, Woon PY, Zhao B, Frengen E, Tateno M, Catanese JJ, de Jong PJ. An improved approach for construction of bacterial artificial chromosome libraries. Genomics. 1998;52:1–8. doi: 10.1006/geno.1998.5423. [DOI] [PubMed] [Google Scholar]
- Potter IC, Rothwell B. The mitotic chromosomes of the lamprey, Petromyzon marinus L. Experientia. 1970;26:429–430. doi: 10.1007/BF01896930. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Iyer LM, Liang L, Glazko GV, Liston VG, Pavlov YI, Aravind L, Pancer Z. Evolution and diversification of lamprey antigen receptors: evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat Immunol. 2007;8:647–656. doi: 10.1038/ni1463. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, Meulemans D, Jones M, Bronner-Fraser M. Ancient evolutionary origin of the neural crest gene regulatory network. Dev Cell. 2007;13:405–420. doi: 10.1016/j.devcel.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Shizuya H, Kim U-G, Mancino V, Slepak T, Tachiiri Y, Simon M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci USA. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker Open-3.0. 2004 http://www.repeatmasker.org.
- Smith JJ, Antonacci F, Eichler EE, Amemiya CT. Programmed loss of millions of base pairs from a vertebrate genome. Proc Natl Acad Sci USA. 2009;106:11212–11217. doi: 10.1073/pnas.0902358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong SJ, Ohta Y, Litman GW, Amemiya CT. Marked improvement of PAC and BAC cloning is achieved using electroelution of pulsed-field gel-separated partial digests of genomic DNA. Nucleic Acids Res. 1997;25:3959–3961. doi: 10.1093/nar/25.19.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington University Genome Sequencing Center. Washington University School of Medicine; 2007. http://genome.wustl.edu/pub/organism/Other_Vertebrates/Petromyzon_marinus/assembly/Petromyzon_marinus-3.0/ [Google Scholar]
- Ye CJ, Liu G, Bremer SW, Heng HH. The dynamics of cancer chromosomes and genomes. Cytogenet Genome Res. 2007;118:237–246. doi: 10.1159/000108306. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]