

Abstract
Fragments of integral membrane proteins have been used to study the physical chemical properties of regions of transporters and receptors. Ste2p(G31-T110) is an 80-residue polypeptide which contains a portion of the N-terminal domain, transmembrane domain 1 (TM1), intracellular loop 1, TM2 and part of extracellular loop 2 of the α-factor receptor (Ste2p) from Saccharomyces cerevisiae. The structure of this peptide was previously determined to form a helical hairpin in lyso-palmitoylphosphatidyl-glycerol micelles (LPPG)[1]. Herein, we perform a systematic comparison of the structure of this protein fragment in micelles and trifluoroethanol(TFE):water in order to understand whether spectra recorded in organic:aqueous medium can facilitate the structure determination in a micellar environment. Using uniformly labeled peptide and peptide selectively protonated on Ile, Val and Leu methyl groups in a perdeuterated background and a broad set of 3D NMR experiments we assigned 89% of the observable atoms. NOEs and chemical shift analysis were used to define the helical regions of the fragment. Together with constraints from paramagnetic spin labeling, NOEs were used to calculate a transiently folded helical hairpin structure for this peptide in TFE:water. Correlation of chemical shifts was insufficient to transfer assignments from TFE:water to LPPG spectra in the absence of further information.
Keywords: GPCR, membrane protein structure, fragment, transmembrane
1. Introduction
Integral membrane proteins (IMPs) are a class of proteins that are difficult to characterize structurally. X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy have been used to study the high-resolution structures of many proteins and the Protein Data Bank currently has more than 69,000 structures of mainly soluble proteins. The number of unique IMP structures deposited towards the end of 2010 was approximately 270, or less than 0.5% of all the structures in the Data Bank [2, 3]. The underrepresentation of IMP structures in the database belies the importance of these proteins. Many membrane proteins are involved in signal transduction, nutrient uptake and various diseases. In fact, 30–60% of the current drugs on the market are targeted to membrane proteins [4–6] and structural information on these proteins is important for the development of more specific drugs that could increase efficacy and decrease side effects.
Crystallization of membrane proteins is complicated by their inherent flexibility and can be hampered by protein-lipid interactions that are required for function. Solution NMR investigations of IMPs require solubilization in a membrane mimetic environment and are hindered by the size of the protein-lipid complex and conformational exchange processes. G protein-coupled receptors (GPCRs) have molecular weights of approximately 50 kDa and size increases in detergent micelles or lipid bicelles result in slow tumbling and short relaxation times with a concomitant loss of signal. Solid state NMR, a technique in which the quality of spectra is less dramatically affected by the size of the protein-lipid complex, has also been used in the structure determination of membrane proteins in lipids [reviewed in 7].
Solution NMR has been very useful when studying protein dynamics and/or protein-ligand interactions. Tools are being developed to better study large protein-lipid complexes in solution[reviewed in 8], but high resolution structures remain elusive[9–11]. Recently, the structure of sensory rhodopsin, a seven transmembrane (TM) GPCR homolog, was determined using a highly deuterated form of the protein which relied on selective introduction of protons into Ile, Leu and Val as well as a protonated protein to determine more methyl group NOEs and aromatic interactions [12, 13]. Although this structure represents an important milestone in GPCR structural biology, sensory rhodopsin is much smaller than most eukaryotic GPCRs, and it is not clear whether this approach can be generalized to this family of molecules. In the interim we are studying fragments of GPCRs to learn more about the folding of these IMPs, to investigate helix-helix interactions, to develop methodologies that may be useful in the assignment of the NMR spectrum of the entire GPCR, and to gain insights into the secondary and tertiary structure of large but discrete domains of these receptors.
To apply this strategy it is important to work with fragments that are large enough to potentially adopt tertiary structure and which may also allow testing of ligand binding. The study of GPCR fragments is thought to be valid because these have been shown to reconstitute to form functional proteins both in vitro and in vivo [14–18]. Furthermore, numerous biological and biochemical studies on Ste2p have been used to elucidate its interaction with the tridecapeptide α-factor ligand and to decipher changes in the structure upon activation of the signal transduction pathway [19–28]. This data will help to place the biophysical analysis of fragments of Ste2p into a biological context.
Polypeptide fragments of Ste2p, including each single TM, loops and the C-terminal tail have been previously studied by solution NMR [29–32] (Bhuiyan unpublished data) and have provided details on the structural tendencies of discrete regions of this GPCR. A high-resolution structure of a peptide containing 2TMs, Ste2p(G31-T110), has been determined in lysopalmitoylphosphatidyl-glycerol (LPPG) micelles [1]. Based on the fact that a dimer of Ste2p(G31-T110) in micelles is expected to have a fairly long correlation time and that the NMR spectral lines were sharp and of good quality we concluded that the peptide is a monomer [1]. Long-range connectivities were observed between hydrophobic residues of TM1 and TM2 when selective methyl group labeling was used. Comparison of the structures of a GPCR fragment in different membrane mimetic media would be useful to ascertain whether micelles and trifluorethanol (TFE):water stabilize similar structures and whether assignments obtained in organic:aqueous media are relevant to the micellar state. If so, the organic:aqueous medium might be extremely valuable in analyzing membrane proteins of increasing size.
Organic:aqueous media have been used for biophysical analysis of hydrophobic peptides for some time, due to their spectroscopic properties which are ideal for circular dichroism and NMR analyses [33–37]. The correlation time of an IMP fragment in an organic:aqueous solvent is much shorter than when embedded in micelles or bicelles resulting in superior spectra. Media such as TFE:water are not considered to be biologically relevant by some investigators [38, 39], although this conclusion has never been systematically evaluated. Indeed, some fragments of both transporters and receptors exhibit highly resolved spectra and some even exhibit long-range contacts in TFE and chloroform:methanol:water and appear to assume folded structures that may be relevant to the biologically active state [31, 40–42]. Intrinsically disordered proteins can be studied in solution, micelles and TFE:water. A protein found in human semen that has been shown to enhance HIV viral infection, SEVI, folds into different conformations dependent on the environment [43]. Conversely, structural intermediates in the folding pathway of α-synuclein have been observed both in TFE:water and in micelles [44, 45] indicating that the folding pathway may be similar in both environments.
Here we describe our efforts aimed at characterizing the structure of Ste2p(G31-T110; TM1-TM2), an 80-residue two-TM containing GPCR fragment, in organic:aqueous media (TFE:water). The structure of TM1-TM2 was assessed using solution NMR and uniformly labeled peptide and peptide selectively protonated on Ile, Val and Leu methyl groups in an otherwise perdeuterated background. To probe for the presence of interhelical contacts we also utilized an analog of TM1-TM2 in which a nitroxide radical was conjugated to an introduced Cys residue. TM1-TM2 assumes distinct helical regions corresponding to the predicted TM domains of the receptor and also shows a tendency to assume a helix in what would be the N-terminal region of the protein. Spin-labeling data indicated that TM1-TM2 undergoes conformational averaging in which the individual helices form mutual transient contacts. Comparison of our results with those from a previous study in LPPG micelles[1] indicates that lipid-protein interactions within the context of bilayer or detergent micelles may be critical for the formation of stable tertiary structure of this 2TM GPCR fragment.
2. Experimental methods
2.1 NMR sample preparation
NMR samples containing 0.5–4 mg of isotopically labeled TM1-TM2 were prepared in 350 μL TFE-d2:water(0.1% TFA) (1:1, v:v) or TFE-d3:D2O(0.1% TFA-d) (1:1, v:v). The samples were prepared by the addition of 175 μL TFE followed by 175 μL water(0.1% TFA). Sample preparation of amide proton-exchanged peptides were first solubilized in a large excess of TFE:D2O(0.1% TFA) and incubated at room temperature overnight or at 50°C for 1 hour to exchange a portion of the amide protons to deuterons and then lyophilized. The resulting peptide was solubilized in TFE-d3:D2O(0.1% TFA-d) as described above.
