

Abstract
Disorders of sex development (DSD) with or without ambiguous genitalia require medical attention to reach a definite diagnosis. Advances in identification of molecular causes of abnormal sex, heightened awareness of ethical issues and this necessitated a re-evaluation of nomenclature. The term DSD was proposed for congenital conditions in which chromosomal, gonadal or anatomical sex is atypical. In general, factors influencing sex determination are transcriptional regulators, whereas factors important for sex differentiation are secreted hormones and their receptors.The current intense debate on the management of patients with intersexuality and related conditions focus on four major issues: 1) aetiological diagnosis, 2) assignment of gender, 3) indication for and timing of genital surgery, 4) the disclosure of medical information to the patient and his/her parents. The psychological and social implications of gender assignment require a multidisciplinary approach and a team which includes ageneticist, neonatologist, endocrinologist, gynaecologist, psychiatrist, surgeon and a social worker. Each patient should be evaluated individually by multidisciplinary approach.
Conflict of interest:None declared.
Keywords: Sex development disorders, gonads, gender assignment
INTRODUCTION
Phenotypic sex results from the differentiation of internal ducts and external genitalia under the influence of sex-determining genes and hormones (1,2,3,4,5,6,7,8). In one of every 4500 births, the genital appearance is abnormal and it is not possible to decide at first glance the sex of the infant. Disorders of sexual development (DSD) are a very important clinical issue with its different aspects relating to diagnosis, treatment and sex of rearing. The classification of ambiguous genitalia in patients is difficult because similar or identical phenotypes may have several aetiologies (1,2,3,4,5).
Physiology of Sex Development
Sex determination is the result of a series of molecular events that direct the undifferentiated bipotential gonad to become either a testis or an ovary. The bipotential gonad develops from the urogenital ridge. By 6-7 weeks of fetal life, fetuses of both sexes have two sets of internal ducts: the Müllerian ducts and the Wolffian ducts. The external genitalia at 6-7 weeks gestation appear female and include a genital tubercle, the genital folds, urethral folds and a urogenital opening. During the bipotential stage, many genes (WT1, SF1, LHX9, LIM1, PAX2, GATA4, EMX2, WNT4) are expressed at similarly low levels in XY and XX gonadal ridges (9,10,11,12,13). However, all are necessary for normal gonadal development in both sexes, with gene dosage and relative expression levels playing an important role in the sexually divergent fate of the gonads. The steroidogenic factor 1 (SF1), Wingless-type gene (WNT4) and Wilms tumor suppressor gene 1 (WT1) are expressed in the urogenital ridge and have a role in the formation of the gonads, kidneys and adrenal cortex. SF1 located on chromosome 9p33, is important for the biosynthesis of adrenal and gonadal steroids and for the development of the ventral nucleus of the hypothalamus. SF1 is expressed in the bipotential gonad of both sexes. Expression of SF1 continues in the developing testis, but is repressed in the ovary. The WT1 gene is located on chromosome 11p13 and encodes a transcription factor necessary for the development of the bipotential gonad and the kidneys. Wilms tumor suppressor gene activates transcription of SRY (15,16). The important event in gonadal differentiation is the commitment of the bipotential gonad to become either an ovary or a testis. The essential genes affecting this process are as follows: WT1, SF1,CBX2, SOX9, fibroblast growth factor 9 (FGF9), prostaglandin D2 (PGD2), DAX1, WNT4, forkhead family transcription factor 2 (FOXL2), R-Spondin 1 (RSPO1) and b-catenin. While SOX9, FGF9 and PGD2 have more testis-promoting activity, DAX1, WNT4, FOXL2, RSPO1 and b-catenin are predominantly ovary-promoting genes (12). SOX9/FGF9 and WNT4/RSPO1 act as antagonistic signals in early gonadal differentiation.
Male Differentiation:The factors involved in testicular differentiation are given in Figure 1(14,15,16,17,18,19,20,21). Male phenotypic development can be viewed as a two-step process: 1) testis formation from the primitive gonad (sexual determination) and 2) internal and external genitalia differentiation by action of hormones secreted by the fetal testis (sexual differentiation) (10). The first step involves the interplay of several transcription factors and signaling cells (14,15,16,17,18,19,20,21).
Figure 1. Figure 1. Factors involved in testes differentation.
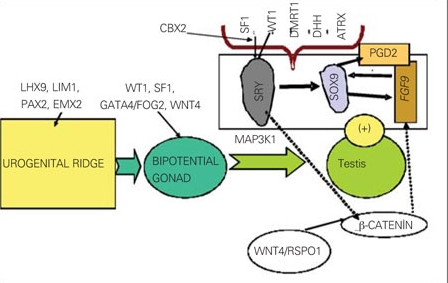
The DAX1 gene region on Xp21 is necessary for gonadal development in both sexes. A single copy of the DAX1 is essential for normal testicular cord formation. However, DAX1 overexpression in an XY fetus impairs testis differentiation. Testis determination occurs at about the sixth week of gestation. SRY gene that is located on the Y-chromosome (Yp11.3) initiates sex determination by downstream regulation of sex-determining factors. Expression of several genes including WT1,CBX2(M33), SF1, GATA4/FOG2 is critical to SRY activation. The SRY gene encodes a unique transcription factor that activates a testis-forming pathway (12,18,19). After expression of SRY, SOX9 (SRY-related HMG box gene) expression is upregulated in the developing testis (20). The SOX9 gene, located on 7q24.3-25.1, is essential for early testis development. SOX9 up-regulates PGD2 and FGF9 genes. FGF9 and PGD2 maintain SOX9 expression, forming a positive feed-forward loop in XY gonads (21). The balance between FGF9 and WNT4/RSPO1/b-catenin signals is shifted in favor of FGF9, establishing the male pathway. On the other hand, DMRT1, ATRX and DHH and several autosomal genes are also involved in testes determination (12).
The second step in male sex differentiation is a more straightforward process. The production of anti-Müllerian hormone (AMH) by Sertoli cells and androgens by Leydig cells in a critical concentration-dependent and time-dependent manner induces male sexual differentiation by means of a hormone–dependent process. AMH acts on its receptor in the Müllerian ducts to cause their regression. Testosterone (T) acts on the androgen receptor (AR) in the Wolffian ducts to induce the formation of epidydimis, deferent ducts and seminal vesicles. The Leydig cells also produce insulin-like factor 3 (INSL3, relaxin-like factor), which causes the testes to descend to the scrotum. T is further reduced to dihydrotestosterone (DHT), which acts on the androgen receptor of the prostate and external genitalia to cause its masculinization. Binding of T or DHT to AR is necessary for androgen effect. Various coregulators interact probably for transcriptional activity of AR. By contrast, coregulators such as corepressors repress transcriptions (22,23).
