

Abstract
Stereoelectronic effects modulate molecular structure, reactivity, and conformation. We find that the interaction between the ester and carboxyl moieties of aspirin has a previously unappreciated quantum mechanical character that arises from the delocalization of an electron pair (n) of a donor group into the antibonding orbital (π*) of an acceptor group. This interaction affects the physicochemical attributes of aspirin, and could have implications for its pharmacology.
INTRODUCTION
Stereoelectronic effects modulate molecular reactivity and conformation.1 For example, the nucleophilic attack at a carbonyl carbon is preferred along the Bürgi–Dunitz trajectory.2 This orientational preference stems from a stereoelectronic effect arising from the delocalization of an electron pair (n) of a nucleophile into an antibonding orbital (π*) of the carbonyl group. Our experimental and theoretical studies have revealed an analogous stereoelectronic effect that affects the conformation of peptides and proteins.3 In these systems conformational stability is provided by delocalization of a lone pair of a carbonyl oxygen into the π* orbital of a proximal carbonyl group. In such an interaction, termed the n→π* interaction, the van der Waals surfaces of the donor oxygen and acceptor carbon interpenetrate.3d,3k,3n,3o In addition to affecting the conformation of peptides and proteins, an n→π* interaction has also been implicated in the origin of life by favorably orchestrating the reactivity and stability of prebiotic molecules4 and in stabilizing conformations of (2S,4S)-4-hydroxyproline, β-alanine, and γ-aminobutyric acid.5 Herein, we report on the existence of an n→π* interaction in one of the oldest of drugs and discuss its implications for structure and reactivity.
The medicinal benefits of salicylic acid have been known since the time of Hippocrates. Its O-acetylated form, “aspirin”, was first synthesized in 18536 and marketed as an analgesic by the German dye manufacturer Friedrich Bayer & Company in 1899.7 This year, daily aspirin was shown to reduce the risk of cancer.8
The molecular structure and reactivity of aspirin have been studied extensively.9 Upon examining the crystal structure of aspirin (Figure 1), we observed that a lone pair of its donor oxygen could be poised to interact with the π* orbital of its ester carbonyl group. Moreover, the key signature of an n→π* interaction—the pyramidalization of the acceptor carbonyl group3k,3o—is evident in this crystal structure. This observation motivated us to perform a detailed conformational analysis of acetylsalicylic acid and its conjugate base. The results reveal that electron delocalization via the n→π* interaction has significant and heretofore unappreciated effects on the physical and chemical properties of aspirin.
Figure 1.

Crystal structure of acetylsalicylic acid9m and its Bürgi– Dunitz parameters d = 2.90 Å and θ = 99.5°, and pyramidalization parameters Δ = 0.011 Å and Θ = 2.49°.
RESULTS AND DISCUSSION
Gas phase geometry optimizations resulted in six different conformations of acetylsalicylic acid, 1–6, with a considerable n→π* interaction (Figure 2). The conformation of lowest energy, 1, has an n→π* interaction between the carbonyl oxygen of the acid and the ester carbonyl group with an interaction energy, as estimated by using second-order perturbation theory as implemented in Natural Bond Orbital 5.0 (NBO 5.0),10 of En→π* = 1.73 kcal/mol (Figure 3; Table 1). In conformation 2, which has the hydroxyl oxygen rather than the carbonyl oxygen of the acid as the electron-pair donor, En→π* = 0.95 kcal/mol. This conformation most resembles that of the crystal structure in Figure 1. Conformation 3 is stabilized by O–H···O hydrogen bond. The notable feature of conformation 4 is its stabilization by two weak n→π* interactions, one involving the acid carbonyl oxygen as the donor and the ester carbonyl group as the acceptor (En→π* = 0.16 kcal/mol), and another with the ester carbonyl oxygen as the donor and the acid carbonyl group as the acceptor (En→π* = 0.28 kcal/mol). Conformations 5 and 6 are similar to conformations 1 and 2, but with the AcO–C bond rotated by nearly 180°. This rotation diminishes the strength of the n→π* interaction (Table 1). Thus, our NBO analyses indicate that most of the low energy conformations of acetylsalicylic acid have a significant n→π* interaction (Table 1).