2.2 NMR Measurements
The NMR experiments were performed on a Varian 600 MHz spectrometer at the College of Staten Island, CUNY, a Bruker Avance 700 MHz spectrometer at the University of Zurich, Switzerland, or a Bruker 900 MHz spectrometer at the New York Structural Biology Center (NYSBC) in New York City. All measurements were conducted at 25°C or 45°C using 0.14 mM to1 mM samples.
2.3 NMR Experiments
The experimental parameters are defined in Supplementary Table S1. Sample stability and initial [15N,1H]-HSQC fingerprinting analysis was performed at 45°C, 35°C, 25°C and 15°C [46]. The backbone resonances were assigned by performing HNCO [47–50], HN(CA)CO [51], HNCA [47–50] and HNCACB [49, 50, 52] experiments at 45°C and 25°C. The 15N-resolved NOESY-HSQC [46, 53] and the 15N-resolved TOCSY-HSQC [46, 53] were performed at 45°C and 25 °C to assign side chain proton chemical shifts and an HCCH-TOCSY experiment was performed at 25°C to assign carbon and proton side chain resonances[54, 55]. The chemical shifts of methyl protons and carbons of the Ile, Leu and Val residues were deduced from HMCMCBCANH and HMCMCANH [56] and ct-[13C,1H]-HSQC [57] experiments performed on [15N,13C,2H(1H(methyl)-Ile, Leu, Val)]-TM1-TM2. Further side chain analysis was performed with 3D 13C-edited TOCSY and NOESY experiments at 25°C. Unlabeled TM1-TM2 was used in 300 or 150 ms NOESY experiments [58] and 60 or 25 ms TOCSY experiments [59, 60]. The [15N,13C,2H(1H(methyl)-Ile, Leu, Val)]-TM1-TM2 peptide was also used in NOESY-ct-[13C,1H]-HSQC [46, 61, 62] and ct-[13C,1H]-HSQC-NOESY-ct-[13C,1H]-HSQC [63] to determine long-range NOEs between the methyl groups of Ile, Leu or Val residues.
Dynamics of [15N]-TM1-TM2 were analyzed by performing [15N,1H]-HSQC versions of CPMG experiments with variable relaxation delays to determine T2 relaxation at 45°C and 25°C. A [15N,1H]-HSQC version of the steady-state NOE was measured to determine the heteronuclear NOE [64]. [15N]-TM1-TM2 was solubilized in TFE-d3:D2O(0.1% TFA-d) and [15N,1H]-HSQC experiments were performed over time to follow the disappearance of signals.
2.4 Structure calculations
Assignments were determined by spectral analysis using the programs NMRView 5 [65] and CARA [66]. NOESY spectra were automatically assigned based on the assigned chemical shift lists using UNIO09 and CYANA [67, 68]. The algorithm was modified to prevent generation of long-range contacts in the initial phase of the structure calculation. Additional backbone dihedral angle restraints derived by TALOS+ from the backbone and Cβ chemical shifts were added [69]. A soft 16 Å restraint was introduced between the γ-oxygen atom of Ser59 and the amide proton of Ala96 to account for the PRE attenuations. Starting from one hundred randomized structures the NMR ensemble was calculated using the standard simulated annealing algorithm in a restrained MD calculation [70] using NOE-derived upper distance bounds, the TALOS+ dihedral angle restraints and the PRE distance restraint. The 20 conformers with the lowest target function values were analyzed using the program MOLMOL [71].
2.5 Automated transfer of chemical shift assignments from TFE:water to LPPG
The two [1H,13C]-HSQC peak lists for the methyl group region of TM1-TM2 in TFE:water or LPPG were matched to each other by minimizing the sum of the normalized distance between corresponding peaks. The normalized distance between two peaks is given by the square root of the squared chemical shift difference for 1H divided by 0.02 ppm plus the squared chemical shift difference for 13C divided by 0.3 ppm. An overall offset of 1.7 ppm was subtracted from all 13C chemical shifts measured in TFE:water. The optimal solution of this rectangular assignment problem was computed using the Hungarian algorithm [72] implemented in CYANA [73].
3. Results
3.1 Assignments of the Backbone Chemical Shifts of Ste2p(G31-T110)
Purified [15N]-Ste2p(G321-T110) was solubilized in TFE:water(+0.1% TFA) (1:1, v:v) as described and the proton-nitrogen correlation map (the [15N,1H]-HSQC spectrum) was recorded at 15°C, 25°C, 35°C and 45°C. All of the expected peaks, 78 out of 80 residues, were observed at each temperature except for 15°C (data not shown). Initial assignments at 45°C were performed with 0.5 mM [15N,13C,2H(1H(methyl)-ILV)]-TM1-TM2 in order to increase the peak resolution and to mimic conditions used during the structure determination of Ste2p(G31-T110) in LPPG micelles [1]. Using triple-resonance experiments that correlate backbone amide moieties with the intra-residue and sequential Cα,Cβ and CO to assign the chemical shifts of the backbone nuclei (HNCA, HNCACB, HNCO and HN(CA)CO) along with the [15N,1H]-HSQC, we were able to assign all of the backbone resonances for this peptide except for the NH of the Gly31 residue and the N of the Pro79 (>99%). Additional experiments were run at 25°C because the reduced temperature may stabilize the formation of interhelical contacts and the peak resolution in the [15N,1H]-HSQC spectra was high (Figure 1). Using a 0.5 mM sample of the [15N,13C]-TM1-TM2, 97% of the backbone nuclei were assigned at 25°C covering essentially the same set of resonances as at 45°C with exception of CO of Thr78 and the Cα and Cβ of Pro79. The chemical shifts have been deposited in the BMRB database under the accession code 17593. The backbone assignments were used in TALOS+ analyses to generate dihedral angle constraints to be used in structure calculations (see below).
Figure 1.

Assignment of the [15N,1H]-HSQC spectrum of Ste2p(G31-T110; TM1-TM2) in TFE:water at 25°C. The boxed region contains crosspeaks due to sidechain amide moieties.
3.2 Assignments of the Sidechain Resonances of Ste2p(G31-T110)
Assignments of the side chain protons at 25°C were determined using 3D 15N-resolved HSQC-NOESY and HSQC-TOCSY NMR experiments [46, 53] to correlate neighboring protons through space and through bonds, respectively, as well the corresponding homonuclear experiments on the unlabeled protein. In this way ~50% of sidechain protons were assigned including all of the protons in the difficult to assign aliphatic Val residues. Many of the chemical shifts that remained unassigned were those of Ile and Leu residues whose high redundancy and very small chemical shift differences complicated the analysis. To facilitate assignment of these methyl groups, which are critical in the determination of long-range interhelical connectivities, the [15N,13C,2H(1H(methyl)-ILV)]-TM1-TM2 was prepared in TFE:water and analyzed by experiments that link the protonated methyl group to the amide proton of the same residue (HMCM(CBCA)NH and HMCM(CA)NH) [1, 56]. Comparison with ct-[13C,1H]-HSQC spectra allowed the complete assignments of the methyl moieties. Together all these experiments resulted in the assignment of 83% of Leu and Ile residues. By inclusion of additional 15N-edited NOESY and 13C-edited TOCSY and NOESY experiments, the overall sidechain assignments for TM1-TM2 in TFE:water was increased to 82% of all of the assignable protons including 92% of the aliphatic residues. Only 62% of the aromatic residues were assigned due to severe overlap of the resonances, in particular of Phe aromatic spin systems. In total, 89% of all of the expected nuclei were assigned for TM1-TM2 in TFE:water at 25°C.