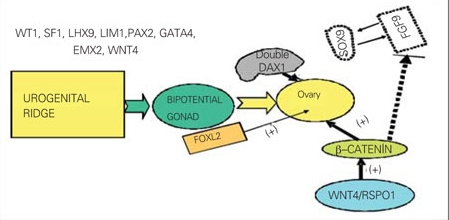
Female Differentiation:The factors involved in ovarian differentiation are given in Figure 2 (24,25,26,27). In the absence of SRY, the support cell precursors differentiate as granulosa cells, thus initiating the ovarian pathway. DAX1 is necessary for both testicular and ovarian development, with a need for precise gene expression dosage. Overexpression in either DAX1 or WNT4/RSPO1 antagonizes testis formation. WNT4/RSPO1/b-catenin pathway blocks FGF9 and promotes the ovarian fate (24). In XX gonads, WNT4 dominates and results in an induction of b-catenin and silencing of FGF9 and SOX9. WNT4-signaling pathway plays a major role in ovarian development and maintenance, regulation of Müllerian ducts formation and ovarian steroidogenesis (26,27,28). WNT4 has been shown to play a critical role in the development of the reproductive system and also in the formation of the kidneys, adrenals, pituitary gland, and mammary tissues. Absence of WNT4 leads to testis-like development within the ovary. Conversely, overexpression of WNT4 in the male leads to female sex reversal. RSPO1 is another gene essential in sex determination responsible for the protein RSPO1, which plays an important role in suppression of the SOX9 gene (27,28). Loss of function mutations in the human RSPO1 gene in mice results in the formation of ovotestes in the XX fetus (28). WNT4, RSPO1 and b-catenin seem to have both pro-ovarian and anti-testicular activities from early embryonic life, while FOXL2 may also have similar actions postnatally.* FOXL2 is expressed early within the genital ridge of the fetal, postnatal and adult ovary and eyelids. FOXL2 is involved in granulosa cell differentiation, follicle development and maintenance during fertile life (29). Müllerian ducts give rise to the fallopian tubes, uterus and the upper two-third of the vagina. In the female, the genital tubercle becomes the clitoris, the labio-scrotal folds become the labia majora, and the urethral folds become the labia minora.
Figure 2. Figure 2. Factors involved in ovarian differentation.
Nomenclature and Definitions
Formerly, intersex disorders were subdivided into three main groups as: associated with gonadal dysgenesis, associated with undervirilization of 46,XY individuals, and conditions associated with prenatal virilization of 46,XX subjects. The nomenclature used to describe atypical sexual differentiation has since changed (2,3,4,5). Instead of using the confusing and/or controversial terms such as "intersex," "hermaphroditism" and "sex reversal", the consensus statement recommended a new taxonomy based on the umbrella term, “DSD” (3). This broad category includes common entities such as Turner syndrome and Klinefelter syndrome as well as rare disorders such as cloacal exstrophy and aphallia. Many DSDs are associated with ambiguous genitalia, however, a few may present with delayed puberty or primary amenorrhea. The Lawson Wilkins Pediatric Endocrine Society (LWPES) and the European Society for Paediatric Endocrinology (ESPE) consensus group proposed the classification of DSDs into:
1) Sex chromosome DSDs (45,X Turner and variants, 47,XXY Klinefelter and variants, 45X/46XY mixed gonadal disgenesis (MGD) and chromosomal ovotesticular DSD “46XX/46XY chimeric type or mosaic type”); 2) 46,XY DSDs (disorders of testicular development or disorders in androgen synthesis/action); and 3) 46,XX DSDs (disorders of ovarian development or fetal androgen excess) (Table 1). Additional categorization based on sex chromosome complement was recommended but not clearly defined (30,31). It is recognized that some conditions do not fit exactly into one specific diagnostic category or may be placed in more than one category (5). The DSD nomenclature has recently divided "ovotesticular DSD" (formerly true hermaphroditism) into 46,XY ovotesticular DSD, 46,XX ovotesticular DSD, and chromosomal ovotesticular DSD (46,XX/46,XY” chimerism or 45,X/46,XY” mosaic type).
Table 1. Table 1. Disorders of sexual development (New DSD nomenclature) (3).
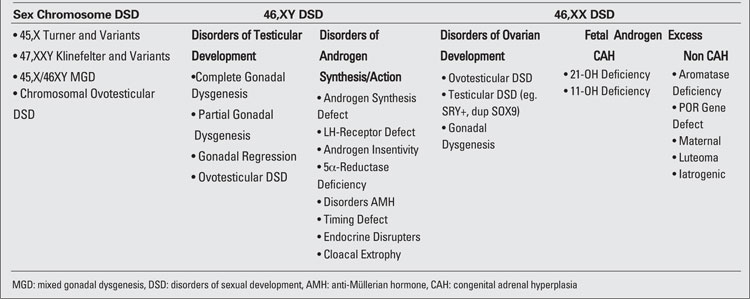
1)Sex chromosome DSDs (45,X Turner and variants, 47,XXY Klinefelter and variants, 45X/46XY MGD, choromosomal ovotesticular DSD (46XX/46XY chimeric type or mosaic type): This type of DSDs is associated with a numerical sex chromosome abnormality leading to abnormal gonadal development (2,3,5,30,31,32,33,34, 35,36,37). Sex chromosome DSD was formerly termed as gonadal dysgenesis in most of the patients in this group (5). If a testis is poorly formed, it is called a dysgenetic testis, and if an ovary is poorly formed, it is called a streak gonad. A patient with a Y chromosome is at high risk of developing a tumor in a streak or dysgenetic gonad. Klinefelter and Turner syndromes (TS) are the most frequently encountered sex chromosomal abnormalities (34,35,36). More than half of girls with TS have chromosomal mosaicism. The most common genotype of Klinefelter syndrome is XXY, although variants exist with different numbers of X chromosome. In patients with 45X/46XY MGD, clinical manifestations are highly variable, ranging from partial virilization and ambiguous genitalia at birth to a completely male or female phenotype. The most common feature of MGD is asymmetric development of the testes, often with a dysgenetic testis on one side and a streak gonad on the other. Asymmetrical external and internal genitalia may also be present. The presence of 45,X cell lines is frequently associated with Y chromosome rearrangements (commonly dicentric and ring Y chromosomes), which may also have an impact on the phenotype. Chromosomal ovotesticular DSD (chimeric type or mosaic type) is associated with ovarian and testicular tissues found in either the same or opposite gonad just as in 46,XX and 46,XY ovotesticular DSD. The genital duct develops according to the ipsilateral gonad.
