

Summary
Activation of p53 by DNA damage results in either cell cycle arrest, allowing DNA repair and cell survival, or induction of apoptosis. As these opposite outcomes are both mediated by p53 stabilization, additional mechanisms to determine this decision must exist. Here we show that glycogen synthase kinase-3 (GSK-3) is required for the p53-mediated induction of the pro-apoptotic BH3 only-protein PUMA, an essential mediator of p53-induced apoptosis. Inhibition of GSK-3 protected from cell death induced by DNA damage and promoted increased long-term cell survival. We demonstrate that GSK-3 phosphorylates serine 86 of the p53-acetyltransferase Tip60. A Tip60S86A mutant was less active to induce p53 K120 acetylation, Histone 4 acetylation and expression of PUMA. Our data suggest that GSK-3 mediated Tip60S86-phosphorylation provides a link between PI3K signaling and the choice for or against apoptosis induction by p53.
Introduction
DNA damage constitutes a potentially deleterious lesion, which triggers signals promoting the stabilization and activation of p53 and the initiation of DNA repair. Activated p53 mediates the arrest of the cell cycle through transcriptional induction of the CDK inhibitor p21WAF/CIP, thereby allowing DNA repair and, if successful, cell survival. In the absence of survival signals however, DNA damage may instead lead to the elimination of a potentially hazardous cell by apoptosis, which also requires stabilization and activation of p53 (Vousden and Lu, 2002).
Apoptosis is induced by p53 through its sequence-specific transcription factor activity, mainly by transcriptional induction of the pro-apoptotic BCL-2 family member PUMA (Nakano and Vousden, 2001; Yu et al., 2001), which is required for p53-induced apoptosis (Jeffers et al., 2003; Villunger et al., 2003). In addition, p53 was found to directly activate BAX in the cytoplasm (Chipuk et al., 2004).
By binding to pro-survival BCL-2 family proteins, PUMA mediates mitochondrial outer membrane permeabilization by release of activators of BAX or BAK (Chipuk et al., 2005; Kuwana et al., 2005; Letai et al., 2002), or BAX/BAK themselves from their inhibitory sequestration by BCL-2, BCL-xL and MCL-1 (Willis et al., 2007). Possibly, PUMA also acts as a direct activator of BAX and BAK (Kim et al., 2006). While the models for cytochrome c release by PUMA are therefore controversial, a key role of PUMA for the elimination of potentially harmful cells by p53-mediated apoptosis is established (Michalak et al., 2008; Villunger et al., 2003).
The molecular basis for the choice between cell cycle arrest and apoptosis induction by p53 is not well understood. As p53 is subject to various posttranslational modifications (Kruse and Gu, 2009), this may provide the level of regulation for or against the choice of cell death through p53. In addition to phosphorylation, mediating p53 stabilization, the acetylation of p53 has recently been shown to be a key signal, promoting its activation upon DNA damage (Kruse and Gu, 2009; Tang et al., 2008). Acetylation at lysine 164 (K164) and six lysines in the C-terminal region of p53 by the acetyltransferase CBP/p300 (Tang et al., 2008) was shown to block repression of p53, mediated by Mdm2- and Mdmx, by preventing their recruitment to target promoters (Kruse and Gu, 2009; Tang et al., 2008). In addition, the pro-apoptotic activity of p53 was demonstrated to depend on its acetylation on lysine 120 (K120) by the acetyltransferases Tip60 and hMOF, demonstrating a role of the acetyltransferase Tip60 for the choice between p53-mediated cell cycle arrest or apoptosis (Sykes et al., 2006; Tang et al., 2006).
Tip60, as part of the evolutionary conserved Tip60/NuA4 complex, was characterized as a histone acetylase involved in gene transcription in yeast and mammalian cells (Allard et al., 1999; Doyon et al., 2004). Further research has extended Tip60 functions as being involved in DNA repair (Kusch et al., 2004; Sun et al., 2005) and required for apoptosis induction upon DNA damage (Ikura et al., 2000). It is now evident that Tip60 acts on multiple levels in gene transcription, the DNA damage response and growth control, by acetylating histone and non-histone proteins (Squatrito et al., 2006).
Importantly, Tip60 was recently characterized as a haplo-insufficient tumor suppressor, as mice lacking a single allele of the Tip60 gene (Htatip) exhibited accelerated myc-induced lymphomagenesis (Gorrini et al., 2007).
A requirement of Tip60 for the p53 pathway was first demonstrated by knock-down and overexpression experiments (Doyon et al., 2004; Legube et al., 2004) and by the identification of Tip60 as a p53 activator in an unbiased large-scale RNAi screening study (Berns et al., 2004). Tip60 was later shown to promote p53-mediated apoptosis (Tyteca et al., 2006). However, while Tip60 modulates p53 activity, the question remains as to how Tip60 acetyltransferase activity is regulated.
It was also proposed that the activity of PI3K/PTEN might contribute to the choice for or against apoptosis upon p53 stabilization (Vousden and Lu, 2002). In support of this idea, mouse embryonic fibroblasts deficient for PTEN were reported to be refractory to p53-induced cell death (Stambolic et al., 2001).
PI3K signaling, induced by growth factor, leads to the inhibition of glycogen synthase kinase-3 (GSK-3). GSK-3 is present in two isoforms, GSK-3α and GSK-3β, which are both repressed by inhibitory phosphorylation through AKT on serine 21 and serine 9, respectively. Accordingly, growth factor stimulation of cells has been shown to reduce GSK-3 activity by 40-50%, while PI3K inhibition increases GSK-3 activity (Cross et al., 1995).
In this study, we set out to investigate the influence of PI3K signaling on p53-mediated apoptosis. We show that p53-induced PUMA, but not p21 expression, requires GSK-3 activity. We have identified the p53-acetyltransferase Tip60 as a novel direct target of GSK-3. GSK-3 phosphorylates S86 of Tip60, and S86 phosphorylation of Tip60 is required for Tip60-mediated acetylation of p53 at K120, H4 acetylation at the puma promoter and the induction of PUMA. These findings demonstrate that Tip60 phosphorylation by GSK-3 contributes to the choice for apoptosis, by promoting the induction of PUMA.
Results
GSK-3 is required to induce PUMA, but not p21 expression
We investigated the role of the PI3K pathway and GSK-3 for the induction of PUMA and apoptosis upon DNA damage. U2OS cells were treated with the PI3K inhibitor LY294002, which results in enhanced GSK-3 activity (Cross et al., 1995; Maurer et al., 2006), combined or not with the potent and specific GSK-3 inhibitor CT98014 as previously described (Cohen and Goedert, 2004; Maurer et al., 2006; Ring et al., 2003). Upon subsequent γ-radiation, p53 and p21 were induced independently of the pharmacological modulation of GSK-3 or PI3K (Fig. 1A, 1B and S1A). However, while we observed some PUMA mRNA induction by inhibition of PI3K, a maximal induction of PUMA mRNA and protein was observed when γ-radiation and the inhibition of PI3K were combined (Fig. 1A, 1C and S1B). Conversely, the pharmacological inhibition of GSK-3 abrogated PUMA, but not p21 expression (Fig. 1A, 1B, 1C and Fig. S1A and S1B). The expression of Noxa mRNA (Fig. S1C) and Bax mRNA and protein (Fig. S1D and S1E) was not largely affected by inhibition of GSK-3, thus indicating a specific requirement of GSK-3 for the induction of PUMA, but not other pro-apoptotic p53 target genes.
Figure 1. PI3K inhibition and DNA damage synergize to induce PUMA expression and apoptosis in a GSK-3-dependent manner.