3.3 Analysis of secondary structure information for TM1-TM2
Circular dichroism data previously indicated that this peptide was highly helical in TFE:water [74]. Chemical shift difference analysis, based on chemical shift indexing [75–77], was used to analyze the secondary structure of TM1-TM2 at the residue level [78–82]. Based on the Cα, Cβ, Hα and CO secondary chemical shifts, TM1-TM2 appears to be highly helical at both 25°C and 45°C (Figure 2 and data not shown, respectively). The helical regions (N-terminal, TM1 and TM2) determined in the structure of TM1-TM2 in LPPG micelles [1] are indicated by boxes in the Figure. Based on the Cα chemical shifts the polypeptide initiates a helical structure at I36 and this helix continues to residue R74. The helix is then interrupted and TM2 spans residues I80 to N105. The Cα shift data indicates a slight perturbation of the helix in the middle of TM1 in the region of the GxxxG motif between residues 56 and 60. Shift data based on Cβ, Hα and CO nuclei are fairly consistent with the Cα conclusions with some differences in the boundary residues. As seen in Figure 2, the helical regions appear to be slightly elongated in TFE:water when compared to the same peptide in LPPG micelles, most likely due to the helix inducing properties of TFE. Furthermore, the region between the N-terminal helix and TM1 in TFE:water is not as well defined as in LPPG micelles, whereas there is a clear break between TM1 and TM2.
Figure 2.

Secondary chemcial shifts of CO, Cα and Cβ nuclei for Ste2p(G31-T110; TM1-TM2) in TFE:water at 25°C. The gray boxes indicates locations of helices in the LPPG structure [1].
3.4 Dynamics of TM1-TM2 in TFE:water
15N relaxation has proven to be a valuable tool to learn about protein backbone dynamics. The 15N{H}-NOE experiment (H-NOE) [83, 84] was performed to determine the relative rigidity of different regions of the peptide backbone. At 25°C, the H-NOE values for many residues are between 0.6 and 0.8 (Figure 3A). Residues near the N- and C-termini have very low H-NOE values indicating lack of secondary structure. The H-NOEs of residues 42–49, 51–61, 65–73, 81–103 are mostly above 0.6 with increased rigidity (H-NOE values >0.75) observed for residues 43–46, 49–51, and 91–93. Residues with lower H-NOE values include residues 62–64, which are C-terminal to the GxxxG region, and the residues of the putative first intracellular loop (residues 74 to 79). These data match the 15N{H}-NOE data determined in LPPG micelles except that the flexible region in TM1 was observed within the GxxxG motif itself (residues 56 and 57; see [1]).
Figure 3.

Dynamics of Ste2p(G31-T110; TM1-TM2) in TFE:water. A) 15N{1H}-NOE of [15N]-TM1-TM2 in TFE:water at 25°C. Values above 0.6 (bottom line) and above 0.75 are indicative of rigidity or increased rigidity, respectively. B) H-D exchange analysis of [15N]-TM1-TM2 in TFE-d3:D2O+0.1%TFA-d. The gray boxes indicates locations of the predicted helices from the LPPG structure [1].
To learn about the presence of hydrogen bonds we used hydrogen-deuterium exchange [85–88]. [15N]-TM1-TM2 was solubilized in fully deuterated TFE-d3:D2O(0.1% TFA-d) and exchange was followed over time by recording a series of [15N,1H]-HSQC experiments performed from 15 min to 10 days after sample preparation (Figure 3B). The fastest exchange occurred at the N- and C-termini. From residue 37 through residue 104, the exchange rate is relatively low (usually 10–1000 fold slower than the terminal residues). The N-terminal helix and TM1 appear to be continuous by this method of analysis as there is no segment with increased exchange in between these regions. The middle of TM1 exchanges more quickly than either end of the helix. There is faster exchange in the loop region and then the exchange slows down in TM2, very much indicative of a tight helix formation.
3.5 Long-range Constraints and Distance Restraints
To probe for interhelical contacts we have investigated the presence of NOEs between the methyl groups of different helices and PRE attenuations. Three-dimensional NOESY experiments were performed using the [15N,13C,2H(1H(methyl)-ILV)]-TM1-TM2 peptide. In contrast to similar experiments using the same peptide in LPPG micelles, only intrahelical connectivities were observed in the 13C-edited NOESY experiments. No unambiguous long-range NOEs could be found under the conditions used.
To probe for the presence of transient interhelical contacts, an MTSL spin-label was attached to a Cys-containing analog (C59) of TM1-TM2 as described in the Supplementary Data (Supplementary Figure S1). At 25°C comparison of [15N,1H]-HSQC spectra recorded on the peptide with the nitroxide radical (MTSL) attached and a control peptide, where the nitroxide radical is replaced by an acetyl group (AcMTSL), exhibited greater than a 60% decrease in intensity for most of the residues spanning Q51 to I71, covering the majority of these TM1 residues (Figure 4, top). This result is expected because residues within this helical region are all within a 10–15Å distance from C59 [89–92](note that the range is somewhat broadened because MTSL is inserted in a somewhat flexible region of TM1 and is attached by a flexible linker). Interestingly, further significant reductions corresponding to normalized intensities of 0.4 to 0.6 were observed for residues I80 to F99 putatively associated with TM2. The side chain NH groups at residues N84 and Q85 were severely broadened (data not shown) and the intensity of A96 was affected the most. At 45°C, the effect of MTSL on the TM2 residues was significantly decreased (Figure 4, bottom).
Figure 4.

Signal attenuation due to the presence of the spin label MTSL conjugated to C59. Relative peak volumes are depicted computed from [15N,1H]-HSQC spectra of [15N]-Met-His6-TM1-TM2-C59-AcMTSL and [15N]-Met-His6-TM1-TM2-C59-MTSL recorded at 25°C (top) and 45°C (bottom). Residues indicated by a star could not be reliably integrated. Locations of helices of TM1-TM2 in LPPG shown are shown as gray boxes. Residues marked by an asterix are partially overlapping and hence difficult to integrate.
3.6 Structure calculations in ATNOS/CANDID of TM1-TM2
The assigned chemical shift list and restraints derived from the PRE measurements were used to calculate an NMR structure for TM1-TM2 in TFE:water at 25°C. The NOEs were automatically assigned using the ATNOS/CANDID algorithm using the CYANA “autoassign” macro [67, 68] and converted to upper distance restraints. Additional dihedral angle restraints as determined from TALOS+ were used for backbone torsion angles. Furthermore, a soft restraint was included at residue A96 based on the paramagnetic relaxation data described above. A summary of intra- and inter-residue contacts is displayed in Figure 5 and the total number of restraints is summarized in Table 1. Based on the presence of both Hα,Hβ i,i+3 and Hα,N i,i+4 NOEs, a short N-terminal α-helix is present from residues 37 to 41. Similar connectivities are observed for residues 42 to 73 which correspond to the TM1 region of the GPCR fragment. The absence of i,i+4 NOEs within TM1 is indicative of a region of flexibility in the center of this transmembrane domain. TM2 is well-defined throughout by i,i+3 and i,i+4 connectivites for residues 80 to 103.
Figure 5.

Characteristic NOE contacts for Ste2p(G31-T110) in TFE:water(0.1% TFA) (1:1,v:v) as used for the structure calculation.
Table 1.