2) 46,XY disorders of sex development (46,XY DSD):
The term “male pseudohermaphrodite” was used to describe the patients with incompletely masculinized external genitalia possessing an XY chromosome.** 46,XY DSD can result either from disorders of testicular development or disorders in androgen synthesis/androgen action (6). These patients are characterized by ambiguous or female external genitalia, caused by incomplete intrauterine masculinization. Male gonad(s) are palpable in the majority of 46,XY DSD patients. Abnormalities in the expression of genes involved in the cascade of testis determination can cause anomalies of gonadal development and consequently, 46,XY DSD (complete or partial forms of gonadal dysgenesis with or without syndromic phenotype, ovotesticular DSD, testicular regression syndrome).
Failure of testis determination results in the development of the female phenotype, while genetic alterations resulting in partial testicular development can give rise to a wide spectrum of incomplete masculinization. 46,XY partial gonadal dysgenesis, characterized by partial testicular differentiation and ambiguous genitalia, is usually observed in the newborn period. Mutation in WT1 gene results in Denys-Drash syndrome (without uterus) or Frasier syndrome (with uterus) characterized by 46,XY partial gonadal dysgenesis and severe renal dysfunction with or without Wilms tumor (16). Complete gonadal dysgenesis in 46,XY individuals (Swyer syndrome) is characterized by a female phenotype with full development of unambiguous female genitalia, normally developed Müllerian structures, and streak gonads. These streak gonads are removed due to their association with gonadoblastoma. In general, these patients present because of delayed puberty. In agonadism (vanishing testes syndrome, testicular regression) boys present with normal male genitalia, indicating that they must have had testicular function in the fetal period, and bilateral anorchia. The defects in peptide hormones and their receptors as well as the timing of hormonal exposure are also critical to appropriate male development. Leydig cell aplasia/hypoplasia, due to abnormalities in hCG/LH receptor, and T biosynthesis defects (STAR deficiency, P450scc deficiency, 3-b hydroxysteroid dehydrogenase type II deficiency, 17a-hydroxylase and 17,20-lyase deficiency, isolated 17,20-lyase deficiency, P450 oxidoreductase ”POR gene” defect, 17b-hydroxysteroid dehydrogenase III deficiency) result in androgen synthesis defect. Severely affected infants for POR gene of both sexes have ambiguous genitalia. The males are undervirilized because of defective 17,20-lyase activity of P450c17. Disorders of AMH and AMH receptors result in persistent Müllerian duct syndrome (PMDS). PMDS is inherited in a sex-limited autosomal recessive manner caused by a mutation in the AMH or AMH-receptor genes. 5a-reductase type 2 deficiency (38,39,40) and complete/partial forms of androgen insensitivity syndromes (CAIS, PAIS) result in disorders of androgen action (22,23).
3) 46,XX disorders of sex development (46,XX DSD): The term female pseudohermaphrodite was used to describe the patients with 46,XX karyotype and with masculinized external genitalia. Currently, these disorders are described as 46,XX DSD.** 46,XX DSD can result either from disorders of ovarian development or fetal androgen excess (6). SRY positivity; WNT4, RSPO1, b-catenin gene defects; and duplication of SOX9 gene lead to testis-like formation within the ovary (streak gonad, dysgenetic testis or ovotestis) in the 46,XX patients. A single copy of the WNT4 gene in females causes Müllerian abnormalities, renal abnormalies (e.g. renal agenesis), and androgen excess. Their phenotype resembles that of patients with the Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. With absence of both copies of this gene, females show male sex and SERKAL syndrome (female to male sex reversal; renal, adrenal and lung dysgenesis) (26). RSPO1 is essential in sex determination and skin differentiation. RSPO1 gene mutations lead to XX sex reversal, palmoplantar hyperkeratosis and predisposition to squamous cell carcinoma of the skin (27,28). FOXL2 mutations result in a variety of phenotypes, from adult ovarian failure to development of streak gonads (29). Mutations in FOXL2 are responsible for blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) and can be associated with premature ovarian failure. Ovarian dysgenesis coexisting with sensorineural deafness is diagnosed as Perrault syndrome. In ovotesticular DSDs, the most common karyotype is 46,XX followed by 46,XX/46,XY chimerism or mosaicism, and 46,XY. Most 46,XX ovotesticular DSDs are SRY-negative, and the genes responsible have not yet been identified. A mutated downstream gene in the sex determination cascade is likely to allow for testicular determination. 46,XX testicular DSD is distinct from XX ovotesticular DSD and is associated with male habitus, small testes, azoospermia and no evidence of uterus or ovaries.
Virilized females with two ovaries, XX karyotype and ambiguous genitalia are usually exposed to external genitalia androgens of fetal origin or to androgens of maternal origin. The majority of virilized 46,XX infants will have congenital adrenal hyperplasia (CAH) (most commonly 21a-hydroxylase and 11b- hydroxylase or rarely 3b-hydroxysteroid dehydrogenase deficiencies). Apparent combined P450c17 and P450c21 deficiency is a rare variant of CAH. Mutations of POR gene cause disordered steroidogenesis with prenatal virilization without worsening of postnatal virilization in female fetuses (41,42). Cytochrome POR is a protein that transfers electrons from NADPH to all microsomal cytochrome P450 enzymes and three steroidogenic enzymes, namely, P450c17 (17a-hydroxylase/17,20 lyase), P450c21 (21-hydroxylase), and P450aro (aromatase). Severely affected female infants for POR gene are virilized because of defective aromatase activity and because of the diversion of 17-hydroxyprogesterone (17OHP) to DHT via the “backdoor pathway” to androgens that bypass dihydroepiandrosterone (DHEA), androstenedione (A) and T (42). Severely affected infants also have the Antley-Bixler skeletal malformation syndrome (ABS) characterized by craniosynostosis and radio-humeral or radio-ulnar synostosis. Rarer causes of fetal androgen excess in XX infants are maternal androgen ingestion, maternal virilizing disease, fetoplacental aromatase deficiency, sulfatase deficiency, virilizing luteoma of pregnancy, glucocorticoid receptor mutation.
Aromatase deficiency is rare in humans (43). Aromatase is the enzyme that catalyzes conversion of androgens into estrogens, and if aromatase is nonfunctional because of an inactivating mutation, estrogen can not be synthesized. If the fetus lacks aromatase activity, DHEA produced by the fetal adrenal glands cannot be converted to estrogen by the placenta, and is converted to T peripherally. This results in virilization of both fetus and mother.
Cystic ovaries and delayed bone maturation can occur during childhood and adolescence in these girls. They may present at pubertal ages with primary amenorrhea, failure of breast development except in partial cases, virilization, and hypergonadotrophic hypogonadism.