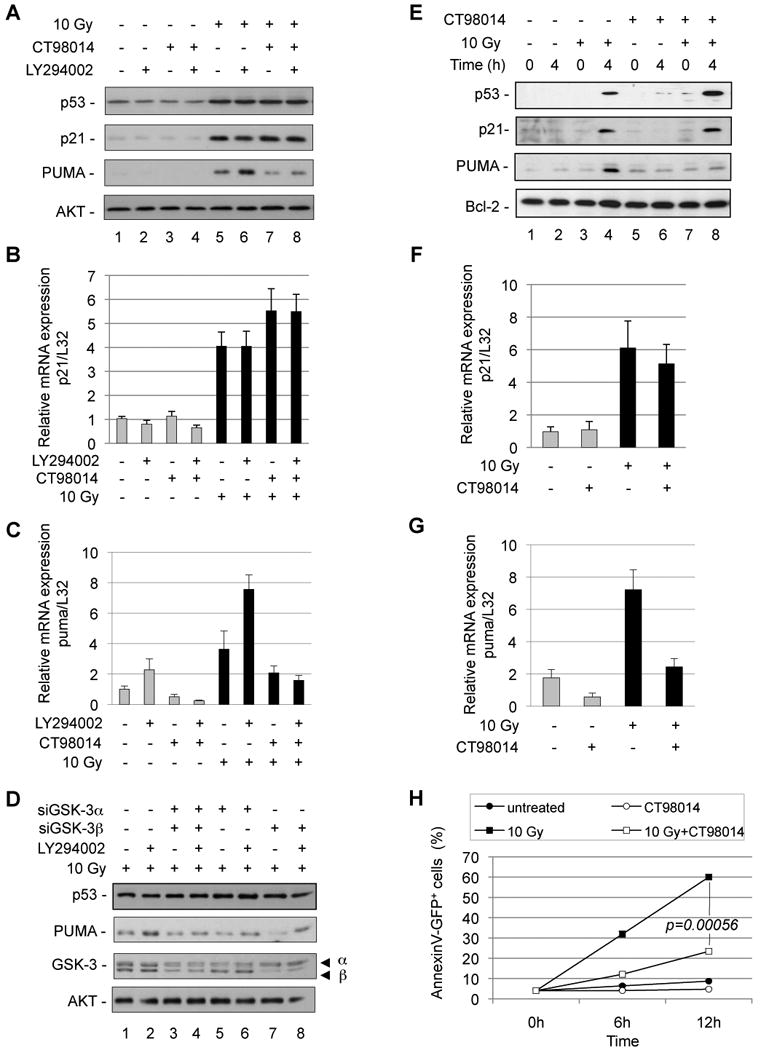
(A) U2OS cells were treated as indicated with LY294002 (10 µM) in the presence or the absence of the GSK3 inhibitor CT98014 (0.75 µM) for 1 h and irradiated (10 Gy). Six hours later, cell lysates were separated on SDS-PAGE and analyzed for p53, p21, PUMA expression and AKT for normalization using specific antibodies. (B) U2OS cells were treated with LY294002 (10 µM) in the presence or absence of GSK-3 inhibitor CT98014 (0.75 µM) for 1 h and subjected to ©-irradiation (10 Gy, black bars). After 3 h, p21 mRNA expression was analyzed and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements. (C) U2OS cells were treated as in B and puma mRNA expression was analyzed and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements. (D) U2OS cells were transfected with a control siRNA, or siRNA knocking down GSK-3〈, ® or both. Fourty-eight hours later, transfected cells were treated as indicated with LY294002 (10 µM) and subjected to irradiation (10 Gy). Six hours after irradiation, cell lysates were analyzed by SDS-PAGE and probed for p53, PUMA, GSK-3 expression and AKT for normalization using specific antibodies. (E) BAF3 cells were maintained in low growth factor (0.05 µg/L IL-3) media for 12 h. Cells were then treated with the GSK3 inhibitor CT98014 (0.75 µM) for 1h and irradiated (10 Gy). Four hours after irradiation, total cell extracts were analyzed for p53, p21, PUMA expression and Bcl-2 for normalization. (F) BAF3 cells were maintained in low IL-3 (0.05 µg/L) for 12 h. Cells were then treated with the GSK-3 inhibitor CT98014 (0.75 µM) for 1 h and subjected to ©-irradiation (10 Gy, black bars). After 2 h, p21 mRNA expression was analyzed and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements. (G) BAF3 cells were treated as in F and PUMA mRNA expression was analyzed and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements. (H) BAF3 cells were treated as in E and stained with Annexin V-GFP and PI to monitor cell death at the indicated time points. Error bars represent standard deviation of triplicate measurements.
To confirm these data, we knocked down GSK-3α, GSK-3β, or both by siRNA or shRNA in U2OS cells. Combined knock-down of GSK-3α and GSK-3β by siRNA substantially diminished PUMA protein (Fig. 1D, compare lanes 2 and 4) and mRNA levels (Fig. S2A), and similar results were obtaind by shRNA-mediated knockdown of GSK-3α and GSK-3β (Fig. S2B). Knock-down of each GSK-3α or GSK-3β expression partly reduced PUMA protein expression (Fig. 1D, lanes 2, 6, 8), indicating that both GSK-3 isoforms contribute to PUMA induction.
We next investigated the regulation of PUMA in growth factor-dependent cells. IL-3 dependent BAF3 cells, which exhibit intact p53 signaling (Canman et al., 1995), were pre-incubated with PI3K inhibitor, GSK-3 inhibitor, or both and subjected to γ-radiation. While PI3K inhibition alone resulted in some PUMA mRNA induction, expression of PUMA mRNA and protein were strongly induced upon combined exposure to γ-radiation and PI3K inhibition (Fig. S3A and S3C). Expression of p21 mRNA and protein did not depend on PI3K or GSK-3 (Fig. S3B and S3C). Accordingly, PI3K inhibition and γ-radiation cooperated to induce apoptosis (Fig. S3D).
In a different approach, IL-3 dependent BAF3 and FL5.12 cells were pre-incubated with a reduced concentration of IL-3, in order to attenuate PI3K signaling and de-repress GSK-3 activity (Frame and Cohen, 2001; Maurer et al., 2006). This treatment, by itself, did not result in the induction of apoptosis. When BAF3 cells were exposed to γ-radiation, we observed an induction of p53, p21 and PUMA (Fig. 1E, lane 4). Inhibition of GSK-3 specifically abrogated PUMA, but not p53 and p21 induction in BAF3 (Fig. 1E, lane 8, Fig. 1F and 1G) and FL5.12 cells (Fig. S4A and S4B). Again, BAX expression did not depend on GSK-3 (Fig. S4C). Consistent with the loss of PUMA induction upon GSK-3 inhibition, the occurrence of apoptosis by γ-radiation was substantially decreased in BAF3 (Fig. 1H) and FL5.12 cells (Fig. S4D).
To extend our observations to primary cells, we investigated the role of GSK-3 in IL-2 dependent activated lymphocytes. The concentration of IL-2 was reduced in order to increase GSK-3 activity and cells were subjected to γ-radiation. While this treatment induced p53 and p21 independently of GSK-3, the pharmacological inhibition of GSK-3 again selectively repressed PUMA protein and mRNA induction (Fig S5A, lanes 3 and 4, and Fig. S5B). Consistent with PI3K and GSK-3 being regulated by growth factor availability, the induction of Puma mRNA upon γ-radiation was repressed by IL-2 in a dose dependent manner, and Puma mRNA expression (Fig. S5C) and apoptosis (Fig. S5D) was prevented by inhibition of GSK-3.
We did not, however, observe a major effect of GSK-3 on the mRNA induction of Noxa and Bax in these cells (Fig. S5E and S5F). Thus, GSK-3 activity appears to specifically promote the expression of PUMA, but not other pro-apoptotic p53 target genes.
Induction of PUMA expression requires GSK-3 activity in vivo
As PUMA is required for lymphocyte apoptosis by DNA damage in vivo (Jeffers et al., 2003; Villunger et al., 2003), we tested the contribution of GSK-3 to DNA damage-mediated PUMA induction in mice. GSK-3 inhibitors CT98014 or CT99021, as described (Bain et al., 2007; Cohen and Goedert, 2004; Maurer et al., 2006; Ring et al., 2003), were injected C57BL/6 in mice, which were subjected to whole body γ-irradiation. Splenocytes from mice which had received the pharmacological GSK-3 inhibitors CT98014 or CT99021 exhibited a reduced induction of Puma mRNA (Fig. 2A) and protein (Fig. S6A). Likewise, thymocytes from mice, which had received CT98014 expressed significantly reduced puma mRNA upon γ-irradiation (Fig. S6B). Thus, pharmacological inhibition of GSK-3 modulates PUMA expression in vivo.
Figure 2. Inhibition of GSK-3 upon γ-irradiation represses induction of Puma in vivo and confers long-term clonogenic survival.