Figure 2.

Low-energy conformations of acetylsalicylic acid with a considerable n→π* interaction.
Figure 3.

n→π* interaction in conformations 1 and 2. (A) Depictions of overlap between n and π* orbitals in conformation 1 (overlap integral: 0.1077). (B) Depictions of overlap between n and π* orbitals in conformation 2 (overlap integral: 0.1031).
TABLE 1.
n→π* Interaction in Conformations 1–8
| Conformation | Relative Energies (kcal/mol) | d (Å) | θ (º) | En→π* (kcal/mol)1 | En→π* (kcal/mol)2 |
|---|---|---|---|---|---|
| 1 | 0.00 | 2.83 | 96.3 | 1.73 | 1.36 |
| 2 | 0.84 | 2.89 | 96.8 | 0.95 | 0.95 |
| 3 | 3.39 | 3.05 | 66.5 | 0.21 | 0.16 |
| 4 | 5.29 | 3.07 3.01 |
92.3 94.8 |
0.16 0.28 |
0.13 0.23 |
| 5 | 5.32 | 3.00 | 113.1 | 1.03 | 0.79 |
| 6 | 5.49 | 2.95 | 110.4 | 0.70 | 0.82 |
| 7 | — | 2.71 | 104.6 | 4.87 | 3.57 |
| 83 | — | 3.19 | 124.6 | 0.52 | 0.32 |
Second-order perturbative estimates.
From deletion analysis.
Less stable than conformation 7 by 1.10 kcal/mol.
A search for low energy conformations of acetylsalicylate revealed two conformers, 7 and 8 (Figure 4). The conformational preferences of acetylsalicylic acid and acetylsalicylate do not overlap. A closer examination of 1 and 7 reveals the differences in their conformational attributes. First, whereas the carboxyl group in 1 is nearly coplanar with the aromatic ring, the carboxylate in 7 is not. Second, the O···C distance between the donor oxygen and acceptor carbon is less in 7 (d = 2.71 Å) than in 1 (d = 2.83 Å). Additionally, the O···C=O angle of the donor oxygen and the acceptor carbonyl group is closer to the optimal Bürgi–Dunitz trajectory in 7 (θ= 104.6°) than in 1 (θ = 96.3°). NBO analyses of 7 reveal substantial conformational stabilization by the n→π* interaction (En→π* = 4.87 kcal/mol, Figure 5). Indeed, the deviation from coplanarity of the carboxylate with the aromatic ring in 7 can be attributed, in part, to its enhanced n→π* interaction. The tilting of the carboxylate plane away from that of the aromatic ring brings the donor oxygen closer to the acceptor carbon, and also reduces the Pauli repulsion between the donor oxygen and the phenolic oxygen.3m We note too that acetylsalicylic acid (pKa 3.48, ref. 11) is a stronger acid than benzoic acid (4.20) or o-methoxybenzoic acid (4.09), consistent with a stronger n→π* interaction in its conjugate base.
Figure 4.

Low-energy conformations of acetylsalicylate.
Figure 5.

n→π* interaction in conformation 7. (A) Overlap of p-rich lone pair with the π* orbital (overlap integral: 0.1423). (B) Overlap of s-rich lone pair with the π* orbital (overlap integral: 0.1050).