Sulfotransferase deficiency is a monogenic cause of hyperandrogenism. DHEA sulfotransferase, known as SULT2A1, converts the androgen precursor DHEA to its inactive sulfate ester, DHEAS, thereby preventing the conversion of DHEA to an active androgen. If this pathway is blocked, more DHEA will be converted to androstenedione and hyperandrogenism may occur (44).
Investigation of DSD Patients
Optimal care of patients with DSD requires a multidisciplinary team and begins in the newborn period. A family history, prenatal history, a general physical examination with attention to any associated dysmorphic features, and an assessment of the genital anatomy are the first steps towards a correct diagnosis. The diagnostic evaluation of DSD includes hormone measurements, imaging, cytogenetic and molecular studies and in some cases endoscopic, laparoscopic and gonadal biopsy (6,7,8,45,46,50,51,52,53,54,55). The genetic evaluation includes karyotype, FISH and, more recently, specific molecular studies to screen the presence of mutations or gene dosage imbalance (AR, SRY, SF1, WT1, CYP21, SOX9, DAX-1, 17b hydroxysteroid dehydrogenase, 5a-reductase-2, and others). However, current molecular diagnosis is limited by cost, accessibility, and quality control. Ultrasonography shows the presence or absence of Müllerian/Wolfian structures and can locate the gonads and their echo texture. Ultrasonography also can identify associated malformations such as renal abnormalities.
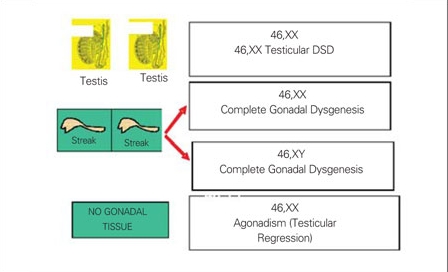
Common findings suggesting DSD are male appearance with associated abnormalities of genitalia including severe hypospadias with bifid scrotum, undescended testis/testes with hypospadias, bilateral non-palpable testes, and micropenis with chordee (47,48) or female appearance with associated abnormalities of genitalia including enlarged clitoris, posterior labial fusion, and an inguinal/labial mass (49). An initial assessment, based on the location of the gonads and presence or absence of a uterus, will provide a provisional clinical diagnosis (45). This information combined with karyotype, will provide the basis for more focussed further investigation. Figures 3a and 3b illustrate chromosomal and gonadal characteristics of DSDs. If no gonads are palpable, all options are possible. Of these, 46,XX DSD (with 2 ovaries) is the most commonly seen, followed by MGD. The presence of a uterus and absence of palpable gonads in a virilized female primarily suggest a clinical diagnosis of 21-hydroxylase deficiency. If one gonad is palpable, 46,XX DSD and complete gonadal dysgenesis are ruled out because ovaries and streak gonads do not descend. MGD, ovotesticular, and 46,XY DSD remain as diagnostic possibilities. If two gonads are palpable, 46,XY DSD and ovotesticular DSD are the most likely diagnoses. Symmetrical external genitalia, with or without palpable gonads, and an absent uterus suggest an undervirilized XY male. The presence of a uterus and asymmetric external genitalia and palpable gonad(s) suggest gonadal dysgenesis with Y and ovotesticular DSD. A gonadal biopsy is required to classify the type of gonadal dysgenesis and ovotesticular DSD, to assess gonadal chromosomal mosaicism and to detect the presence of a gonadal tumor.
Figures 3a. Figure 3a. Gonadal and chromosomal characteristics of DSD DSD: disorders of sexual development.
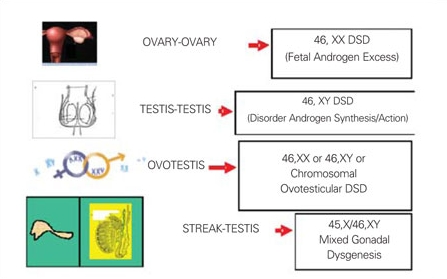
3b. Figure 3b. Gonadal and chromosomal characteristics of DSD DSD: disorders of sexual development.
Hormone measurements should be interpreted in relation to specific assay characteristics and also considering normal values for gestational and chronological age. In some cases serial measurements may be needed. The results of decision making algorithms are available to guide further investigation. These include hCG and ACTH stimulation tests to assess testicular and adrenal steroid biosynthesis. The endocrine evaluation of patients with 46,XY DSDs and sex chromosome DSDs include assessment of testicular function by basal measurement of LH, FSH, inhibin B, T, DHT, AMH, A, and DHEAS. In patients with T synthesis defects, neonatal and post pubertal diagnosis is made based on basal steroid levels. The stimulation of T production by hCG is used to pinpoint abnormalities in T biosynthesis and to detect functioning testicular tissue. Testosterone, DHT, A should be measured at baseline and 72 hours after hCG stimulation. The T increment should be at least threefold (56). A failure to respond to hCG in combination with elevated LH/FSH levels and low/undetectable value of AMH is consistent with anorchia or gonadal dysgenesis. Androgen insensitivity should be considered in individuals with a 46,XY karyotype and with normal T biosynthesis. The diagnosis of androgen insensitivity is difficult in absence of a defined androgen receptor mutation. Patients with 5a-reductase deficiency have normal T levels, low or normal DHT levels and a high T/DHT ratio after hCG stimulation test. The diagnosis of 17bHSD deficiency is made when a 10-15-fold elevation is observed in the ratio of A/T. Minimum A/T ratio in a cohort of 24 individuals with a confirmed mutation was 0.7 (57). Inhibin B and AMH are useful markers for the presence of Sertoli cells and their assessment could help in the diagnosis of testis determination disorders. In boys with bilateral cryptorchidism, serum AMH and inhibin B correlate with the presence of testicular tissue and undetectable values are highly suggestive of absence of testicular tissue (58). In XY patients, AMH was found low when the intersex condition was caused by abnormal testicular determination (including complete and partial gonadal dysgenesis) but was normal or elevated in patients with impaired T secretion, whereas serum T was low in both groups. AMH was also elevated during the first year of life and at puberty in intersex states caused by androgen insensitivity. In 46,XX patients with ambiguous genitalia, a serum AMH level higher than 75 pmol/L is indicative of the presence of testicular tissue and correlates with the mass of functional testicular parenchyma. In conclusion, serum AMH determination is a powerful means to assess Sertoli cell function in children with intersex , and helps to distinguish between defects of abnormal testicular determination and of isolated impairment of T secretion or action.