NMR Characterization of Ste2p(G31-T110) in TFE:water
| Distance restraints | Total | 1628 |
|---|---|---|
| Intra-residual | 342 | |
| Sequential (|i-j|=1) | 533 | |
| Short-range (|i-j|≤1) | 875 | |
| Medium (1≤|i-j|<5) | 738 | |
| Long-range (|i-j|≥5) | 15 | |
| Distance angle restraints | Total | 753 |
| RMSD (Å) | ||
|
| ||
| 33–41 backbone | 0.02±0.02 | |
| 33–41 all heavy atoms | 0.30±0.11 | |
| 45–74 backbone | 2.50±1.29 | |
| 45–74 all heavy atoms | 2.82±1.28 | |
| 81–106 backbone | 1.13±0.43 | |
| 81–106 all heavy atoms | 1.81±0.53 | |
| 45–106 backbone | 4.49±2.57 | |
| 45–106 all heavy atoms | 4.94±2.72 | |
| Structure check | ||
|
| ||
| Ramachandran statistics | Favored | 88.6% |
| Additionally allowed | 11.4% | |
| Generously allowed | 0% | |
| Disallowed | 0% | |
The superposition of 20 energy-minimized structures is shown in Figure 6A. The structures generated are predominantly α-helical in which TM2 is observed throughout in all conformers. In contrast, the putative segment of TM1 does not display a continuous helix. Instead the contact region of TM1 with TM2 serves as a hinge region, dividing TM1 into two parts with the C-terminal end sampling a larger conformational space. The presence of the rather stable TM2 helix and N-terminal helix with a split TM1 is in agreement with the secondary chemical shifts and the dynamics data presented above. In all of the structures, the N-terminal helix begins at residue 33 and ends at residue 41. When comparing the calculated structures with those generated in LPPG micelles (Figure 6B), the N-terminal helix is the same length, but is located closer to the N-terminus of the peptide. Conversely, TM1 is longer in TFE:water (residues 45 to 74) than in LPPG (residues 49 to 72). There is a flexible region near the GxxxG motif involving residues 58 through 63 which is also observed in the micellar environment [1]. This region has been shown to be relevant for α-factor binding [23, 26–28]. Finally, TM2 is defined to begin at residue 81 and extends to residue 106 which is longer than in LPPG micelles (residues 81 to 103). The RMSDs for individually superimposing backbone atoms of the NT- helix, TM1 and TM2 are 0.02±0.02Å, 2.50±1.29Å and 1.13±0.43Å, respectively. The overall structure, when aligning residues 44–106, lacks convergence to a single structure and has a backbone RMSD of 4.49±2.57Å.
Figure 6.

Calculated structures of Ste2p(G31-T110; TM1-TM2) in TFE:water and LPPG micelles. A) Superposition of TM1-TM2 in TFE:water(+01.% TFA) for backbone atoms of residues 45–74. The lowest energy structure is displayed as a ribbon from a side view and bottom view. B) The calculated structure of TM1-TM2 in LPPG micelles (Neumoin et al., 2009, Figure 5 [1]).
3.7 Comparison of chemical shifts in organic solvent and detergent micelles
An important aim of this work was to compare chemical shifts obtained in the TFE:water mixture with those measured in micelles which we have previously assigned [1]. The backbone assignment was fairly complete but significant difficulties in LPPG micelles prevented complete sidechain assignments.
A detailed comparison of CO, Cα and Cβ chemical shifts of TM1-TM2 in LPPG micelles and in TFE:water is depicted in Figure 7. In general, large deviations are observed, but most of the differences seem to stem from a systematic offset (~2.7 ppm for Cβ; ~2.7 ppm for Cα and ~1.1 ppm for CO) of the chemical shift. While the reason for this offset is unclear to us presently, and may be due to differences in calibration in the two different mimetics, we expect that it can be corrected. Furthermore, the spectra were acquired at two different temperatures. In LPPG, the NMR experiments were performed at 45°C to increase the peak sharpness, whereas the NMR was performed at 25°C for the TFE:water experiments in order to increase the chances for interhelical contact. Even after the subtraction of the offset, significant differences remain. In the case of the carbonyl chemical shifts, a somewhat oscillatory behavior is observed. The residual differences of Cα and Cβ chemical shifts between the two environments for most internal residues are relatively small. Larger differences are observed for residues close to the termini of the N-terminal helix. This is in agreement with the secondary chemical shifts that indicate that the helix location is slightly shifted in the two environments. In case of the Cα shifts, an increase in the chemical shift difference is present in the loop between TM1 and TM2 as well as for the beginning of TM2. The former is most likely related to the destabilization of a particular loop conformation due to the more flexible orientation of TM1 relative to TM2 in organic solvents. The origin of the difference in the beginning of TM2 is less obvious but may be due to different orientations of the two TM domains relative to each other in the two membrane mimetic environments. Furthermore, larger differences when compared to the rest of TM1 are observed for the central region of TM1, for which a destabilized helix is observed in TFE:water. The differences observed in the Cβ shifts are more consistent, except for large deviations in the N-terminal region and at the beginning of TM1, and seem to follow trends observed for the Cα shifts. We noticed that the standard deviations for the chemical shifts of each class of nuclei in LPPG are in the same range as their offset-corrected differences between the two environments, and therefore we conclude that these shifts, unfortunately, are of limited predictive power.
Figure 7.

Chemical shift differences of CO,Cα and Cβ nuclei of Ste2p(G31-T110) {Δδ13C(TFE:water-LPPG)}. The locations of helices as determined in the LPPG structure are indicated by the shaded background.
As an example of remote sidechain moieties we have also compared the positions of methyl group peaks of the selectively methyl protonated sample in the proton-carbon correlation spectra. A comparison of the constant-time [13C,1H]-HSQC spectra in both membrane mimetics is depicted in Figure 8, top. In general, most of the methyl peaks from the same residues are in similar positions. Again, an overall offset of ~1.9 ppm seems to exist, but there are some residues that are shifted more than others. A more detailed analysis shows that standard deviations of proton and carbon chemical shifts are in the same range as the average difference between the two environments (data not shown). Interestingly, we have not observed that differences in proton or carbon chemical shifts are larger for those methyl groups that are involved in the formation of interhelical contacts in LPPG.
Figure 8.
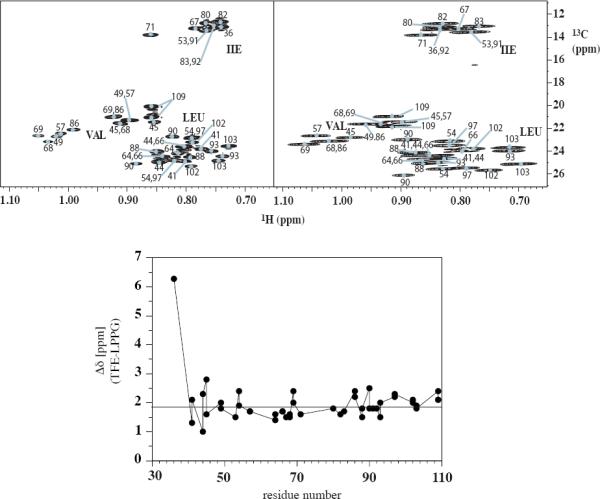
Top: Comparison of [13C,1H]-HSQC spectra of [15N,13C,2H(1H(methyl)-ILV)]-TM1-TM2 in LPPG micelles (45°C, left) and TFE:water (25°C, right). Bottom: 13C chemical shift differences of methyl carbon chemical shifts {Δδ 13C(TFE:water-LPPG)}.
To a large extent differences in proton chemical shifts are due to a collapse in chemical shift dispersion in TFE:water, and therefore cannot be simply corrected by a linear calibration term. We have tried to automatically transfer the assignments from TFE:water to the LPPG spectra using an algorithm that minimizes the sum of the normalized differences of the corresponding proton and carbon chemical shifts (see Methods). Using this algorithm 15 out of the 42 methyl groups were correctly assigned. Many of the correctly assigned peaks were relatively isolated, while automatic assignments failed to be successful for crowded regions of the spectrum.