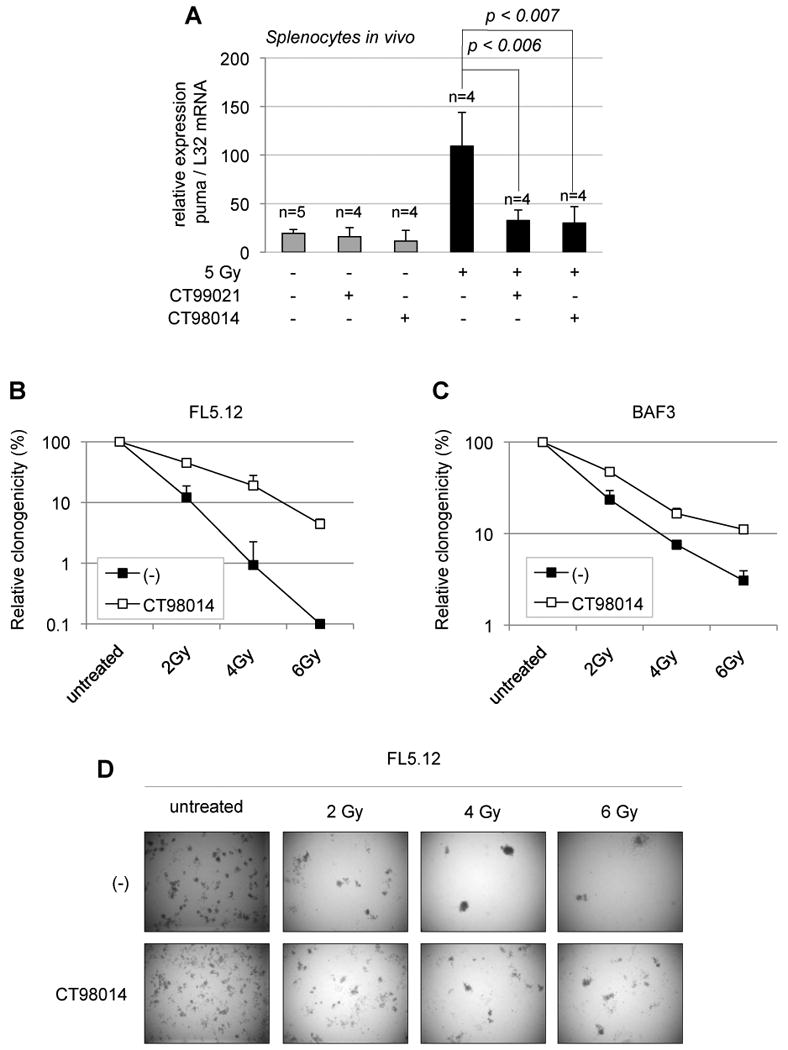
(A) C57BL/6 mice were injected with two different GSK-3 inhibitors, either CT98014 or CT99021 (30 mg/kg) or vehicle and subjected to whole-body irradiation (5 Gy) (black bars) or not (gray bars). Two and a half hours later, splenocytes were isolated and PUMA mRNA expression was analyzed by real-time PCR and normalized to L32 mRNA. (n= mouse number). Error bars represent standard deviation of data obtained from indicated number of mice, with each measurement done in triplicate. (B) FL5.12 cells were maintained on low IL-3 concentration (0.05 µg/L) for 12 h in order to activate GSK-3 at a concentration of 0.75×106/ml. Cells were then treated or not (-) with GSK-3 inhibitor CT98014 (0.75 µM) for 1 h and γ-irradiated (2, 4 or 6 Gy) or left untreated. Eight hours after, cells were plated in methylcellulose containing IL-3 (1 µg/L). Clonogenicity was calculated relative to the colony number in the untreated condition, with or without GSK-3 inhibitor, respectively, as a percentage. Error bars represent standard deviation of triplicate measurements. (C) BAF3 cells were treated as in B and relative clonogenicity was determined. Error bars represent standard deviation of triplicate measurements. (D) FL5.12 cells were treated as in B. A representative picture is shown for each condition.
Inhibition of GSK-3 confers long-term survival to irradiated cells
To assess whether inhibition of GSK-3 promotes sustained cell survival after γ-irradiation, we performed clonogenic assays in methylcellulose with growth factor-dependent FL5.12 and BAF3 cells. As described above, cells were initially maintained for 12 h in reduced IL-3 in order to decrease PI3K activity (de-repressing GSK-3 activity), and then were either left untreated, or subjected to doses of γ-irradiation of 2, 4 and 6 Gy, either in presence or absence of the GSK-3 inhibitor. Eight hours after irradiation, cells were plated in methylcellulose containing IL-3. After 7 days, FL5.12 and BAF3 cells, which had been irradiated in presence of the GSK-3 inhibitor, showed substantially elevated relative clonogenicity, demonstrating that inhibition of GSK-3 during the period of γ-irradiation promotes increased long term cell survival (Fig. 2B, 2C and 2D).
p53-dependent, DNA damage-induced apoptosis requires GSK-3 and acetylation of p53 on K120
To investigate whether the role of GSK-3 in damage-induced cell death depends on p53, we subjected activated lymphocytes generated from p53+/+ and p53-/- mice to γ-irradiation, after maintaining them for 12 h in reduced growth factor. In order to achieve DNA damage-induced cell death in absence of p53, a high radiation dose of 25 Gy was applied, in presence or absence of the GSK-3 inhibitor. After 12 h of irradiation, about 50% of p53+/+ lymphocytes were apoptotic, which was, despite the high radiation dose, partially suppressed by inhibition of GSK-3, while p53-/- cells were all viable (Fig. 3A). In p53-/- lymphocytes, a similar extent of cell death was reached only after 48 h (when p53+/+ cells were all dead). This was, however, barely changed by the presence of the GSK-3 inhibitor (Fig. 3B). Thus, the promotion of DNA damage-induced cell death by GSK-3 in activated lymphocytes is to a large part dependent on p53.
Figure 3. DNA-damage-induced cell death depends on p53 and GSK-3.
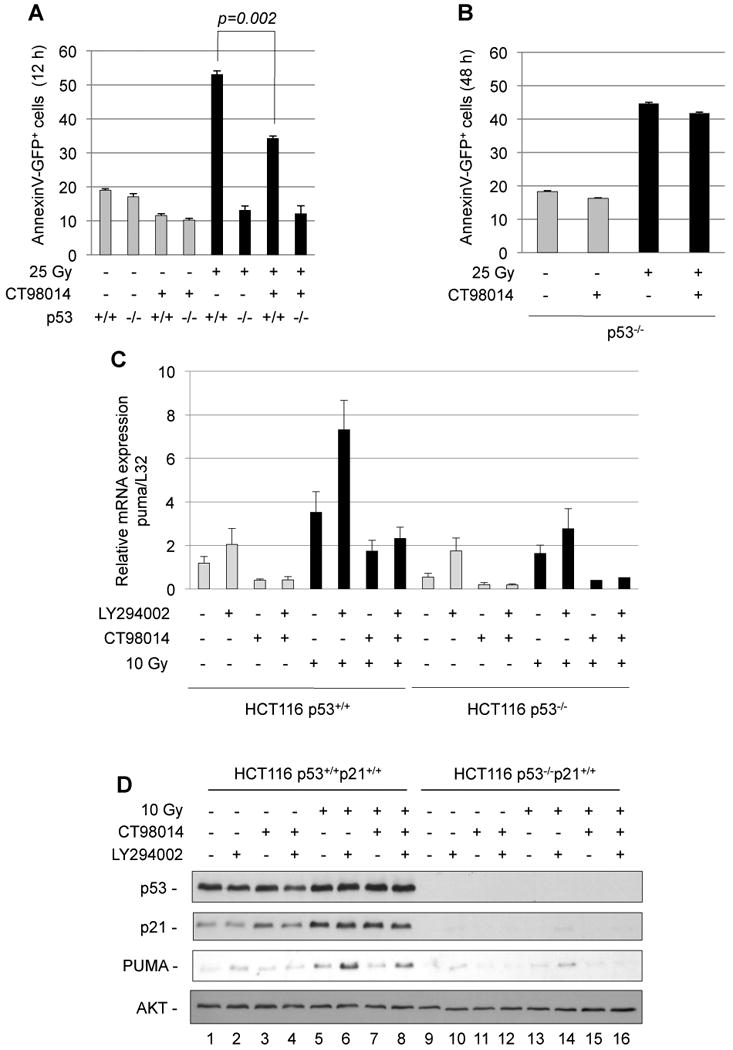
(A) IL-2-dependent lymphocytes generated from p53+/+ or p53-/- mice were maintained in low IL-2 (5 U/ml) for 12 h in order to activate GSK-3. After pre-treatment with the GSK-3 inhibitor (CT98014, 1.5 µM, 1 h) as indicated, cells were ©-irradiated (25 Gy). After 12 h, apoptosis was analyzed by flow cytometry analysis of AnnexinV+ cells. Error bars represent standard deviation of triplicate measurements. (B) Assessment of cell death of IL-2-dependent p53-/- lymphocytes at 48 h. Error bars represent standard deviation of triplicate measurements. (C) HCT116 p53+/+ and HCT116 p53-/- cells were treated with the PI3K inhibitor LY294002 (10 µM) the GSK-3 inhibitor CT98014 (1.5 µM) for 1 h as indicated and γ-irradiated (10 Gy). After 3 h, puma mRNA expression was analyzed by realtime RT-PCR and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements. (D) HCT116 p53+/+ and HCT116 p53-/- cells were treated as in C and 5 h after ©-radiation, total cell extracts were analyzed for p53, p21, PUMA expression and AKT for normalization.