The diagnosis of 21-hydroxylase deficiency in 46,XX DSDs with two ovaries relies on the detection of elevated 17-OHP levels either as a basal measurement or after a short ACTH stimulation test. High concentration of 11-deoxycortisol and deoxycortisol (DOC) with low levels of plasma renin activity (PRA) will help differentiate 11- from 21-hydroxylase deficiency.
Supported by good facilities for investigation, clinicians can follow an algorithm that in many cases will lead to an aetiological diagnosis, but with the spectrum of findings and diagnosis, no single evaluation protocol can be recommended in all circumstances 59,60).
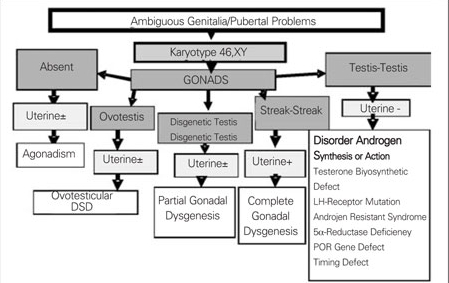
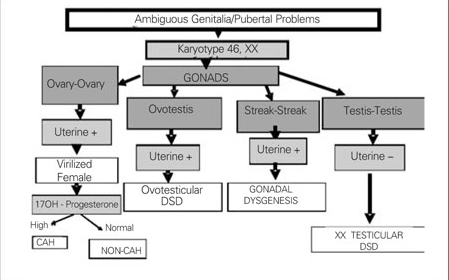
Chromosomal characteristics, gonadal histology and presence or absence uterus are taken into consideration in the classification of DSDs. In our department, we use the diagnostic algorithm prepared in accordance with the new classification for 46,XY DSD and for 46,XX DSD, as given in Figures 4 and 5.
Figures 4. Figure 4. Diagnostic algorithm of 46,XY DSD for new classification DSD: disorders of sexual development .
5. Figure 5. Diagnostic algorithm of 46,XX DSD for new classification DSD: disorders of sexual development, CAH: congenital adrenal hyperplasia .
Management
The psychological and social implications of gender assignment and those relating to treatment are very important and require a multidisciplinary approach with the inclusion of geneticists, neonatologists, endocrinologists, gynaecologists, psychiatrists, surgeon and social workers in the team. The members of such a team should have a special interest in DSD and possess sufficient experience with this group of patients. The current intense debate on the management of patients with intersexuality and related conditions focuses on four major issues, namely, aetiological diagnosis, assignment of gender, indications for and timing of genital surgery, and disclosure of medical information to the patient (61,62,63,64,65,66,67,68, 69,70,71,72,73). Gender identity is a multifactorial process involving both prenatal and postnatal variables. Psychosexual development is influenced by multiple factors such as exposure to androgens, sex chromosome genes, social circumstances and family dynamics. Outcomes can be influenced by timing, dose and type of androgen exposure, receptor availability, and modification by the social environment. Pre- and postnatal hormonal conditions, sex rearing, timing of sex reassignment and corrective surgery appear to be important components for the development of gender-role behavior and gender identity in DSD patients. Karyotype, gonadal function, phenotype, internal genitalia (i.e. presence of uterus), potential for fertility and sexuality, risk of future malignancy, and prenatal brain virilization are some of the many factors which must be taken into account when assessing gender in a child with DSD. Each patient should be evaluated individually by a multidisciplinary approach. Gender assignment should be done after completion of the diagnostic process, including full clinical, genetic, biochemical and psychiatric investigation. The whole procedure should be fully explained to the parents and they should partake in the discussions and decisions.
The traditional gender assignments for some medical conditions leading to DSDs are as follows: assignment of female gender for 46,XX DSD resulting from fetal androgen exposure, CAIS, and 46,XY complete gonadal dysgenesis; assignment of male gender for 46,XY cloacal exstrophy; assignment of male or female gender for PAIS, 5a-reductase deficiency, ketoreductase deficiency, 46,XY partial gonadal dysgenesis, MGD and ovotesticular DSD. Underlying endocrine disturbances are present in most cases and usually require long-term medication. The current recommendation for a girl with CAH is to rear her as a female and perform a feminizing genitoplasty depending on the degree of masculinization. However, male sex assignment may be mandatory in severely virilized 46,XX CAH patients in whom the diagnosis was not made in the early stage. Recommendations for sex of rearing especially in infants with genital ambiguity, testicular differentiation disorders and Y chromosome continue to be challenging. Genital masculinization is a poor predictor of the masculinization of the brain (63). Patients with PAIS, 5a-reductase and ketoreductase deficiency, partial gonadal dysgenesis, and MGD may be female or predominantly female at birth and generally are raised as females, but they change their social sex to male at puberty. Sex assignment is more problematic in this group. The decision on sex of rearing in ovotesticular DSD should be based on gonadal and internal ductal formation.
Surgical techniques of 'feminization' and 'masculinization' and their outcomes have also evolved over time (72,73). Surgeons should have both pediatric training and expertise in DSD surgery. Functional outcome should be taken into consideration rather than a strictly cosmetic appearance. In children assigned a male sex, hypospadias or chordee, if present, requires early surgical correction. Scrotal testis/testes should be monitored and, if needed, biopsied, to rule out malignancy. A streak ovary should be removed. The testis/testes may also need to be removed if there is any risk of malignancy on clinical suspicion or biopsy specimens. T replacement may be required at puberty. The testes in patients with DSD raised as females should be removed to prevent malignancy in pre-puberty or after puberty. Feminizing surgery has three main aims: reducing the size of the enlarged masculinized clitoris, reconstructing the female labia, and increasing the opening and if possible, the length of the vagina. Early surgery should only be considered in cases of severe virilization (Prader III-V) and should be carried out in conjunction, when appropriate, with repair of the common urogenital sinus. As orgasmic function and erectile sensation may be disturbed by clitoral surgery, the surgical procedure should be anatomically based to preserve erectile function and the innervation of the clitoris. Consensus conferences recommend not to perform genital surgery between 12 months and adolescence (except for compelling medical indications). Vaginal dilatation should not be recommended before adolescence.