If an n→π* interaction is an important conformational determinant in aspirin, then altering its strength should alter the distance between the donor oxygen and acceptor carbon. The strength of the n→π* interaction can be altered by the introduction of electron-withdrawing groups (EWGs)3g or electron-donating groups (EDGs) on the aryl ring. Towards this end, we optimized several geometries of acetylsalicylate derivatives, installing a nitro or cyano group as an EWG, and a methoxy or dimethylamino group as an EDG. All EWGs and EDGs were located either para or meta to the carboxylate group (Figure 6). In the meta position, EWGs are expected to strengthen the n→π* interaction by lowering the energy of the π* orbital of the ester carbonyl group;3g conversely, EDGs should attenuate the n→π* interaction by increasing the energy of that orbital. Thus, the order of En→π* for the meta-substituted acetylsalicylate is expected to be: nitro (9) > cyano (10) > acetylsalicylate (7) > methoxy (11) > dimethylamino (12). Conversely, EDGs at the para position are expected to strengthen the n→π* interaction by increasing the energy of the lone pair, whereas the EWGs should attenuate the n→π* interaction. Thus, the order of En→π* for the para-substituted acetylsalicylate is expected to be opposite of the meta substituted: dimethylamino (16) > methoxy (15) > acetylsalicylate > cyano (14) > nitro (13). The strength of the n→π* interaction in the low energy conformations of these molecules follows this trend exactly (Table 2). As expected, shorter donor– acceptor distances (d) are observed for conformations with a larger En→π*.
Figure 6.
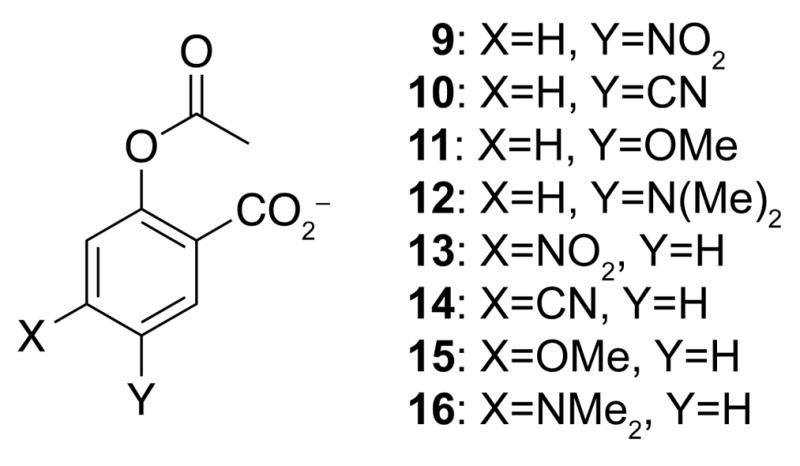
Compounds 9–16.
TABLE 2.
Computational Data for Compounds 9–16
| X | Y | Frequency (cm−1) | Eπ* (Hartrees)1 | d (Å) | θ (°) | En→π* (kcal/mol) | |
|---|---|---|---|---|---|---|---|
| 9 | H | NO2 | 1805 | 0.12064 | 2.65 | 103.5 | 6.32 |
| 10 | H | CN | 1803 | 0.12270 | 2.67 | 103.7 | 5.76 |
| 11 | H | OMe | 1792 | 0.13994 | 2.72 | 104.3 | 4.67 |
| 12 | H | NMe2 | 1791 | 0.14122 | 2.72 | 104.6 | 4.62 |
| 13 | NO2 | H | 1806 | 0.11854 | 2.82 | 104.2 | 3.26 |
| 14 | CN | H | 1803 | 0.12222 | 2.76 | 104.3 | 4.14 |
| 15 | OMe | H | 1794 | 0.14074 | 2.66 | 103.8 | 6.07 |
| 16 | NMe2 | H | 1793 | 0.14171 | 2.66 | 103.7 | 6.21 |
Energy of the π* acceptor orbital.
In addition to the correlation between En→π* and the donor–acceptor distance, we also considered the effects of the EWGs and EDGs on the vibrational stretching frequency of the ester carbonyl group. In the meta- and para-substituted systems, this frequency can be influenced by the strength of the n→π* interaction and the electronic effects exerted by the EDGs and EWGs.12 The n→π* interaction should lower the frequency. The perturbation of the ester stretching frequency by other electronic effects of the EDGs and EWGs should be less significant in the para position (which is meta to the ester). Hence, groups in the para position are expected to affect the ester stretching frequency primarily via the n→π* interaction, and the order of carbonyl stretching frequencies should be: nitro (13) > cyano (14) > acetylsalicylate (7) > methoxy (15) > dimethylamino (16). Indeed, the calculated frequencies of the para derivatives follow this trend (Table 2).