4. Discussion
4.1 The structure of TM1-TM2 in TFE:water and its biological significance
Our NMR analysis of TM1-TM2, a two transmembrane fragment of the GPCR Ste2p in TFE:water at 25°C employed a variety of labeling strategies [1, 74, 93] to obtain assignment for 89% of the observable resonances. Secondary chemical shifts defined regions of helicity that were slightly longer but in fairly good agreement with the boundaries predicted by various computational tools including PHD [94] and MPex [95], and with those found in an NMR analysis of this same fragment in LPPG micelles [1]. When compared to the rhodopsin-templated model [96], the helical boundaries are extended in TFE:water, but are similar to those observed by solvent accessibility functional analysis of Ste2p [97]. In addition, we observed flexibility in the middle of TM1 at a GxxxG kink and a helical region in the N-terminal portion of this polypeptide in TFE:water similar to what was observed in LPPG micelles [1].
Although more than 1600 NOEs and additional dihedral angle constraints were used, the calculated NMR structures of TM1-TM2 in TFE:water converged poorly (Figure 6A). However the helices pertaining to the N-terminal region, TM1 or TM2 were locally well-defined. Based on the PRE data, the polypeptide is likely adopting many conformations with transient contacts being formed around residues C59 and A96. The PRE data clearly indicate that the tertiary contact is less stable at 45°C compared to 25°C. Similar transient effects have been observed in the folding and unfolding of α-synuclein [98]. These data indicate that the interhelical contacts are unstable, but that the sampled conformational space contains significant contributions from helical hairpin-like structures.
4.2 Structure comparison in different membrane mimetic environments
An important conclusion of the present study is that the tertiary structure is different inc TFE:water and LPPG micelles and that the nature of the membrane mimetic environment affects the stability of the hairpin formed by TM1-TM2. The choice of TFE:water in a 1:1 ratio (v:v) was based on comparisons of the predicted helicity of the peptide region and the results from the CD analysis [74]. The helicities at TFE contents larger than 50% (v:v) were all very similar, but significantly decreased when the TFE content was 25% (v:v). TFE:water (1:1, v:v) was chosen so that the minimal amount of organic solvent was present in the sample. The initial screenings by both CD and [15N,1H]-HSQC did not probe for the presence of helix-helix interactions and therefore are not predictive of tertiary structure.
The NMR structure of TM1-TM2 in LPPG micelles had an overall RMSD of 2.36±0.97 Å for residues 49–103 which encompass the two TM domains of the hairpin that is formed (Figure 6B). In comparison, the structure of TM1-TM2 in TFE:water has an RMSD of 4.49±2.57 Å for residues 45–106. Flexibility at the center of TM1 would lead to a larger spread of the conformational space sampled by this helical element of the hairpin. In both membrane mimetics increased flexibility within the center of TM1 is supported by secondary chemical shifts and 15N relaxation data, and coincides with the contact point region with TM2. The secondary structures assumed by TM1-TM2 are similar in both membrane mimetics. However, although the PRE data is consistent with some contact between the TM1 and TM2 helices the structures calculated from the NMR data for TM1-TM2 in TFE:water do not converge. We conclude that in TFE:water transient tertiary conformations exist but that the conformational distribution is significantly more disperse than in the micellar environment of LPPG. Nevertheless, it is reasonable to conclude that the TM1-TM2 domain of Ste2p has an intrinsic tendency to fold and that this tendency is manifested even in TFE:water mixtures.
The fact that the tertiary structures differ in the two membrane mimetics can be attributed to the essentially isotropic nature of the organic:aqueous solvent. Therefore, TFE:water cannot reproduce the change in hydrophobicity that a peptide chain experiences when traversing a lipid bilayer. Interhelical contacts are often mediated by interactions of polar groups, i.e. Ser residues, that are deeply buried within the bilayer. In the TFE:water solvent system the water molecules can solvate the hydroxyl group and hence the driving force for helix-helix contact to compensate for the unsolvated Ser hydroxyl in the hydrophobic lipid core is lowered substantially. Another difference between the organic and detergent membrane mimetics is the presence of the acyl chains in the LPPG micelles. The long hydrocarbon side chains of palmitic acid of LPPG may lead to multiple interactions that stabilize the hydrophobic core of the TM helices. We have found that the length of the acyl chains is very important to the formation of the secondary structure as observed by CD (Cohen and Naider, unpublished data). From this perspective it is noteworthy that recent crystallographic studies of GPCRs have revealed the presence of specific contacts of lipids or cholesterol with the TM helices [99–102].
4.3 Correlation of chemical shifts derived from TM1-TM2 in LPPG micelles and TFE:water
Chemical shifts are influenced by various parameters. For example, amide proton shifts depend on many factors, with torsion angles making one contribution, but hydrogen bonds, ring current shifts and other local anisotropies influence the shifts as well. 13C chemical shifts are more predictable. The extent to which the shift is influenced by ϕψ torsion angles has been estimated to be 50, 25 and 10 % for CO, Cα and Cβ resonances [103]. Chemical shift differences in TFE:water and LPPG of the Cα shifts of TM1-TM2 seem to correlate loosely with changes in secondary structure, and the same is true, albeit to a smaller extent, for Cβ shifts. For the methyl groups in the selectively ILV protonated sample, coincidence of chemical shifts is slightly better, most likely due to the fact that the change in the environment is smaller. These methyl groups serve as important spectroscopic probes in membrane proteins and the Nietlispach group has used them to make assignments in the sensory rhodopsin receptor, which led to a high-resolution structure [13].
Given the greater efficiency of making resonance assignments for membrane proteins in TFE:water compared to detergent micelles, one objective of this study was to evaluate whether one could use such assignments in order to accelerate structure calculations in detergents. We investigated whether automated algorithms could help transferring chemical shift assignments from TFE:water to detergent micelles. Using exclusively 2D [13C, 1H]-HSQC spectra, preliminary tests in which an automatic transfer of the assignments of the methyl groups was attempted by globally minimizing the differences in chemical shifts in the two environments we had, however, only a moderate success rate (approx. 35%). Considering that spectra of the entire receptor contain many more signals from methyl moieties than TM1-TM2, it is likely that the automatic transfer would be even less successful for an intact GPCR. It is, however, possible that the reliability of the transfer procedure can be improved by taking more spectral data into account, e.g. if a comparison of the strips from a 13C-resolved NOESYs were added to the analysis. At best, assignments derived from TFE:water environments presently seem to be useful to provide initial suggestions for assignments in large IMPs, which then need to be verified from other data.
In conclusion, herein we have determined that in TFE:water (1:1,v:v) Ste2p(G31-T110), a two transmembrane domain fragment of a GPCR, assumes a distribution of structures. In this membrane mimetic, the peptide assumes helical domains, which were in reasonable agreement with predictions and conclusions from NMR studies in LPPG micelles. Notably, TM1-TM2 forms a transient helical hairpin at 25°C in the organic aqueous medium. An analysis of chemical shift data in TFE:water and LPPG indicates that, perhaps due to differences in the secondary and tertiary structure, there is no simple correlation between main chain or side chain chemical shifts,. Given these observations it will be interesting to explore whether an increase in the number of TM domains will stabilize the tertiary structure in organic:aqueous media, and thereby improve the agreement of the chemical shifts. This would clearly increase the usefulness of organic:aqueous media as a membrane mimetic environment for solution NMR analysis of large fragments of GPCRs.
Supplementary Material
Highlights
-
➢
NMR analysis of a double transmembrane fragment of a GPCR, Ste2p(G31-T110).
-
➢
Ste2p(G31-T110) in TFE:water(1:1) resulted in a transiently folded helical hairpin.
-
➢
Comparison of NMR structures of Ste2p(G31-T110) in micelles and TFE:water.
-
➢
Chemical shift correlation analysis of shifts derived in two membrane mimetics.