References
- 1.Allen L. Disorders of sexual development. Gynecol. Clin.North Amer. 2009;36:25–45. doi: 10.1016/j.ogc.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Dreger AD, Chase C, Sausa A, Gruppusa A, Frader J. Changing nomenclature/ Toxonomy for intersex , scientific and clinical rationale. J Pediatr Endocrinol Metab. 2005;18:729–733. doi: 10.1515/jpem.2005.18.8.729. [DOI] [PubMed] [Google Scholar]
- 3.Hughes IA, Houk C, Ahmed SF, Lee PA LWPES Consensus Group; ESPE Consensus Group. Consensus Statement on Management of Intersex Disorders. Pediatrics. 2006;118:488–500. doi: 10.1542/peds.2006-0738. [DOI] [PubMed] [Google Scholar]
- 4.Hughes IA. Disorders of sexual differentiation. Horm Res. 2007;67(suppl 1):91–95. [Google Scholar]
- 5.Houk CP, Lee PA. Consensus statement on terminology and management: Disorders of sex development. Sex Dev. 2008;2:172–180. doi: 10.1159/000152032. [DOI] [PubMed] [Google Scholar]
- 6.Damian D, Paulo S. Disorder of sexual development: Still a big challenge. J Pediatr Endocrinol Metab. 2007;20:749–750. doi: 10.1515/jpem.2007.20.7.749. [DOI] [PubMed] [Google Scholar]
- 7.Carillo AA, Damian M, Bercovitz G. Disorders of sexual differentiation. From Pediatric Endocrinology Fifth ed. Vol 2. Ed by Lifshitz F. London: İnforma Health Care; 2007. pp. 365–413. [Google Scholar]
- 8.Hughes IA. Testes: Disorders of sexual differantiation and puberty in male. From Texbook Pediatric Endocrinology. Third edition. Ed by Sperling MA. Saunders comp: Pittsburg; 2008. pp. 662–685. [Google Scholar]
- 9.Arboleda VA, Flaming AA and Vilain E. Disorders of sex development from texbook of Genetic Diagnosis of Endocrine Disorders. Ed by Weiss RE, and Retetoff S. London: Press in Elsevier; 2010. pp. 227–243. [Google Scholar]
- 10.MacLaughlin DT, Donahoe PK. Sex determination and differentiation. N Engl J Med. 2004;350:367–378. doi: 10.1056/NEJMra022784. [DOI] [PubMed] [Google Scholar]
- 11.Biason-Lauber A. Control of sex development. Best Pract Res Endocrinol Metab. 2010;24:163–186. doi: 10.1016/j.beem.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Houmart B, Small C, Yang L, Naluai–Cecchini T, Cheng E, Hassold T, Griswold M. Global gene expression in human fetal testis or ovary. Biol Reproduct. 2009;81:438–443. doi: 10.1095/biolreprod.108.075747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brennan J, Capel B. One tissue two fates: molecular genetic events that underlie testis versus ovary development. Nat Rev Genet. 2004;5:509–521. doi: 10.1038/nrg1381. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed SF, Hughes A. The genetics of male undermasculinization. Clinical Endocrinol. 2002;56:1–10. doi: 10.1046/j.1365-2265.2002.01430.x. [DOI] [PubMed] [Google Scholar]
- 15.Willhelm D, and Englerd C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of SF1. Genes and Development. 2002;16:1839–1851. doi: 10.1101/gad.220102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Çetinkaya E, Öçal G, Berberoğlu M, Adıyaman P, Ekim M, Yalçınkaya F, Orun E. Association of partial gonadal dysgenesis, nephropathy and WT1 gene mutation without Wilm’s tumor: Incomplete Denys-Drash syndrome. J Pediatr Endocrinol Metab. 2001;14:561–564. doi: 10.1515/jpem.2001.14.5.561. [DOI] [PubMed] [Google Scholar]
- 17.Biason-Lauber A, Konrad D, Meyer M, deBauford C, Schoenle EJ. Ovaries and female phenotype in a girl with 46,XY karyotype and mutation CBX2 gene. New York: Horm Res LSPES/ESPE 8th Joint Meeting Abstract book; 2009. pp. 53–54. [Google Scholar]
- 18.Palanco JC, Koopman P. SRY and hesistant begining of male development. Dev Biol. 2007;302:13–24. doi: 10.1016/j.ydbio.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 19.Sekido R, Lovel-Badge R. Sex determination and SRY: down to a wink and a nudge? Trends in Genetics. 2009;25:19–29. doi: 10.1016/j.tig.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Morais da Silva S, Hacker A, Harley V, Goodfellow P, Swain A, Lovell-Badge R. SOX9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nature Genetics. 1996;14:62–68. doi: 10.1038/ng0996-62. [DOI] [PubMed] [Google Scholar]
- 21.Hiramatsu R, Harikae K, Tsunekawa N, Kurohmaru M, Matsuo Kanai Y. FGF signaling directs a center-to-pole expansion of tubulogenesis in mouse testis differentiation. Development. 2010;137:303–312. doi: 10.1242/dev.040519. [DOI] [PubMed] [Google Scholar]
- 22.Oakes MB, Eyvazzadeh AD, Quint E, Smith YR. Complete androgen insensitivity syndrome—a review. J Pediatr Adolesc Gynecol. 2008;21:305–310. doi: 10.1016/j.jpag.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Choi JH, Kim GH, Seo EJ, Kim KS, Kim SH, Yoo HW. Molecular analysis of the AR and SRD5A2 genes in patients with 46,XY disorders of sex development. J Pediatr Endocrinol Metab. 2008;21:545–553. [PubMed] [Google Scholar]
- 24.Yao HH. The pathway to femaleness: current knowledge on embryonic development of the ovary. Mol Cell Endocrinol. 2005;230:87–93. doi: 10.1016/j.mce.2004.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edson MA, Nagaraja AK, Matzuk MM. The mammalian ovary from genesis to revelation. Endocr Rev. 2009;30:624–712. doi: 10.1210/er.2009-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vainio S, Heikkil AM, Kispert A, Chin N, McMohon AP. Female development in mammals is regulated by WNT4signalling. Nature. 1999;4:405–409. doi: 10.1038/17068. [DOI] [PubMed] [Google Scholar]
- 27.Biason-Lauber A: WNT4 and R-Spontin1 signalling in ovarian development. Highlights. 46th Meeting ESPE Praq; 2010. [Internet] http://www.eurospe.org/ [Google Scholar]