We next addressed the requirement of p53 for the regulation of PUMA, employing HCT116p53+/+ or HCT116p53-/- cells. While PI3K inhibition alone resulted in low induction of PUMA mRNA and protein in both HCT116 p53+/+ and HCT116 p53-/- cells (which was not prevented by kockdown of the p53-relative p73 [Fig. S7A and B]), Puma mRNA and protein were strongly induced after a combination of PI3K inhibition and γ-irradiation in HCT116 cells retaining p53, which was reduced by inhibition of GSK-3 (Fig. 3C, 3D and Fig. S7C).
Foxo3a has recently been reported to be a transcriptional inducer of PUMA upon growth factor withdrawal (You et al., 2006), and we interrogated its contribution to PUMA induction upon DNA damage, combined with attenuated PI3K signaling. Using activated lymphocytes from wild-type (Foxo3a+/+) and Foxo3a-deficient (Foxo3atrap/trap) lymphocytes (Lin et al., 2004), observed similar PUMA and apoptosis induction by DNA damage and maintenance in low growth factor (Fig. S7D, E and F). This suggests that GSK-3, rather than Foxo3a, determines PUMA induction and apoptosis upon low PI3K signaling.
Recently, the importance of p53 acetylation at lysine 120 (K120) by the acetyltransferase Tip60 was demonstrated for the pro-apoptotic function of p53 (Sykes et al., 2006; Tang et al., 2006). We investigated the requirement of the acetylation of K120 of p53 for the cooperation of inhibition of PI3K signaling and DNA damage to induce PUMA. HCT116 p53-/- cells, infected with retrovirus encoding either p53wtERtam or K120-acetylation defective p53K120RERtam were treated with etoposide and 4-hydroxytamoxifen (4-OHT) in presence or absence of LY294002. Consistent with the observations described before, high induction of PUMA was observed in cells infected with p53ERtam after addition of etoposide and 4-OHT only when PI3K was inhibited. This effect was considerably diminished in cells expressing p53K120RERtam, while only a slight decrease of p21 protein expression was observed (Fig. 4A, lanes 8 and 16).
Figure 4. PUMA expression upon PI3K inhibition and DNA damage depends on p53 acetylation on K120.
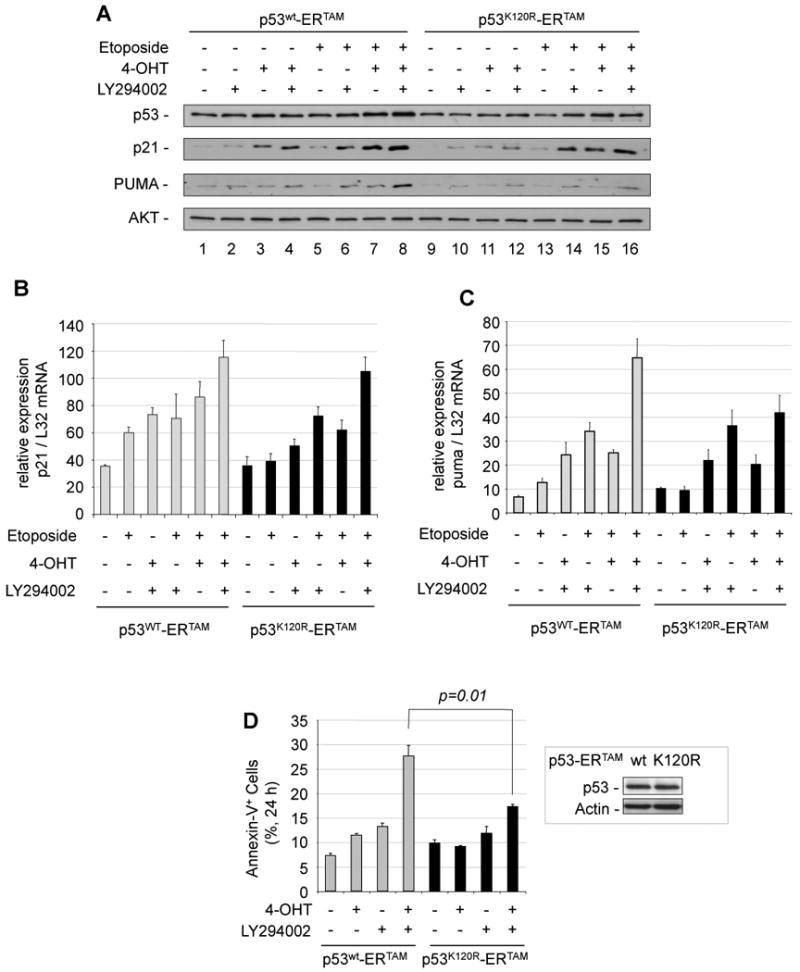
(A) HCT116 p53-/- cells overexpressing p53wtERtam or p53K120RERtam were treated with LY294002 (10 µM) and/or Etoposide (100 µM) with or without 4-OHT (100 nM) for 6 h. Total cell extracts were analyzed for p53, p21, PUMA expression and AKT for normalization. (B) HCT116 p53-/- cells overexpressing p53wtERtam or p53K120RERtam were treated as in A. Two hours after ©-radiation, p21 mRNA expression was analyzed and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements. (C) HCT116 p53-/- cells overexpressing p53wtERtam or p53K120RERtam were treated as in B and puma mRNA expression was analyzed and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements. (D) H1299 p53null cells overexpressing p53wtERtam (gray bars) or p53K120RERtam (black bars) were treated with LY294002 (10 µM) and/or 4-OHT (100 nM) for 24 h and AnnexinV+ cells were analyzed by flow cytometry. Error bars represent standard deviation of triplicate measurements. Overexpressed p53wtERtam and p53K120RERtam proteins are shown in the inset.
Likewise, Puma mRNA induction by the same treatment was decreased in cells expressing the K120R mutant, while p21 mRNA induction was similar (Fig. 4B and 4C). These data suggest that K120 acetylation of p53 contributes to PUMA induction by PI3K inhibition and DNA damage.
Consistently, in p53-null H1299 cells expressing p53wtERtam, the inhibition of the PI3K pathway (activating GSK-3) enhanced cell death induced by 4-OHT-mediated p53wtERtam activation. In contrast, cells expressing the K120-acetylation-deficient mutant p53K120R and treated with 4-OHT and PI3K inhibitor exhibited diminished apoptosis (Fig. 4D).
Together, these results show that full PUMA induction after DNA damage depends on GSK-3 and p53 K120-acetylation.
GSK-3 phosphorylates Tip60 on S86 in vitro and in vivo
Recent reports have shown that p53 acetylation on K120 is mediated by the lysine-acetyltransferase Tip60 (Sykes et al., 2006; Tang et al., 2006). As the presence of K120 of p53 was required to induce PUMA expression after PI3K inhibition and DNA damage, we investigated the possibility that GSK-3 and the p53 K120-acetyltransferase Tip60 are part of the same pathway.
We therefore asked whether inhibition of PI3K creates a pro-apoptotic signal, acting on K120 of p53, by way of an activating phosphorylation of Tip60 by GSK-3. In support of this idea, Tip60 contains an evolutionary conserved GSK-3 phosphorylation site (S86XXXS90, Fig. 5A).
Figure 5. Tip60 is phosphorylated by GSK-3 on Serine 86 in vitro and in vivo.