Acknowledgements
Fred Naider is the Leonard and Esther Kurtz Term Professor at the College of Staten Island, City University of New York. This work was supported by research grants GM22086 (F.N.) and GM22087 (J.M.B.) from the National Institutes of Health. P.G. acknowledges financial support by the Lichtenberg program of the Volkswagen Foundation, the Deutsche Forschungsgemeinschaft (DFG), and the Japan Society for the Promotion of Science (JSPS) and O.Z. general support by the Swiss Science Foundation. Professor Naider is a member of the New York Structural Biology Center, which is a STAR center supported by the New York State Office of Science, Technology, and Academic Research. The 900 MHz spectrometer at the New York Structural Biology Center was purchased with funds from the National Institutes of Health, USA; the Keck Foundation, New York State; and the New York City Economic Development Corp.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Neumoin A, Cohen LS, Arshava B, Tantry S, Becker JM, Zerbe O, Naider F. Structure of a double transmembrane fragment of a G-protein-coupled receptor in micelles. Biophys J. 2009;96:3187–3196. doi: 10.1016/j.bpj.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].White S. Membrane Proteins of Known 3D Structure. Stephen White Laboratory; 2011. [Google Scholar]
- [3].Raman P, Cherezov V, Caffrey M. The Membrane Protein Data Bank. Cell Mol Life Sci. 2006;63:36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nature reviews. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- [5].McCusker EC, Bane SE, O'Malley MA, Robinson AS. Heterologous GPCR expression: a bottleneck to obtaining crystal structures. Biotechnology progress. 2007;23:540–547. doi: 10.1021/bp060349b. [DOI] [PubMed] [Google Scholar]
- [6].Arinaminpathy Y, Khurana E, Engelman DM, Gerstein MB. Computational analysis of membrane proteins: the largest class of drug targets. Drug Discov Today. 2009;14:1130–1135. doi: 10.1016/j.drudis.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Durr UH, Waskell L, Ramamoorthy A. The cytochromes P450 and b5 and their reductases--promising targets for structural studies by advanced solid-state NMR spectroscopy. Biochimica et biophysica acta. 2007;1768:3235–3259. doi: 10.1016/j.bbamem.2007.08.007. [DOI] [PubMed] [Google Scholar]
- [8].Wider G. NMR techniques used with very large biological macromolecules in solution. Methods in enzymology. 2005;394:382–398. doi: 10.1016/S0076-6879(05)94015-9. [DOI] [PubMed] [Google Scholar]
- [9].Tian C, Breyer RM, Kim HJ, Karra MD, Friedman DB, Karpay A, Sanders CR. Solution NMR spectroscopy of the human vasopressin V2 receptor, a G protein-coupled receptor. Journal of the American Chemical Society. 2005;127:8010–8011. doi: 10.1021/ja051161b. [DOI] [PubMed] [Google Scholar]
- [10].Tian C, Breyer RM, Kim HJ, Karra MD, Friedman DB, Karpay A, Sanders CR. Solution NMR spectroscopy of the human vasopressin v2 receptor, a g protein-coupled receptor. Journal of the American Chemical Society. 2006;128:5300. doi: 10.1021/ja051161b. j. Am. Chem. Soc. 2005, 127, 8010–8011. [DOI] [PubMed] [Google Scholar]
- [11].Van Horn WD, Kim HJ, Ellis CD, Hadziselimovic A, Sulistijo ES, Karra MD, Tian C, Sonnichsen FD, Sanders CR. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science. 2009;324:1726–1729. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gautier A, Kirkpatrick JP, Nietlispach D. Solution-state NMR spectroscopy of a seven-helix transmembrane protein receptor: backbone assignment, secondary structure, and dynamics. Angewandte Chemie. 2008;47:7297–7300. doi: 10.1002/anie.200802783. International ed. [DOI] [PubMed] [Google Scholar]
- [13].Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol. 2010;17:768–774. doi: 10.1038/nsmb.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kobilka BK, Kobilka TS, Daniel K, Regan JW, Caron MG, Lefkowitz RJ. Chimeric alpha 2-,beta 2-adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science. 1988;240:1310–1316. doi: 10.1126/science.2836950. [DOI] [PubMed] [Google Scholar]
- [15].Schoneberg T, Liu J, Wess J. Plasma membrane localization and functional rescue of truncated forms of a G protein-coupled receptor. J Biol Chem. 1995;270:18000–18006. doi: 10.1074/jbc.270.30.18000. [DOI] [PubMed] [Google Scholar]
- [16].Ridge KD, Lee SS, Abdulaev NG. Examining rhodopsin folding and assembly through expression of polypeptide fragments. J Biol Chem. 1996;271:7860–7867. doi: 10.1074/jbc.271.13.7860. [DOI] [PubMed] [Google Scholar]
- [17].Martin NP, Leavitt LM, Sommers CM, Dumont ME. Assembly of G protein-coupled receptors from fragments: identification of functional receptors with discontinuities in each of the loops connecting transmembrane segments. Biochemistry. 1999;38:682–695. doi: 10.1021/bi982062w. [DOI] [PubMed] [Google Scholar]
- [18].Overton MC, Blumer KJ. The extracellular N-terminal domain and transmembrane domains 1 and 2 mediate oligomerization of a yeast G protein-coupled receptor. J Biol Chem. 2002;277:41463–41472. doi: 10.1074/jbc.M205368200. [DOI] [PubMed] [Google Scholar]
- [19].Dosil M, Giot L, Davis C, Konopka JB. Dominant-negative mutations in the G-protein-coupled alpha-factor receptor map to the extracellular ends of the transmembrane segments. Molecular and cellular biology. 1998;18:5981–5991. doi: 10.1128/mcb.18.10.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee BK, Lee YH, Hauser M, Son CD, Khare S, Naider F, Becker JM. Tyr266 in the sixth transmembrane domain of the yeast alpha-factor receptor plays key roles in receptor activation and ligand specificity. Biochemistry. 2002;41:13681–13689. doi: 10.1021/bi026100u. [DOI] [PubMed] [Google Scholar]
- [21].Henry LK, Khare S, Son C, Babu VV, Naider F, Becker JM. Identification of a contact region between the tridecapeptide alpha-factor mating pheromone of Saccharomyces cerevisiae and its G protein-coupled receptor by photoaffinity labeling. Biochemistry. 2002;41:6128–6139. doi: 10.1021/bi015863z. [DOI] [PubMed] [Google Scholar]
- [22].Celic A, Martin NP, Son CD, Becker JM, Naider F, Dumont ME. Sequences in the intracellular loops of the yeast pheromone receptor Ste2p required for G protein activation. Biochemistry. 2003;42:3004–3017. doi: 10.1021/bi0269308. [DOI] [PubMed] [Google Scholar]
- [23].Son CD, Sargsyan H, Naider F, Becker JM. Identification of ligand binding regions of the Saccharomyces cerevisiae alpha-factor pheromone receptor by photoaffinity cross-linking. Biochemistry. 2004;43:13193–13203. doi: 10.1021/bi0496889. [DOI] [PubMed] [Google Scholar]
- [24].Hauser M, Kauffman S, Lee BK, Naider F, Becker JM. The first extracellular loop of the Saccharomyces cerevisiae G protein-coupled receptor Ste2p undergoes a conformational change upon ligand binding. J Biol Chem. 2007;282:10387–10397. doi: 10.1074/jbc.M608903200. [DOI] [PubMed] [Google Scholar]
- [25].Bajaj A, Connelly SM, Gehret AU, Naider F, Dumont ME. Role of extracellular charged amino acids in the yeast alpha-factor receptor. Biochimica et biophysica acta. 2007;1773:707–717. doi: 10.1016/j.bbamcr.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huang LY, Umanah G, Hauser M, Son C, Arshava B, Naider F, Becker JM. Unnatural amino acid replacement in a yeast G protein-coupled receptor in its native environment. Biochemistry. 2008;47:5638–5648. doi: 10.1021/bi701866e. [DOI] [PubMed] [Google Scholar]