For the meta derivatives, the effect on the stretching frequency of the ester carbonyl group is expected to be an interplay between the mesomeric effect exerted by a substituent in the meta position (which is para to the ester) and the n→π* interaction. For the EWGs, the mesomeric effect is expected to increase the frequency but the increased n→π* interaction is expected to decrease the frequency; for the EDGs, the mesomeric effect is expected to decrease the frequency but the decreased En→π* is expected to increase the frequency. We found the order of the stretching frequencies for the meta derivatives to be: nitro (9) > cyano (10) > acetylsalicylate (7) > methoxy (11) > dimethylamino (12) (Table 2), as with the para derivatives. Apparently, the mesomeric effect is greater than the n→π* interaction in modulating the frequency of vibration.
Next, we explored another signature of the n→π* interaction. The two canonical structures of a carboxylate contribute equally to the resonance hybrid (Figure 7A). Due to the n→π* interaction, however, the analogous resonance structures in acetylsalicylate are not equivalent (Figure 7B). In other words, an n→π* interaction should cause the breakdown of equivalency of the two resonance structures of the carboxylate in acetylsalicylate. To confirm this hypothesis, we measured the bond lengths of the two C–O bonds in the carboxylate of 7. We find that the two C–O bonds do indeed differ in length, by 0.01 Å. To test that this difference was real and not an artifact of optimization, we optimized the geometry of benzoate at the same level of theory. In the optimized geometry of benzoate, the two C–O bond lengths differ by <0.0001 Å. To validate this hypothesis further, we repeated the analysis with a nitro group instead of a carboxylate group as an electron-pair donor. We find that non-equivalency of the canonical structures is observed with the nitro group as well.
Figure 7.
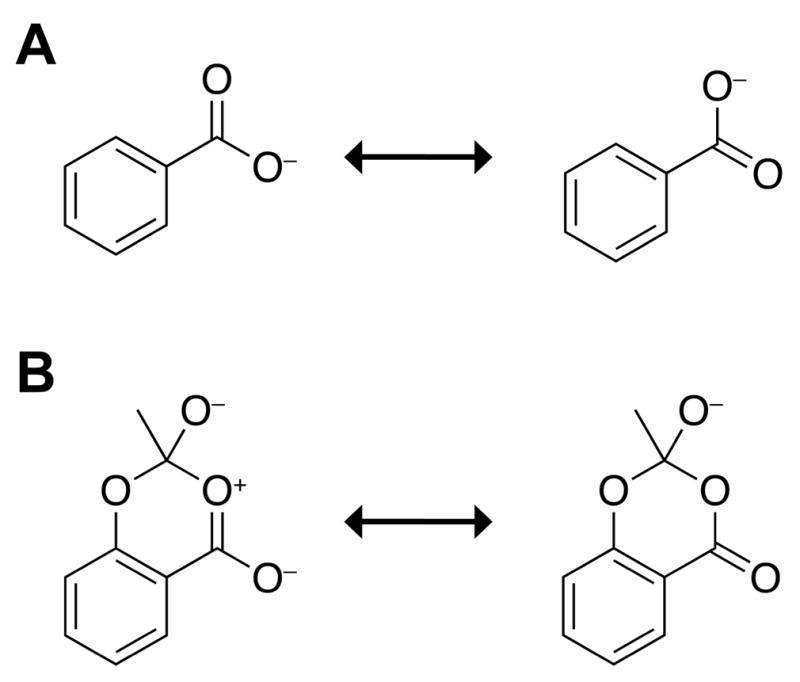
Breakdown of resonance equivalence. (A) Equivalent resonance structures of benzoate. (B) Nonequivalent resonance structures of acetylsalicylate.