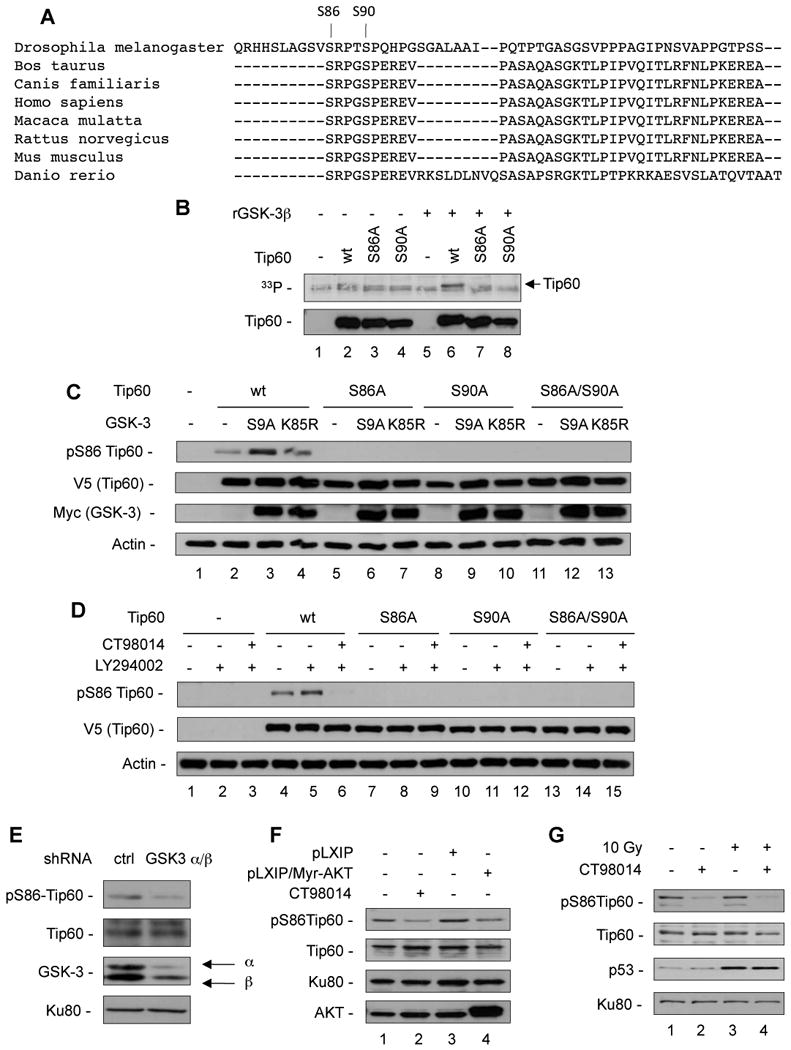
(A) Alignment of the potential Tip60 GSK-3 target site in different species using the multiple sequence alignment MAFFT. The S86 and S90 residues are indicated. (B) V5/His-tagged Tip60wt, Tip60S86A and Tip60S90A were expressed and purified from 293T cells and incubated for 20 min in the presence of 33P©-ATP (12.5 µCi) with or without recombinant GSK3® (0.1 µg/reaction). After blotting, the membrane was exposed on a film for 12 h, and subsequently probed with the anti-Tip60 antibody. (C) 293T cells were transiently transfected with V5-tagged Tip60wt and mutants along with myc-tagged constitutively active GSK-3S9A or kinase-inactive GSK-3K85R. Whole cell lysates were analyzed for pS86Tip60, V5-Tip60, myc-GSK-3 and Actin. (D) BAF3 cells were transiently transfected with empty vector, V5-tagged Tip60wt and mutants. Twenty-four hours later, transfected cells were treated with LY294002 (10 µM) with or without CT98014 (0.75 µM) for 4 h. Total cell extracts were analyzed by western blot using anti-pS86Tip60, anti-V5 and anti-Actin antibodies. (E) U2OS cells were infected with either control lentivirus or lentiviruses encoding shRNA targeting GSK-3〈 and GSK-3®. After 72 h, cells were subjected to nuclear fractionation and nuclear extracts were analyzed for phosphoS86-Tip60, Tip60, GSK-3 and Ku80 for normalization. (F) FL5.12wt cells and Fl5.12 cell infected with a control retrovirus (pLXIP) or retrovirus encoding myristoylated AKT (Myr-AKT) were maintained in low IL-3 concentrations (0.05 µg/L) for 12 h to activate GSK-3, FL5.12wt were treated with CT98014 as indicated, and nuclear extracts were analyzed for pS86-Tip60, Tip60, Ku80 and AKT. (G) BAF3 cells were maintained in low IL-3 (0.05 µg/L) for 12 h and subjected to ©-irradiation (10 Gy). Four hours later, nuclear extracts were analyzed for phosphoS86-Tip60, Tip60, p53 and Ku80 for normalization.
We investigated the phosphorylation of Tip60 by GSK-3 by an in vitro kinase assay. In order to phosphorylate its substrates, GSK-3 requires a priming phosphorylation, located four amino acids C-terminal of the serine to be phosphorylated by GSK-3 (Cohen and Frame, 2001). Tip60wt, the GSK-3 phosphorylation mutant Tip60S86A or the priming phosphorylation mutant Tip60S90A were subjected to a kinase assay with recombinant GSK-3β as described before (Maurer et al., 2006). While wild-type Tip60 was phosphorylated by recombinant GSK-3β, phosphorylation was absent in the GSK-3 phosphorylation mutant Tip60S86A and in the priming phosphorylation mutant Tip60S90A (Fig. 5B). In order to investigate S86-phosphorylation of Tip60 in cells, we generated a phospho-S86Tip60-specific antibody, which specifically recognized phosphoS86Tip60 (Fig. S8A and S8B).
We expressed wild-type Tip60, as well as the mutants Tip60S86A, Tip60S90A and Tip60S86A/S90A along with constitutively active GSK-3β (GSK-3βS9A) or kinase-inactive GSK-3β (GSK-3βK85R) in 293T cells. The presence of GSK-3βS9A, but not GSK-3βK85R, resulted in the phosphorylation of S86 of wild-type Tip60, while no signal for S86-phosphorylation was detected with any of the mutants (Fig. 5C, lanes 3, 6, 9, and 12).
To explore if Tip60 phosphorylation depended on PI3K signaling, we expressed Tip60wt, as well as Tip60S86A, Tip60S90A and Tip60S86A/S90A in BAF3 cells and incubated them with the PI3K inhibitor LY294002 (increasing GSK-3 activity) with or without the GSK-3 inhibitor (CT98014). Tip60wt was phosphorylated on S86 at a basal level, while PI3K inhibition further increased S86 phosphorylation of Tip60. Inhibition of GSK-3 completely abolished LY294002-induced S86 phosphorylation of Tip60. Again, none of the mutants were phosphorylated upon GSK-3 activation (Fig. 5D).
These data not only indicate that GSK-3 phosphorylates Tip60 on S86, but also that phosphorylation of Tip60 by GSK-3 requires the priming phosphorylation of S90, as demonstrated before for other GSK-3 substrates (Cohen and Frame, 2001).
We next addressed the phosphorylation of endogenous Tip60 in nuclear extracts of HCT116 cells. Inhibition of PI3K enhanced the phosphorylation of endogenous Tip60 at S86, which was completely lost upon inhibition of GSK-3 (Fig. S8C).
Importantly, S86-phosphorylation of endogenous Tip60 was largely reduced in U2OS cells which had been transfected with siRNA specific for GSK-3α and β, but not in cells transfected with a siRNA control, confirming the data obtained by pharmacological inhibition of GSK-3 (Fig. 5E).
PI3K signaling leads to activation of AKT, which suppresses GSK-3 activity by inhibitory phosphorylation (Cross et al., 1995). We therefore investigated the effect of AKT on Tip60 phosphorylation by in FL5.12 cells expressing constitutively active (myristoylated) AKT (Maurer et al., 2006), which had been maintained in reduced growth factor permitting GSK-3 activity. Consistent with a suppression of GSK-3 activity by AKT (Cross et al., 1995), we found that expression of myrAKT in these cells prevented S86-phosphorylation of endogenous Tip60 (Fig. 5F) and that myrAKT prevented induction of PUMA mRNA (Fig. S8D), while having no suppressive effect on p21 expression (Fig. S8E).
Consistently, in activated lymphocytes, maintained in decreasing concentrations of IL-2, the extent of Tip60S86 phosphorylation depended on growth factor availability and was correlated with the extent of DNA damage-induced apoptosis after 24 h (Fig. S9A and B).
To validate our results in vivo, we administered the GSK-3 inhibitors CT98014 and CT99021 to C57BL/6 mice and analyzed Tip60 S86-phosphorylation in splenocytes. The Tip60 α and β isoforms (Ran and Pereira-Smith, 2000) were expressed in murine splenocytes, and were both phosphorylated on S86. Tip60 S86-phosphorylation was strongly reduced 90 min after injection of the GSK-3 inhibitos, corroborating our results in vivo (Fig. S9C).
We observed that Tip60S86 phosphorylation was independent of DNA damage induced by γ-radiation (Fig. 5G, Fig. S8B). Also, it was not influenced by cdc2/CDK1, which had been reported to phosphorylate S90 of Tip60 in vitro, representing the priming site for GSK-3 (Lemercier et al., 2003) (Fig. S10A and B).
Phosphorylation of Tip60 on S86 by GSK-3 is required for PUMA induction after PI3K inhibition and DNA damage
We next asked whether S86-phosphorylation of Tip60 is required for the induction of PUMA by DNA damage. We stably knocked down endogenous Tip60 in U2OS cells by lentiviral shRNA, while re-introducing retrovirus encoding shRNA-resistant wild-type Tip60 or the Tip60S86A mutant, respectively.
Upon γ-radiation and inhibition of PI3K, PUMA was strongly induced in cells where Tip60wt was re-expressed. U2OS cells expressing Tip60S86A, however, displayed largely reduced PUMA induction (Fig. 6A, lanes 4 and 6).
Figure 6. GSK-3-phosphorylation of Tip60 on Serine 86 is required for optimal PUMA induction.