- [27].Umanah GK, Son C, Ding F, Naider F, Becker JM. Cross-linking of a DOPA-containing peptide ligand into its G protein-coupled receptor. Biochemistry. 2009;48:2033–2044. doi: 10.1021/bi802061z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tantry S, Ding FX, Dumont M, Becker JM, Naider F. Binding of fluorinated phenylalanine alpha-factor analogues to Ste2p: evidence for a cation-pi binding interaction between a peptide ligand and its cognate G protein-coupled receptor. Biochemistry. 2010;49:5007–5015. doi: 10.1021/bi100280f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arshava B, Liu SF, Jiang H, Breslav M, Becker JM, Naider F. Structure of segments of a G protein-coupled receptor: CD and NMR analysis of the Saccharomyces cerevisiae tridecapeptide pheromone receptor. Biopolymers. 1998;46:343–357. doi: 10.1002/(SICI)1097-0282(199811)46:6<343::AID-BIP1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- [30].Arshava B, Taran I, Xie H, Becker JM, Naider F. High resolution NMR analysis of the seven transmembrane domains of a heptahelical receptor in organic-aqueous medium. Biopolymers. 2002;64:161–176. doi: 10.1002/bip.10157. [DOI] [PubMed] [Google Scholar]
- [31].Estephan R, Englander J, Arshava B, Samples KL, Becker JM, Naider F. Biosynthesis and NMR analysis of a 73-residue domain of a Saccharomyces cerevisiae G protein-coupled receptor. Biochemistry. 2005;44:11795–11810. doi: 10.1021/bi0507231. [DOI] [PubMed] [Google Scholar]
- [32].Neumoin A, Arshava B, Becker J, Zerbe O, Naider F. NMR studies in dodecylphosphocholine of a fragment containing the seventh transmembrane helix of a G-protein-coupled receptor from Saccharomyces cerevisiae. Biophys J. 2007;93:467–482. doi: 10.1529/biophysj.106.103770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Goodman M, Naider F, Rupp R. Conformations of Alanine Oligopeptides in Solution. Bioorganic Chemistry. 1971;1:310–328. [Google Scholar]
- [34].Naider F, Goodman M. Conformational Analysis of Oligopeptides by Spectral Techniques. In: Tamelen EEV, editor. Bioorganic Chemistry III, Macro and Multimolecular Systems. Academic Press, Inc; 1977. p. 177. [Google Scholar]
- [35].Nelson JW, Kallenbach NR. Persistence of the alpha-helix stop signal in the S-peptide in trifluoroethanol solutions. Biochemistry. 1989;28:5256–5261. doi: 10.1021/bi00438a050. [DOI] [PubMed] [Google Scholar]
- [36].Bruch MD, Gierasch LM. Comparison of helix stability in wild-type and mutant LamB signal sequences. J Biol Chem. 1990;265:3851–3858. [PubMed] [Google Scholar]
- [37].Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Quarterly reviews of biophysics. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- [38].White SH, Ladokhin AS, Jayasinghe S, Hristova K. How membranes shape protein structure. J Biol Chem. 2001;276:32395–32398. doi: 10.1074/jbc.R100008200. [DOI] [PubMed] [Google Scholar]
- [39].Krueger-Koplin RD, Sorgen PL, Krueger-Koplin ST, Rivera-Torres IO, Cahill SM, Hicks DB, Grinius L, Krulwich TA, Girvin ME. An evaluation of detergents for NMR structural studies of membrane proteins. Journal of biomolecular NMR. 2004;28:43–57. doi: 10.1023/B:JNMR.0000012875.80898.8f. [DOI] [PubMed] [Google Scholar]
- [40].Girvin ME, Rastogi VK, Abildgaard F, Markley JL, Fillingame RH. Solution structure of the transmembrane H+-transporting subunit c of the F1F0 ATP synthase. Biochemistry. 1998;37:8817–8824. doi: 10.1021/bi980511m. [DOI] [PubMed] [Google Scholar]
- [41].Schwaiger M, Lebendiker M, Yerushalmi H, Coles M, Groger A, Schwarz C, Schuldiner S, Kessler H. NMR investigation of the multidrug transporter EmrE, an integral membrane protein. European journal of biochemistry / FEBS. 1998;254:610–619. doi: 10.1046/j.1432-1327.1998.2540610.x. [DOI] [PubMed] [Google Scholar]
- [42].Ma D, Liu Z, Li L, Tang P, Xu Y. Structure and dynamics of the second and third transmembrane domains of human glycine receptor. Biochemistry. 2005;44:8790–8800. doi: 10.1021/bi050256n. [DOI] [PubMed] [Google Scholar]
- [43].Brender JR, Nanga RP, Popovych N, Soong R, Macdonald PM, Ramamoorthy A. The amyloidogenic SEVI precursor, PAP248–286, is highly unfolded in solution despite an underlying helical tendency. Biochimica et biophysica acta. 2011;1808:1161–1169. doi: 10.1016/j.bbamem.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Anderson VL, Ramlall TF, Rospigliosi CC, Webb WW, Eliezer D. Identification of a helical intermediate in trifluoroethanol-induced alpha-synuclein aggregation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18850–18855. doi: 10.1073/pnas.1012336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Giehm L, Oliveira CL, Christiansen G, Pedersen JS, Otzen DE. SDS-induced fibrillation of alpha-synuclein: an alternative fibrillation pathway. Journal of molecular biology. 2010;401:115–133. doi: 10.1016/j.jmb.2010.05.060. [DOI] [PubMed] [Google Scholar]
- [46].Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. Journal of the American Chemical Society. 1992;114:10663–10665. [Google Scholar]
- [47].Ikura M, Kay LE, Bax A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- [48].Grzesiek S, Bax A. Improved 3D Triple-Resonance NMR Techniques Applied to a 31-kDa Protein. J Magn Reson. 1992;96:432–440. [Google Scholar]
- [49].Muhandiram D, Kay LE. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J Magn Reson B. 1994;103:203–216. [Google Scholar]
- [50].Kay LE, Xu GY, Yamazaki T. Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J Magn Reson A. 1994;109:129–133. [Google Scholar]
- [51].Yamazaki T, Lee W, Arrowsmith CH, Muhandiram D, Kay LE. A suite of triple resonance NMR experiments for the backbone assignment of 15N, 13C, 2H labeled proteins with high sensitivity. Journal of the American Chemical Society. 1994;116:11655–11666. [Google Scholar]
- [52].Wittekind M, Muller L. HNCACB, a high sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonanaces with alpha- and beta-carbon resonances in proteins. J Magn Reson B. 1993;101:201–205. [Google Scholar]
- [53].Zhang O, Kay LE, Olivier JP, Forman-Kay JD. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- [54].Bax A, Clore GM, Gronenborn AM. H-1-H-1 correlation via isotropic mixing of C-13 magnetization, a new 3-dimensional approach for assigning H-1 and C-13 spectra of C-13-enriched proteins. J Magn Reson. 1990;88:425–431. [Google Scholar]
- [55].Kay LE, Xu G, Singer A, Muhandiram D, Formankay J. A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. Journal of Magnetic Resonance, Series B. 1993;101:333–337. [Google Scholar]
- [56].Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. Journal of the American Chemical Society. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- [57].Vuister GW, Bax A. Resolution enhancement and spectral editing of uniformly 13C-enriched proteins by homonuclear broadband 13C decoupling. Journal of Magnetic Resonance. 1992;98:428–435. [Google Scholar]
- [58].Thrippleton MJ, Keeler J. Elimination of Zero-Quantum Interference in Two-Dimensional NMR Spectra Angewandte Chemie. 2003;42:3938–3941. doi: 10.1002/anie.200351947. International Edition. [DOI] [PubMed] [Google Scholar]
- [59].Levitt MH, Freeman R, Frenkiel T. Broadband heteronuclear decoupling. Journal of Magnetic Resonance. 1982;47:328–330. [Google Scholar]
- [60].Bax A, Davis DG. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. Journal of Magnetic Resonance. 1985;65:355–360. [Google Scholar]
- [61].Palmer AG, III, Cavanagh J, Wright PE, Rance M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. Journal of Magnetic Resonance. 1991;93:151–170. [Google Scholar]