The existence of the n→π* interaction was validated further by conformational analysis of yet another aspirin derivative. This derivative, 17, has a second carboxylate group ortho to the ester, resulting in a potential plane of symmetry (Figure 8). Electron delocalization from an n→π* interaction has the potential to raise the energy of the π* orbital of the ester carbonyl group, making it less able to participate in an n→π* interaction with a second donor (Figure 8). In other words, the two carboxylate groups cannot partake in equally strong n→π* interactions. This inequality could lead to the breakdown of symmetry, which would be reflected in the optimized geometry of 17. Indeed, the optimized geometry of 17 lacks a plane of symmetry— one of the carboxylate oxygen donors is in a short contact with the ester carbonyl group (d = 2.86 Å, En→π* = 1.55 kcal/mol), the other is much more distal (d = 3.31 Å, En→π* = 0.22 kcal/mol).13
Figure 8.

Breakdown of symmetry and notional perturbation of the energy of the π* orbital by an n→π* interaction in compound 17.
Chemical Implications
The n→π* interaction likely plays a role in the reactivity of aspirin. The van der Waals surfaces of the donor oxygen and the acceptor carbon interpenetrate by as much as 0.5 Å in low-energy conformation 7. Such short contacts are disfavored by steric effects and would not occur in the absence of the n→π* interaction.3m Aspirin is known to have the character of a mixed anhydride that can arise from transfer of the acyl group from the phenolic oxygen to the carboxylate group.14 We postulate that the early events in the acyl transfer reaction might be promoted by the n→π* interaction.
The n→π* interaction is also likely to play a role in the pKa of aspirin. The greater acidity of aspirin (pKa 3.48, ref. 11) compared with p-acetoxybenzoic acid (4.38) has been attributed to the “ortho effect” that arises from sterically induced non-coplanarity of the carboxylate group with the aromatic ring.15,9k A close examination of low energy conformation 7 reveals, however, that coplanarity of the carboxylate and the aromatic ring would attenuate the n→π* interaction by increasing the distance between the donor and acceptor groups. Thus, the n→π* interaction could contribute to the anomalously low pKa of aspirin by enforcing non-coplanarity.
Biological Implications
A number of physiological effects of aspirin, such as pain relief, stem from its ability to inhibit cyclooxygenases. Aspirin acetylates a serine residue located near the active site of cyclooxygenases, thereby rendering them inactive. Before doing so, aspirin must traverse a paraffin-like channel with no persistent hydration sites.16 By shielding the donor oxygen from solvent, an intramolecular n→π*interaction, like an intramolecular hydrogen bond in other systems, could facilitate the passage of aspirin through this hydrophobic channel.
CONCLUSION
We have discovered previously unrecognized electron delocalization in a well-known organic compound, aspirin. We found that the intimate interaction between the ester and carboxyl moieties of aspirin has substantial quantum mechanical character, arising from the delocalization of an electron pair (n) of a donor (e.g., a carboxyl or carboxylate oxygen) into the antibonding orbital (π*) of an acceptor (e.g., the ester carbonyl group). We propose that this interaction modulates the physical, chemical, and pharmacological properties of aspirin.
THEORETICAL PROCEDURES
Hybrid density functional theory as implemented in Gaussian ’0317 was employed to determine the conformational preferences of acetylsalicylic acid, acetylsalicylate, and its derivatives. The initial conformations of acetylsalicylic acid corresponded to the geometries optimized by Glaser.9k Gas phase full geometry optimizations and frequency calculations were performed at the B3LYP/6-311+G(2d,p) level of theory18 employing a Berny algorithm and tight SCF convergence criteria. Frequency calculations of the optimized structures yielded no imaginary frequencies, indicating the attainment of a true stationary point on the potential energy surface. The resulting self-consistent field (SCF) energies were corrected by the zero-point vibrational energy (ZPVE) determined in the frequency calculations, and are listed in Table S1. Optimized geometries were analyzed by NBO 5.0 (ref. 10) at the B3LYP/6-311+G(2d,p) level of theory.