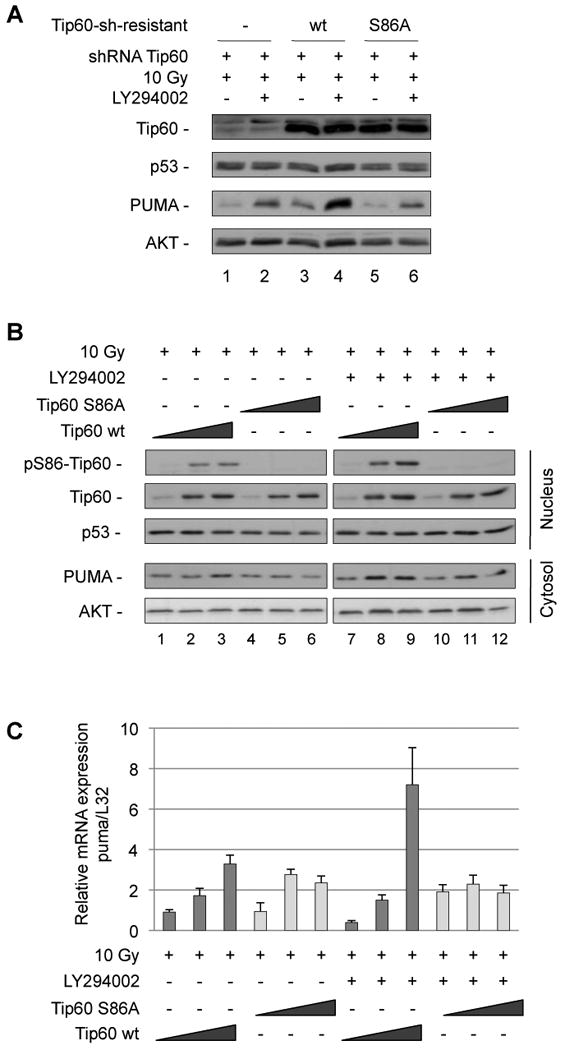
(A) U2OS cells were infected with virus encoding sh-resistant Tip60wt, Tip60S86A or Tip60S90A mutants along with lentivirus encoding control shRNA or shRNA to knockdown endogenous Tip60. Infected cells were incubated with the PI3K inhibitor LY294002 (10 µM) and γ-irradiated (10 Gy). Four hours later, cells were lysed and analyzed for Tip60, p53, PUMA and actin expression. (B) HCT116p53+/+ cells were transiently transfected with increasing doses (0.1 µg, 0.5 µg and 1 µg) of plasmid DNA encoding Tip60wt or Tip60S86A mutant. Twenty-four hours later, cells were treated with LY294002 (10 µM) and irradiated (10 Gy). Five hours later, nuclear and cytosolic extracts were analyzed for pS86Tip60, Tip60, p53 and PUMA and AKT, respectively. (C) HCT116p53+/+ cells were transfected and treated as in B. Three hours after irradiation, expression of puma mRNA was analyzed by real-time RT-PCR and normalized to L32 mRNA. Error bars represent standard deviation of triplicate measurements.
In a different approach, increasing amounts of Tip60wt or Tip60S86A were transiently transfected into HCT116 cells before treatment with γ-radiation and PI3K inhibitor. PUMA protein and mRNA induction was higher in cells transfected with wildtype Tip60, compared to those expressing Tip60S86A (Fig. 6B, compare lanes 8 and 9 to lanes 11 and 12 and Fig. 6C).
This demonstrates the importance of Tip60S86-phosphorylation for the induction of PUMA by p53, when DNA damage is combined with loss of PI3K signaling.
Acetylation of p53 and H4 acetylation at the puma promoter depend on Tip60 S86-phosphorylation and GSK-3
Two separate functions of Tip60 were previously described to play a role for DNA-damage mediated pro-apoptotic signaling: Tip60 was shown to directly acetylate p53 on K120 (Sykes et al., 2006; Tang et al., 2006), and to mediate acetylation of histone H4 (Ikura et al., 2000). H4 acetylation at the puma promoter was shown to depend on p53K120 acetylation and involved p53-dependent recruitment of Tip60 to the puma promoter (Tang et al., 2006).
Therefore, we investigated how Tip60 phosphorylation affected both the p53 and the histone acetyltransferase activities of Tip60.
We first explored whether phosphorylation of Tip60 on S86 influenced the p53K120 acetyltransferase activity of Tip60. We observed that p53 was acetylated at K120 only upon co-expression of Tip60wt, but much less by the phosphorylation-defective Tip60S86A mutant (Fig. 7A). This demonstrates that the absence of S86 phosphorylation substantially diminishes the p53K120-acetyltransferase activity of Tip60.
Figure 7. Serine-86 phosphorylation of Tip60 modulates p53K120 and H4 acetyltransferase activity.
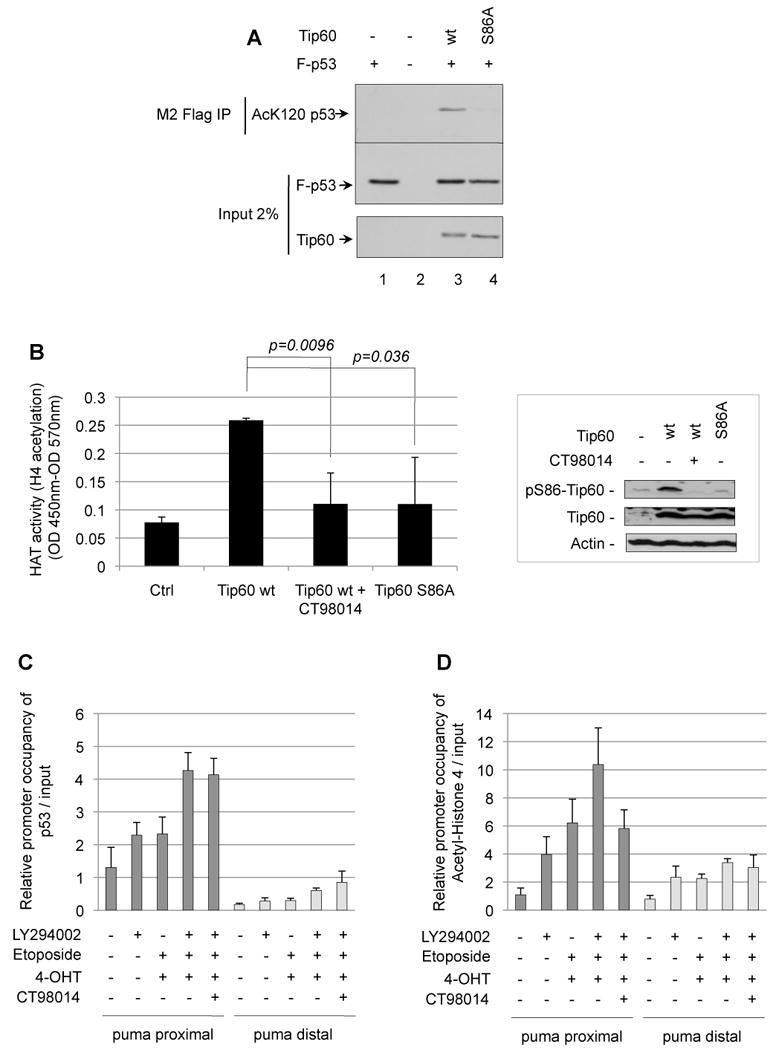
(A) H1299 cells were transfected with plasmid DNA expressing Flag-p53 and Tip60wt or the S86A phosphorylation mutant. Cells were treated with HDAC inhibitors TSA (1 ⎧M) and nicotinamide (5 mM) 4 h before harvesting. To immunoprecipitate p53 protein, cells were lysed and total cell extracts were immunoprecipitated with M2 Flag beads. Total cell extracts and the M2 immunoprecipitation were analyzed by Western blot using the anti-Tip60 (CLHF), anti-AcK120-p53, and anti-p53 (DO-1) antibodies, respectively. (B) Control vector (Ctrl) or V5/His-tagged Tip60wt, Tip60S86A and Tip60S90A were expressed, and purified from 293T cells and subjected to in vitro a HAT assay using a H4 peptide as a substrate. Right panel shows expression and S86-phosphorylation of Tip60. Error bars represent standard deviation of triplicate measurements. (C and D) HCT116p53-/- cells expressing p53wtERtam were treated with a combination of 4-OHT (100 nM) and etoposide (100 µM) with or without LY294002 (10 µM) in the presence or absence of GSK-3 inhibitor CT98014 (1.5 µM) for 5 h and subjected to ChIP using anti-p53 or anti-AcH4. Immunoprecipitated material and input were subjected to quantitative real-time PCR using primers annealing in the puma promoter proximal or distal from the p53 binding site. Promoter occupancy by either p53 (C) or Ac-H4 (D) was determined relative to input. Error bars represent standard deviation of triplicate measurements.
We next analyzed the histone acetyltransferase (HAT) activity of Tip60. Tip60wt, expressed and purified from 293T cells in absence of the GSK-3 inhibitor, mediated H4 acetylation in a HAT ELISA, using a H4 peptide as a substrate. Tip60 expressed in 293T upon inhibition of GSK-3 displayed compromised Tip60 HAT activity, as did the S86A mutant (Fig. 7B). Thus, S86-phosphorylation, mediated by GSK-3, modulates Tip60 HAT activity.