- [62].Davis AL, Keeler J, Laue ED, Moskau D. Experiments for recording pure-absorption heteronuclear correlation spectra using pulsed field gradients. Journal of Magnetic Resonance. 1992;98:207–216. [Google Scholar]
- [63].Diercks T, Coles M, Kessler H. An efficient strategy for assignment of cross-peaks in 3D heteronuclear NOESY experiments. Journal of biomolecular NMR. 1999;15:177–180. doi: 10.1023/A:1008367912535. [DOI] [PubMed] [Google Scholar]
- [64].Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- [65].Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. Journal of biomolecular NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- [66].Keller R. In: The Computer Aided Resonance Assignment. Verlag C, editor. Goldau; Switzerland: 2004. [Google Scholar]
- [67].Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. Journal of biomolecular NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- [68].Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. Journal of molecular biology. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- [69].Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. Journal of biomolecular NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. Journal of molecular biology. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- [71].Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. Journal of molecular graphics. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- [72].Munkres J. Algorithms for the assignment and transporation problems. Journal of the Society for the Industrial and Applied Mathematics. 1957;5:32–38. [Google Scholar]
- [73].Schmucki R, Yokoyama S, Guntert P. Automated assignment of NMR chemical shifts using peak-particle dynamics simulation with the DYNASSIGN algorithm. Journal of biomolecular NMR. 2009;43:97–109. doi: 10.1007/s10858-008-9291-x. [DOI] [PubMed] [Google Scholar]
- [74].Cohen LS, Arshava B, Estephan R, Englander J, Kim H, Hauser M, Zerbe O, Ceruso M, Becker JM, Naider F. Expression and biophysical analysis of two double-transmembrane domain-containing fragments from a yeast G protein-coupled receptor. Biopolymers. 2008;90:117–130. doi: 10.1002/bip.20950. [DOI] [PubMed] [Google Scholar]
- [75].Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. Journal of molecular biology. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- [76].Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- [77].Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. Journal of biomolecular NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- [78].Dutta K, Cox CJ, Alexandrov A, Huang H, Basavappa R, Pascal SM. Letter to the Editor: Sequence-specific chemical shift assignment and chemical shift indexing of murine apo-Mts1. Journal of biomolecular NMR. 2002;22:181–182. doi: 10.1023/a:1014275610386. [DOI] [PubMed] [Google Scholar]
- [79].Jones CT, Ma L, Burgner JW, Groesch TD, Post CB, Kuhn RJ. Flavivirus capsid is a dimeric alpha-helical protein. Journal of virology. 2003;77:7143–7149. doi: 10.1128/JVI.77.12.7143-7149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Burton RA, Tsurupa G, Medved L, Tjandra N. Identification of an ordered compact structure within the recombinant bovine fibrinogen alphaC-domain fragment by NMR. Biochemistry. 2006;45:2257–2266. doi: 10.1021/bi052380c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ahmed MA, Bamm VV, Harauz G, Ladizhansky V. The BG21 isoform of Golli myelin basic protein is intrinsically disordered with a highly flexible amino-terminal domain. Biochemistry. 2007;46:9700–9712. doi: 10.1021/bi700632x. [DOI] [PubMed] [Google Scholar]
- [82].Libich DS, Harauz G. Backbone dynamics of the 18.5 kDa isoform of myelin basic protein reveals transient alpha-helices and a calmodulin-binding site. Biophys J. 2008;94:4847–4866. doi: 10.1529/biophysj.107.125823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- [84].Teng Q. Structural Biology: Practical NMR Applications. Springer; New York: 2005. [Google Scholar]
- [85].Santoro J, Gonzalez C, Bruix M, Neira JL, Nieto JL, Herranz J, Rico M. High-resolution three-dimensional structure of ribonuclease A in solution by nuclear magnetic resonance spectroscopy. Journal of molecular biology. 1993;229:722–734. doi: 10.1006/jmbi.1993.1075. [DOI] [PubMed] [Google Scholar]
- [86].Rodriguez JC, Wilks A, Rivera M. Backbone NMR assignments and H/D exchange studies on the ferric azide- and cyanide-inhibited forms of Pseudomonas aeruginosa heme oxygenase. Biochemistry. 2006;45:4578–4592. doi: 10.1021/bi0600188. [DOI] [PubMed] [Google Scholar]
- [87].Cavanagh J, Fairbrother WJ, Palmer AG, III, Rance M, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Second Edition ed. Elsevier, Inc; 2007. [Google Scholar]
- [88].Werner K, Lehner I, Dhiman HK, Richter C, Glaubitz C, Schwalbe H, Klein-Seetharaman J, Khorana HG. Combined solid state and solution NMR studies of alpha,epsilon-15N labeled bovine rhodopsin. Journal of biomolecular NMR. 2007;37:303–312. doi: 10.1007/s10858-007-9143-0. [DOI] [PubMed] [Google Scholar]
- [89].Teilum K, Kragelund BB, Poulsen FM. Transient structure formation in unfolded acyl-coenzyme A-binding protein observed by site-directed spin labelling. Journal of molecular biology. 2002;324:349–357. doi: 10.1016/s0022-2836(02)01039-2. [DOI] [PubMed] [Google Scholar]
- [90].Liang B, Bushweller JH, Tamm LK. Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. Journal of the American Chemical Society. 2006;128:4389–4397. doi: 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Teriete P, Franzin CM, Choi J, Marassi FM. Structure of the Na,K-ATPase regulatory protein FXYD1 in micelles. Biochemistry. 2007;46:6774–6783. doi: 10.1021/bi700391b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hildebrand PW, Preissner R, Frommel C. Structural features of transmembrane helices. FEBS Lett. 2004;559:145–151. doi: 10.1016/S0014-5793(04)00061-4. [DOI] [PubMed] [Google Scholar]
- [93].Cohen LS, Becker JM, Naider F. Biosynthesis of peptide fragments of eukaryotic GPCRs in Escherichia coli by directing expression into inclusion bodies. J Pept Sci. 2010;16:213–218. doi: 10.1002/psc.1222. [DOI] [PubMed] [Google Scholar]
- [94].Rost B, Sander C, Schneider R. PHD--an automatic mail server for protein secondary structure prediction. Comput Appl Biosci. 1994;10:53–60. doi: 10.1093/bioinformatics/10.1.53. [DOI] [PubMed] [Google Scholar]
- [95].Snider C, Jayasinghe S, Hristova K, White SH. MPEx: a tool for exploring membrane proteins. Protein Sci. 2009;18:2624–2628. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Eilers M, Hornak V, Smith SO, Konopka JB. Comparison of class A and D G protein-coupled receptors: common features in structure and activation. Biochemistry. 2005;44:8959–8975. doi: 10.1021/bi047316u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lin JC, Duell K, Konopka JB. A microdomain formed by the extracellular ends of the transmembrane domains promotes activation of the G protein-coupled alpha-factor receptor. Molecular and cellular biology. 2004;24:2041–2051. doi: 10.1128/MCB.24.5.2041-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science. 2008 doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Alexandrov AI, Mileni M, Chien EY, Hanson MA, Stevens RC. Microscale fluorescent thermal stability assay for membrane proteins. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- [103].Wishart DS, Case DA. Use of chemical shifts in macromolecular structure determination. Methods in enzymology. 2001;338:3–34. doi: 10.1016/s0076-6879(02)38214-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.