Supplementary Material
Acknowledgments
We thank B. R. Caes and Dr. M. D. Shoulders for contributive discussions. K. J. K. was supported by a Barry M. Goldwater Scholarship and a Hilldale Undergraduate/Faculty Research Fellowship. This work was supported by Grant R01 AR044276 (NIH).
Footnotes
The computed energies and coordinates of conformations 1–8 and compounds 9–17. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kirby AJ. Stereoelectronic Effects. Oxford University Press; New York: 1996. [Google Scholar]; (b) Eliel EL, Wilen SH. Stereochemistry of Organic Compounds. Wiley Interscience Publication; New York: 1996. [Google Scholar]
- 2.Bürgi HB, Dunitz JD, Shefter E. Acta Cryst. 1974;B30:1517–1527. [Google Scholar]
- 3.(a) Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT. J Am Chem Soc. 2001;123:777–778. doi: 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]; (b) Jenkins CL, Raines RT. Nat Prod Rep. 2002;19:49–59. doi: 10.1039/a903001h. [DOI] [PubMed] [Google Scholar]; (c) DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL. J Am Chem Soc. 2002;124:2497–2505. doi: 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]; (d) Hinderaker MP, Raines RT. Protein Sci. 2003;12:1188–1194. doi: 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jenkins CL, Lin G, Duo J, Rapolu D, Guzei IA, Raines RT, Krow GR. J Org Chem. 2004;69:8565–8573. doi: 10.1021/jo049242y. [DOI] [PubMed] [Google Scholar]; (f) Horng JC, Raines RT. Protein Sci. 2006;15:74–83. doi: 10.1110/ps.051779806. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hodges JA, Raines RT. Org Lett. 2006;8:4695–4697. doi: 10.1021/ol061569t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Shoulders MD, Guzei IA, Raines RT. Biopolymers. 2008;89:443–454. doi: 10.1002/bip.20864. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kotch FW, Guzei IA, Raines RT. J Am Chem Soc. 2008;130:2952–2953. doi: 10.1021/ja800225k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Shoulders MD, Raines RT. Ann Rev Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Choudhary A, Gandla D, Krow GR, Raines RT. J Am Chem Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Bartlett GJ, Choudhary A, Raines RT, Woolfson DN. Nat Chem Biol. 2010;6:615–620. doi: 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Jakobsche CE, Choudhary A, Raines RT, Miller SJ. J Am Chem Soc. 2010;132:6651–6653. doi: 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Choudhary A, Pua KH, Raines RT. Amino Acids. 2011;41:181–186. doi: 10.1007/s00726-010-0504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Choudhary A, Raines RT. Protein Sci. 2011;20:1077–1081. doi: 10.1002/pro.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choudhary A, Kamer KJ, Powner MW, Sutherland JD, Raines RT. ACS Chem Biol. 2010;5:655–657. doi: 10.1021/cb100093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Lesarri A, Cocinero EJ, López JC, Alonso JL. J Am Chem Soc. 2005;127:2572–2579. doi: 10.1021/ja045955m. [DOI] [PubMed] [Google Scholar]; (b) Sanz ME, Lesarri A, Peña MI, Vaquero V, Cortijo V, López JC, Alonso JL. J Am Chem Soc. 2006;2006:3812–3817. doi: 10.1021/ja058194b. [DOI] [PubMed] [Google Scholar]; (c) Blanco S, López JC, Mata S, Alonso JL. Angew Chem Int Ed. 2010;49:9187–9192. doi: 10.1002/anie.201002535. [DOI] [PubMed] [Google Scholar]
- 6.Gerhardt C. Ann Chem Pharm. 1853;87:149–179. [Google Scholar]