- 28.Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, Guerra L, Schedl A. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nature Genetics. 2006;38:1304–1309. doi: 10.1038/ng1907. [DOI] [PubMed] [Google Scholar]
- 29.Ottolenghi C, Omari S, Garcia-Ortis JE, Uda M, Crisponi L, Foraboska A, Pilia G, Schlessinger D. FOXL2 is required for commitment to ovary differentiation. Hum Mol Genet. 2005;14:205–205. doi: 10.1093/hmg/ddi210. [DOI] [PubMed] [Google Scholar]
- 30.Aaronson IA. Terminology for disorders of sex development: clarity or confusion. 2011;185:388-389. 2011;185:388–389. doi: 10.1016/j.juro.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Barthold JS. Disorders of sex differentiation: a pediatric urologist's perspective of new terminology and recommendations. J Urol. 2011;185:393–400. doi: 10.1016/j.juro.2010.09.083. [DOI] [PubMed] [Google Scholar]
- 32.Wijchers PJ, Yandim C, Panousopoulou E, Ahmad M, Harker N, Saveliev A, Burgoyne PS, Festenstein R. Sexual dimorphism in mammalian autosomal gene regulation is determined not only by SRY but by sex chromosome complement as well. Dev Cell. 2010;19:477–484. doi: 10.1016/j.devcel.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Nishi MY, Costa EM, Oliveira SB, Mendonca BB, Domenice S. The role of SRY mutations in the etiology of gonadal dysgenesis in patients with 45,X/46,XY disorder of sex development and variants. Horm Res Paediatr. 2011;75:26–31. doi: 10.1159/000316536. [DOI] [PubMed] [Google Scholar]
- 34.Oliveira RM, Verreschi IT, Lipay MV, Eça LP, Guedes AD, Bianco B. Y chromosome in Turner syndrome: review of the literature. Sao Paulo Med J. 2009;127:373–378. doi: 10.1590/S1516-31802009000600010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khandelwal A, Agarwal A, Jiloha RC. A 47,XXY female with gender identity disorder. Arch Sex Behav. 2010;39:1021–1023. doi: 10.1007/s10508-010-9628-x. [DOI] [PubMed] [Google Scholar]
- 36.Siklar Z, Berberoğlu M, Adiyaman P, Salih M, Tükün A, Cetinkaya E, Aycan Z, Evliyaoğlu O, Ergur AT, Ocal G. Disorders of gonadal development: a broad clinical, cytogenetic and histopathologic spectrum. Pediatr Endocrinol Rev. 2007;4:210–217. [PubMed] [Google Scholar]
- 37.Balkan M, Akbas H, Isi H, Oral D, Turkyilmaz A, Kalkanli S, Simsek S, Fidanboy M, Alp MN, Gedik A, Budak T. Cytogenetic analysis of 4216 patients referred for suspected chromosomal abnormalities in Southeast Turkey. Genet Mol Res. 2010;11:1094–1103. doi: 10.4238/vol9-2gmr827. [DOI] [PubMed] [Google Scholar]
- 38.38. Maimoun L, Philibert P, Cammas B, Audran F, Bouchard P, Fenichel P, Cartigny M, Pienkowski C, Polak M, Skordis N, Mazen I, Ocal G, Berberoglu M, Reynaud R, Baumann C, Cabrol S, Simon D, Kayemba-Kay's K, De Kerdanet M, Kurtz F, Leheup B, Heinrichs C, Tenoutasse S, Van Vliet G, Grüters A, Eunice M, Ammini AC, Hafez M, Hochberg Z, Einaudi S, Al Mawlawi H, Nuñez CJ, Servant N, Lumbroso S, Paris F CJ, Servant N, Lumbroso S, Paris F, Sultan C. Phenotypical, biological, and molecular heterogeneity of 5α-reductase deficiency: an extensive international experience of 55 patients. J Clin Endocrinol Metab. 2011;96:296–307. doi: 10.1210/jc.2010-1024. [DOI] [PubMed] [Google Scholar]
- 39.Maimoun L, Philibert P, Bouchard P, Ocal G, Leheup B, Fenichel P, Servant N, Paris F, Sultan C. Primary amenorrhea in four adolescents revealed 5α-reductase deficiency confirmed by molecular analysis. Fertil Steril. 2011;95:804–804. doi: 10.1016/j.fertnstert.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Ocal G, Adıyaman P, Berberoğlu M, Çetinkaya E, Akar N, Uysal A, Duman T, Evliyaoğlu O, Aycan Z, Lumbroso S, Sultan C. Mutation of the 5 alpha steroid reductase type 2 gene in six unrelated patients from unrelated families and a large pedigree of an isolated Turkish village. J Pediatr Endocrinol Metab. 2002;15:411–421. doi: 10.1515/jpem.2002.15.4.411. [DOI] [PubMed] [Google Scholar]
- 41.Miller WL. P450 oxireductase deficiency: a new disorder of steroidogenesis with multiple clinical manifestation. Trends Endocrinol Metab. 2004;15:311–315. doi: 10.1016/j.tem.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Arlt W, Walker EA, Draper N, Ivison HE, Ride JP, Hammer F, Chalder SM, Borucka-Mankiewicz M, Hauffa BP, Malunowicz EM, Stewart PM, Shackleton CH. Congenital adrenal hyperplasia caused by mutant P450 oxireductase and human androgen synthesis: analytical studies. Lancet. 2004;363:2128–2135. doi: 10.1016/S0140-6736(04)16503-3. [DOI] [PubMed] [Google Scholar]
- 43.Carani C, Simpson ER. Recognizing rare disorders: aromatase deficiency. Nat Clin Pract Endocrinol Metab. 2007;3:414–421. doi: 10.1038/ncpendmet0477. [DOI] [PubMed] [Google Scholar]
- 44.Noordam C, Dhir V, McNelis JC, Schlereth F, Hanley NA, Smeitink JA, Smeets R, Sweep FC, Claahsen-van der Grinten HL, Arlt W. Inactivating PAPSS2 mutations in a patient with premature pubarche. NEJM. 2009;360:2310–2318. doi: 10.1056/NEJMoa0810489. [DOI] [PubMed] [Google Scholar]
- 45.Lee PA. A perspective on the approach to the intersex child born with genital ambiquity. J Pediatr Endocrinol Metab. 2004;17:133–140. doi: 10.1515/jpem.2004.17.2.133. [DOI] [PubMed] [Google Scholar]
- 46.Brown J, and Warner G. Practical manegement of the intersex infants. J Pediatr Endocrinol Metab. 2005;18:3–23. doi: 10.1515/jpem.2005.18.1.3. [DOI] [PubMed] [Google Scholar]
- 47.Guerra-Junior G, Marciel-Guerra AT. The role of the pediatrician in the management of children with genital ambiquities. J de Pediatria. 2007;83:184–191. doi: 10.2223/JPED.1706. [DOI] [PubMed] [Google Scholar]
- 48.Ouigley CA, De-Bellis A, Marschke KB, el-Awady MK, Wilso M, French FS. Androgen receptör defects. Endocr Rev. 1995;16:271–321. doi: 10.1210/edrv-16-3-271. [DOI] [PubMed] [Google Scholar]