We did not find, however, that the interaction of Tip60 with p53 was dependent on S86 phosphorylation of Tip60 (Fig. S11).
Finally, we questioned whether PI3K and GSK-3 influence p53 binding to the puma promoter, and histone H4 acetylation at the puma promoter, respectively, by Chromatin-Immunoprecipitation (ChIP) followed by quantitative real-time PCR.
HCT116 p53-/- cells expressing p53wtERtam were treated with 4-OHT/etoposide, PI3K inhibitor, or combined, and subjected to ChIP employing antibodies specific for p53 and AcH4 and primers, annealing proximal or distal from the p53-binding site in the puma promoter (Wang et al., 2007). We found that inhibition of PI3K, along with induction of p53 by 4-OHT/etoposide, increased the association of p53 with the puma promoter, as assessed by quantitative real-time PCR using the proximal primers, but not to a region distal from the p53 binding site. The binding of p53 to the puma promoter was, however, not reduced by inhibition of GSK-3 (Fig. 7C).
The puma promoter-specific H4 acetylation proximal, but not distal to the p53 binding site was also promoted by the combination of PI3K inhibition and induction of p53 by 4-OHT/etoposide. Inhibition of GSK-3, however, reduced H4 acetylation at the puma promoter, which is consistent with a suppression of Tip60-histone acetyltransferase activity upon GSK-3 inhibition (Fig. 7D). Thus, by modulating Tip60 phosphorylation, GSK-3 regulates H4 acetylation at the puma promoter, while it did not influence p53 binding to the puma promoter.
Discussion
The data presented here show that GSK-3 determines p53-mediated PUMA expression and apoptosis. We show that the underlying mechanism is the phosphorylation of Tip60 on S86 by GSK-3, which is enhancing the acetyltransferase activity of Tip60.
Tip60 was shown to promote PUMA induction and apoptosis by acetylating p53 on K120 (Sykes et al., 2006; Tang et al., 2006). Moreover, H4 acetylation at the Puma promoter depended on p53K120 acetylation and was associated with the recruitment of Tip60 to the Puma promoter (Tang et al., 2006).
Our results show that S86 phosphorylation of Tip60 influences both the p53K120 acetyltransferase activity as well as the H4 HAT activity of Tip60. We further show that H4 acetylation at the puma promoter, proximal to the p53 binding site, was reduced by inhibition of GSK-3. This is consistent with the reduced HAT activity of Tip60 upon inhibition of GSK-3 we find, and with an earlier study showing that Tip60 histone acetylase activity was attenuated due to the absence of the phosphorylation site S86/S90 (Lemercier et al., 2003). The observation that GSK-3 dependent H4 acetylation occurred proximal, but not distal to the p53 binding site, indicates that H4 acetylation was dependent on p53.
Together, while not ruling out additional ways of PI3K to influence p53 signaling, our data show that the PI3K signaling pathway, via GSK-3 and Tip60, and the DNA damage pathway converge on p53-mediated transcriptional regulation of PUMA.
While GSK-3 facilitated p53-dependent PUMA induction and death, it was not required for p53-mediated p21 expression. Interestingly, we observed mostly in cytokine-dependent cells, that in contrast to the repression of PUMA induction, GSK-3 inhibition leads to an increase of p53 and p21 expression upon DNA damage. Likewise, elevation of p21 mRNA was also observed in cells expressing constitutively active AKT, which inactivates GSK-3. This may be explained by GSK-3-mediated MDM2 phosphorylation, which has been shown to contribute to p53 degradation (Kulikov et al., 2005). Thus, the state of GSK-3 activation contributes to the choice as to whether p53 induces cell cycle arrest or apoptosis.
Our data suggest that the activity of GSK-3 increases PUMA induction, but not the expression of BAX or NOXA, both pro-apoptotic p53 target genes (Miyashita and Reed, 1995; Oda et al., 2000). Thus, GSK-3 exhibits a selective enhancement of PUMA as a pro-apoptotic p53 target. It is unclear how this specificity is mediated, but it is possible that p53 and pS86Tip60 interact with a factor specific for the PUMA promoter.
An earlier report has shown that IL-3 dependent cells, when treated with γ-radiation, undergo cell cycle arrest in presence of IL-3 and rapid apoptosis upon deprivation of the growth factor (Canman et al., 1995). As IL-3 regulates AKT and GSK-3 signaling (Maurer et al., 2006), our results suggest that this effect is mediated, at least in part, by GSK-3.
In contrast, Foxo3a which also has been shown to be a PUMA regulator induced by cytokine-withdrawal (You et al., 2006) did not influence PUMA expression induced by DNA damage and low growth factor, indicating that PUMA, under these conditions, is controlled in a GSK-3-dependent, but Foxo3a-independent manner.
We observed a low PUMA induction upon PI3K inhibition or growth factor reduction alone, which also was GSK-3 dependent. As p53 is not stabilized in the absence of DNA damage, this observation seems incompatible with a GSK-3-promoted mechanism involving p53K120 acetylation to induce PUMA.
There is, however, evidence that p53 plays a role for apoptosis by growth factor withdrawal, independent of DNA damage. A recent study, exploring PUMA induction in IL-3 dependent and primary hematopoietic progenitor cells, respectively, demonstrated that growth factor withdrawal-induced up-regulation of PUMA occurred in the absence of detectable p53 stabilization, but not in p53-/- cells, therefore being dependent on p53 (Jabbour et al., 2010). This confirms an earlier study, reporting that p53-/- bone marrow cells show enhanced viability and increased colony formation in conditions of limited growth factor (Lotem and Sachs, 1993). Likewise, PUMA protein induction was shown to depend on p53 in activated lymphocytes deprived from IL-2 (Zhao et al., 2008). Interestingly, this study also showed that only PUMA, but not BAX or p21 was p53-dependently induced by growth factor withdrawal, which is consistent with our data. Thus, the GSK-3 dependent induction of PUMA upon growth factor withdrawal or PI3K inhibition we have observed, may be mediated by Tip60-dependend K120 acetylation of low levels of p53 (possibly a consequence of stress-induced, moderate p53 activation due to in vitro-cell culture conditions), which contribute to PUMA induction. Indeed, by Western blotting, we observed low but detectable levels of p53 in the absence of DNA damage in U2OS and FL5.12 cells. In addition, Tip60 S86-phosphorylation was independent of DNA damage, permitting its activation as a transcriptional co-activator for PUMA in the absence of DNA damage response signaling.
We did not find that GSK-3 substantially facilitates DNA damage-induced apoptosis of p53-deficient, IL-2 dependent activated lymphocytes, indicating that the promotion of apoptosis by GSK-3 required p53 in this system. We observed, however, a low induction of PUMA in HCT116p53-/- cells upon activation of GSK-3 by inhibition of PI3K, which obviously cannot be explained by GSK-3 promoting PUMA induction through p53. It is conceivable, however, that another transcription factor, acting PI3K/GSK-3 dependently in cooperation with Tip60 as well, accounts for this observation.
GSK-3 substrates require a priming phosphorylation four amino acids C-terminal of the GSK-3 target site (Frame and Cohen, 2001; Frame et al., 2001). CDK-1 was reported to phosphorylate the GSK-3 priming site S90 of Tip60 in vitro (Lemercier et al., 2003). Our data show however, that under the conditions we investigated, CDK-1 is unlikely to provide the priming phosphorylation of Tip60 on S90, since no change in GSK-3 mediated phosphorylation of Tip60-S86 was observed.
Axin, which interacts with GSK-3 in the Wnt signaling pathway, has been shown to play an important role for p53-mediated apoptosis in a signaling complex with Tip60 and HIPK2 (Li et al., 2009). It is an important question how Axin-dependent p53 signaling and the mechanism described here are interconnected.
We think that our results potentially have therapeutic implications: It has been shown that the p53-mediated pathological response to DNA damage, causing massive cell death of bone marrow cells and small intestine epithelium, has no tumor suppressive function (Christophorou et al., 2006). In contrast, after acute DNA damage, when rare oncogene activations occured, p53 was required for a protection from tumorigenesis (Christophorou et al., 2006). Thus, sparing bone marrow cells and small intestine epithelium from cell death during acute DNA damage (inflicted by chemo-or radiation therapy) by turning off p53-mediated cell death would potentially be beneficial for a patient, without the risk of increased malignancy. This could be achieved by transient pharmacological inhibition of GSK-3 during the period of the insult, as we demonstrate that application of GSK-3 inhibitors suppress PUMA induction in vivo. Subsequent discontinuation of pharmacological GSK-3 inhibition after the insult would reinstate p53-induced apoptosis and thus, response to potential oncogene activation. Consistent with this idea, a recent study showed that administration of pharmacological GSK-3 inhibitors to mice substantially increased survival after whole-body γ-irradiation (Thotala et al., 2010).