- 7.Sneader W. Br Med J. 2000;321:1591–1594. doi: 10.1136/bmj.321.7276.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothwell PM, Fowkes FGR, Belch JFF, Ogawa H, Warlow CP, Meade TW. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 9.(a) Edwards LJ. Trans Faraday Soc. 1950;46:723–735. [Google Scholar]; (b) Bender ML. Chem Rev. 1960;60:53–113. [Google Scholar]; (c) Wheatley PJ. J Chem Soc. 1964:6036–6048. [Google Scholar]; (d) Fersht AR, Kirby AJ. J Am Chem Soc. 1967;89:4853–4857. doi: 10.1021/ja00995a006. [DOI] [PubMed] [Google Scholar]; (e) Fersht AR, Kirby AJ. J Am Chem Soc. 1967;89:4857–4863. doi: 10.1021/ja00995a007. [DOI] [PubMed] [Google Scholar]; (f) St Pierre T, Jencks WP. J Am Chem Soc. 1968;90:3817–3827. doi: 10.1021/ja01016a040. [DOI] [PubMed] [Google Scholar]; (g) Kemp DS, Thibault TD. J Am Chem Soc. 1968;90:7154–7155. [Google Scholar]; (h) Kim Y, Machida K, Taga T, Osaki K. Chem Pharm Bull. 1985;33:2641–2647. doi: 10.1248/cpb.33.2641. [DOI] [PubMed] [Google Scholar]; (i) Binev IG, Stamboliyska BA, Binev YI. J Mol Struc. 1996;378:189–197. [Google Scholar]; (j) Payne RS, Rowe RC, Roberts RJ, Charlton MH, Docherty R. J Comput Chem. 1999;20:262–273. [Google Scholar]; (k) Glaser R. J Org Chem. 2001;66:771–779. doi: 10.1021/jo001241s. [DOI] [PubMed] [Google Scholar]; (l) Ouvrard C, Price SL. Cryst Growth Des. 2004;4:1119–1127. [Google Scholar]; (m) Vishweshwar P, McMahon JA, Oliveira M, Peterson ML, Zaworotko MJ. J Am Chem Soc. 2005;127:16802–16803. doi: 10.1021/ja056455b. [DOI] [PubMed] [Google Scholar]; (n) Bond AD, Boese R, Desiraju GR. Angew Chem Int Ed. 2007;46:618–622. doi: 10.1002/anie.200603373. [DOI] [PubMed] [Google Scholar]
- 10.(a) Weinhold F. In: Encyclopedia of Computational Chemistry. Schleyer PvR, Allinger NL, Clark T, Gasteiger J, Kollman PA, Shaefer HF, III, Schreiner PR., editors. Vol. 3. John Wiley & Sons; Chichester, UK: 1998. pp. 1792–1811. [Google Scholar]; (b) Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F. NBO 5.0. 2001. [Google Scholar]; (c) Weinhold F, Landis CR. Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective. Cambridge University Press; Cambridge, UK: 2005. [Google Scholar]
- 11.Habibi-Yangjeh A, Esmailian M. Chin J Chem. 2008;26:875–885. [Google Scholar]
- 12.(a) Bromilow J, Brownlee RTC, Craik DJ, Fiske PR, Rowe JE, Sadek MJ. J Chem Soc Perkin Trans. 1981;2:753–759. [Google Scholar]; (b) Neuvonen H, Neuvonen K, Koch A, Kleinpeter E, Pasanen P. J Org Chem. 2002;67:6995–7003. doi: 10.1021/jo020121c. [DOI] [PubMed] [Google Scholar]
- 13.The breakdown of symmetry in compound 17 is reminiscent of Jahn–Teller distortion (Jahn H, Teller E. Proc R Soc Lond A. 1937;161:220–235.).
- 14.Davidson D, Auerbach L. J Am Chem Soc. 1953;75:5984–5986. [Google Scholar]
- 15.Fujita T, Nishioka T. In: Progress in Physical Organic Chemistry. Taft RW, editor. John Wiley & Sons; New York: 1976. pp. 49–89. [Google Scholar]
- 16.(a) Picot D, Loll PJ, Garavito RM. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]; (b) Smith WL, DeWitt DL, Garavito RM. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]; (c) Young T, Abel R, Kim B, Berne BJ, Friesner RA. Proc Natl Acad Sci USA. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.RC, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 18.(a) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (b) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.