- 49.Sinnecker GH., Hiort , Dibbelt L, Albers N, Dörr HG, et al. Phenotypic classification of male pseudohermaphroidism due to steroid 5 alfa reductase 2 deficiency. Am J Med Genet. 1996;63:223–230.. doi: 10.1002/(SICI)1096-8628(19960503)63:1<223::AID-AJMG39>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 50.Prader A. Der genital befund be im pseudohermaphroditismus femininus des kongenitalen androgenitalen syndroms. Helvetica Paediatr Acta. 1954;3:231–248. [PubMed] [Google Scholar]
- 51.Öcal G, Berberoğlu M, Şıklar Z, Bilir P. Uslu R, Yağmurlu A, Tükün A, Akar N, Soygür T, Gültan S, Toykukuk G. Disorders Sexual Development: an overviev of 18 years experience in Pediatric Endocrinology Department of Ankara University. J Pediatr Endocrinol Metab. 2010;23:1123–1132. doi: 10.1515/jpem.2010.177. [DOI] [PubMed] [Google Scholar]
- 52.Al–Agha AE, Thomset MJ, Batch JA. The child of uncertain sex: 17 years of experiance. J Pediatr Child Health. 2001;37:348–351. doi: 10.1046/j.1440-1754.2001.00669.x. [DOI] [PubMed] [Google Scholar]
- 53.Mazen I., Hiort O, Bassiouny R, El-Gammal M. Differential diagnosis of disorders of sex development in Egypt. Horm Res. 2008;70:118–123. doi: 10.1159/000137657. [DOI] [PubMed] [Google Scholar]
- 54.Al- Muttair A, Igbal MA, Ashwal A. Cytogenetic and etiology of ambiquous genitaliain 120 pediatric patients. Ann Sauidi Med. 2004;37:348–51. [Google Scholar]
- 55.Zdravkovic D, Milankovic T, Sedlecki K, Guc-Scekic M, Rajic V, Banicevic M. Causes of ambiquous genitalia in neonates. Srp Arh Celok Lek. 2001;129:57–60. [PubMed] [Google Scholar]
- 56.Kolon TF, Miller OF. Comparison of single versus multible dose regimens for the human chorionic gonadotropin stimulation test. J Urol. 2001;166:1451–1454. [PubMed] [Google Scholar]
- 57.Simard J, Moisan AM, Michel LC, Morel Y. Males with 17β-hydroxysteroiddehydrogenase deficiency. Endocrinologist. 2003;13:195–200. [Google Scholar]
- 58.Grinspon RP, Rey RA. Anti-Müllerian hormone and Sertoli cell function in paediatric male hypogonodism. Horm Res. 2010;73:81–92. doi: 10.1159/000277140. [DOI] [PubMed] [Google Scholar]
- 59.Garel L. Abnormal sex differentiation: who, how and when to image. Pediatr Radiol. 2008;38(Supll 3):508–511. doi: 10.1007/s00247-008-0836-7. [DOI] [PubMed] [Google Scholar]
- 60.Katorza E, Pinhas-Hamail O, Mazkereth R, Gilboa Y, Achiron R. Sex differentiation disorders (SDD) prenatal sonographic diagnosis, genetic and hormonal work-up. Pediatric Endocrinol Rev. 2009;7:11–21. [PubMed] [Google Scholar]
- 61.Money C. Amative orientation. The hormonal hypothesis rexemined. J Pediatr Endocrinol Metab. 2002;15:951–957. doi: 10.1515/jpem.2002.15.7.951. [DOI] [PubMed] [Google Scholar]
- 62.Houk CP, Dayner J, Lee PA. Genital ambiquity with Y choromosome: Des gender assigment matter? J Pediatr Endocrinol Metab. 2004;17:825–839. doi: 10.1515/jpem.2004.17.6.825. [DOI] [PubMed] [Google Scholar]
- 63.Ahmet SF, Morrison S, Hughes IA. Intersex and gender assignment ; the third way? Arch Dis Child. 2004;89:847–850. doi: 10.1136/adc.2003.035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyer-Bahlburg HFL. Gender assignment and reassignment in 46,XY pseudohermaphroditism and related condition. J Clin Endocrinol Metab. 1999;84:3455–3458. doi: 10.1210/jcem.84.10.6099. [DOI] [PubMed] [Google Scholar]
- 65.Reiner WG. Gender identity and sex-of–rearing in children with disorders sexual differentation. J Pediatr Endocrinol Metab. 2005;18:549–553. doi: 10.1515/jpem.2005.18.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reiner WG. Case study: Sex re-assigment in a teenage girl. Am Acad Child. Adolesc Psychiatry. 1996;35:799–803. doi: 10.1097/00004583-199606000-00020. [DOI] [PubMed] [Google Scholar]
- 67.Birnbacher R, Marberger M, Weissenbacher G, Schober E, Frisch H. Gender identity reversal in an adolescent with mixed gonadal dysgenesis. J Pediatr Endocrinol Metab. 1999;12:687–690. doi: 10.1515/jpem.1999.12.5.687. [DOI] [PubMed] [Google Scholar]
- 68.Ocal G, Berberoğlu M, Siklar Z, Bilir P. Gender Dysphoria and Gender Change in an Adolescent with 45,X/46,XY Mixed Gonadal Dysgenesis. Exp Clin Endocrinol Diabetes. 2009;117:301–304. doi: 10.1055/s-0028-1085999. [DOI] [PubMed] [Google Scholar]
- 69.Uslu R, Oztop D, Ozcan O, Yilmaz S, Berberoğlu M, Adiyaman P, Cakmak M, Kerimoğlu E, Ocal G. Factors contributing to sex assignment and reassignment decisions in Turkish children with 46,XY disorders of sex development. J Pediatr Endocrinol Metab. 2007;20:1001–1015. doi: 10.1515/jpem.2007.20.9.1001. [DOI] [PubMed] [Google Scholar]
- 70.DeVries ALC, Doreleijers TAH, Cohen-Cettenis PT. Disorders of sex development and gender identity outcome in adolescence ande adulthood: Understandig gender identity development and its clinical implications. Pediatr Endocrinol Rew. 2007;4:343–351. [PubMed] [Google Scholar]
- 71.Daaboul J. and Frader J. Ethics and the management of the patient with intersex: A middle way. J Pediatr Endocrinol Metab. 2001;14:1575–1583. doi: 10.1515/jpem.2001.14.9.1575. [DOI] [PubMed] [Google Scholar]
- 72.Crouch NS, Creighton SM. Minimal surgical intervention in the manegement of intersex conditions. J Pediatr Endocrinol Metab. 2004;17:1591–1596. doi: 10.1515/jpem.2004.17.12.1591. [DOI] [PubMed] [Google Scholar]
- 73.Vidal I, Gorduza DB, Haraux E, Gay CL, Chatelain P, Nicolino M, Mure PY, Mouriquand P. Surgical options in disorders of sex development (DSD) with ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010;24:311–324. doi: 10.1016/j.beem.2009.10.004. [DOI] [PubMed] [Google Scholar]