Experimental procedures
Cell fractionation, Immunoblotting and antibodies
Cells were subjected to nuclear fractionation as described previously (Charvet et al., 2006). Whole cells lysis was described previously (Maurer et al., 2006). Proteins (20-60µg) were separated by SDS-PAGE and transferred on nitrocellulose membranes. The membranes were then probed with anti-Puma (#3043, Prosci Incorporated, San Diego, Califiornia), anti-GSK-3 (sc-56913), anti-p53 (DO-1, sc-126), and anti-Tip60 (N-17, sc-5725) (all Santa Cruz, Santa Cruz, California), anti-p21 (#556430, BD Pharmingen, San Diego, California), anti-myc (#2276, 9B11), anti-Ku80 (#2753) and anti-Akt (#9272, all from Cell Signaling, Danvers, Massachusetts) anti-Bcl-2 (10C4, Zymed, San Francisco, California), anti-actin (A2026, Sigma, Taufkirchen, Germany), anti-V5 (#R960-25, Invitrogen, Carlsbad, California), and anti-FOXO3a (#07-702, Millipore, Billerica, Massachusetts) antibodies. Anti-AcK120p53 antibody was described (Tang et al., 2006). The anti-phosphoSerine86 Tip60 was generated using the immunogenic peptide CGGNGLPGpS86RPG (ProSci Incorporated, San Diego, California).
For co-immunoprecipitation of p53 and Tip60, HCT116p53-/- cells were transfected with Flag-p53 and CMV-Tip60 using Lipofectamine™ 2000 and lysed with the BC100 buffer and mild sonication according to the protocol described previously (Tang et al., 2006).
Colony assay
FL5.12 or BAF3 cells were maintained in low IL-3 medium (0.05 µg/L) for 12 h and subjected to different doses of γ-irradiation (2, 4 or 6 Gy) or left untreated, in presence or absence of CT98014 (0.75 µM). Eight hours after γ-irradiation, 103 cells were plated in methylcellulose-based media (Methocult™) containing recombinant IL-3 (1 µg/L). After 7 days, relative clonogenicity was calculated from the number of colonies for each condition relative to the number of colonies from the untreated condition (with or without GSK-3 inhibitor, respectively), defined as 100%.
In vivo acetylation of p53 at K120 by Tip60 phosphorylation mutants
H1299 cells were transiently transfected with Flag-p53 along with Tip60wt or Tip60S86A as indicated. Cells were treated with deacetylase inhibitors TSA (1 µM) and nicotinamide (5 mM) for the last 4 h of culture. To immunoprecipitate Flag-p53, total cell extracts were incubated with M2 flag beads (Sigma, Taufkirchen, Germany) overnight. Beads were washed five times with Flag lysis buffer (Tang et al., 2006) and the bound materials were eluted using Flag peptides (Sigma, Taufkirchen, Germany). Total cell extracts and the M2 immunoprecipitated materials were analyzed by western blot using the anti-Tip60, anti-AcK120-p53 and anti-p53 antibodies (Tang et al., 2006).
Apoptosis assays
Apoptosis was determined by staining with recombinant GFP-coupled Annexin V (25 µM, made in our laboratory) for 10 min and propidium iodide (5 µg/ml) and analyzed by flow cytometry.
HAT ELISA
The HAT ELISA was performed essentially according to the manufacturer's protocol (Millipore, Billerica, MA). Briefly, HIS-tagged Tip60wt (+/- CT98014 for the last 1 h before harvest) and Tip60S86A were expressed in 293T cells. Proteins were purified by Ni2+ affinity, and HAT activity was determined with a H4 peptide as a substrate by HAT ELISA.
Real-time PCR
Relative expression of PUMA was determined by real-time PCR in comparison to the L32 housekeeping gene as described before (Maurer et al., 2006). Primers sequences are in the supplemental table.
Statistics
Statistical significance was analyzed by 2-tailed Student's t test. Unless indicated otherwise, data represent the mean ± SD.
Supplementary Material
Highlights.
PUMA and apoptosis induction by p53 require GSK-3
GSK-3 phosphorylates the acetyltransferase Tip60 on Serine 86
Tip60S86A exhibits diminished acetylation of p53 at K120, H4, and expression of PUMA
Acetylation of H4 at the puma promoter depends on GSK-3 and p53
Acknowledgments
We thank Simon Wöhrle and Andreas Hecht for crucial advice with the Chromatin-IP, Karin Neubert and Monika Lutterbeck for technical assistance, Pat Fitzgerald, Martin Schuler, Marcel Deckert and Bert Vogelstein for reagents, Clemens Schmitt and Taro Fukao for p53-/- mice, Stanford L. Peng for Foxo3atrap/trap mice, Stéphane Bécart for support and Valentina Savich (ProSci Incorporated, San Diego) for the generation of the phosphoS86-specific Tip60 antibody. This study was supported by grants 107397 and 109199 from the Deutsche Krebshilfe and Ma1967/1-1 from the Deutsche Forschungsgemeinschaft to U.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Cote J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. Embo J. 1999;18:5108–5119. doi: 10.1093/emboj/18.18.5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Canman CE, Gilmer TM, Coutts SB, Kastan MB. Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes Dev. 1995;9:600–611. doi: 10.1101/gad.9.5.600. [DOI] [PubMed] [Google Scholar]
- Charvet C, Canonigo AJ, Becart S, Maurer U, Miletic AV, Swat W, Deckert M, Altman A. Vav1 promotes T cell cycle progression by linking TCR/CD28 costimulation to FOXO1 and p27kip1 expression. J Immunol. 2006;177:5024–5031. doi: 10.4049/jimmunol.177.8.5024. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature. 2006;443:214–217. doi: 10.1038/nature05077. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448:1063–1067. doi: 10.1038/nature06055. [DOI] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Jabbour AM, Daunt CP, Green BD, Vogel S, Gordon L, Lee RS, Silke N, Pearson RB, Vandenberg CJ, Kelly PN, et al. Myeloid progenitor cells lacking p53 exhibit delayed up-regulation of Puma and prolonged survival after cytokine deprivation. Blood. 2010;115:344–352. doi: 10.1182/blood-2009-07-230730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov R, Boehme KA, Blattner C. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol. 2005;25:7170–7180. doi: 10.1128/MCB.25.16.7170-7180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, 3rd, Abmayr SM, Washburn MP, Workman JL. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Legube G, Linares LK, Tyteca S, Caron C, Scheffner M, Chevillard-Briet M, Trouche D. Role of the histone acetyl transferase Tip60 in the p53 pathway. J Biol Chem. 2004;279:44825–44833. doi: 10.1074/jbc.M407478200. [DOI] [PubMed] [Google Scholar]
- Lemercier C, Legube G, Caron C, Louwagie M, Garin J, Trouche D, Khochbin S. Tip60 acetyltransferase activity is controlled by phosphorylation. J Biol Chem. 2003;278:4713–4718. doi: 10.1074/jbc.M211811200. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Li Q, Lin S, Wang X, Lian G, Lu Z, Guo H, Ruan K, Wang Y, Ye Z, Han J, Lin SC. Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nat Cell Biol. 2009;11:1128–1134. doi: 10.1038/ncb1927. [DOI] [PubMed] [Google Scholar]
- Lin L, Hron JD, Peng SL. Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21:203–213. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Lotem J, Sachs L. Hematopoietic cells from mice deficient in wild-type p53 are more resistant to induction of apoptosis by some agents. Blood. 1993;82:1092–1096. [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–1029. doi: 10.1038/cdd.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Ran Q, Pereira-Smith OM. Identification of an alternatively spliced form of the Tat interactive protein (Tip60), Tip60(beta) Gene. 2000;258:141–146. doi: 10.1016/s0378-1119(00)00410-8. [DOI] [PubMed] [Google Scholar]
- Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, Ma ST, Reeder JW, Samuels I, Slabiak T, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16:433–442. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thotala DK, Geng L, Dickey AK, Hallahan DE, Yazlovitskaya EM. A new class of molecular targeted radioprotectors: GSK-3beta inhibitors. Int J Radiat Oncol Biol Phys. 2010;76:557–565. doi: 10.1016/j.ijrobp.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyteca S, Vandromme M, Legube G, Chevillard-Briet M, Trouche D. Tip60 and p400 are both required for UV-induced apoptosis but play antagonistic roles in cell cycle progression. EMBO J. 2006;25:1680–1689. doi: 10.1038/sj.emboj.7601066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci U S A. 2007;104:4054–4059. doi: 10.1073/pnas.0700020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J Biol Chem. 2008;283:36344–36353. doi: 10.1074/jbc.M803580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.