

Abstract
The CpG dinucleotide is disproportionately represented in human genetic variation due to the hypermutability of 5-methyl-cytosine (5mC). We exploit this hypermutability and a novel codon substitution model to identify candidate functionally important exonic nucleotides. Population genetic theory suggests that codon positions with high cross-species CpG frequency will derive from stronger purifying selection. Using the phylogeny-based maximum likelihood inference framework, we applied codon substitution models with context-dependent parameters to measure the mutagenic and selective processes affecting CpG dinucleotides within exonic sequence. The suitability of these models was validated on >2,000 protein coding genes from a naturally occurring biological control, four yeast species that do not methylate their DNA. As expected, our analyses of yeast revealed no evidence for an elevated CpG transition rate or for substitution suppression affecting CpG-containing codons. Our analyses of >12,000 protein-coding genes from four primate lineages confirm the systemic influence of 5mC hypermutability on the divergence of these genes. After adjusting for confounding influences of mutation and the properties of the encoded amino acids, we confirmed that CpG-containing codons are under greater purifying selection in primates. Genes with significant evidence of enhanced suppression of nonsynonymous CpG changes were also shown to be significantly enriched in Online Mendelian Inheritance in Man. We developed a method for ranking candidate phenotypically influential CpG positions in human genes. Application of this method indicates that of the ∼1 million exonic CpG dinucleotides within humans, ∼20% are strong candidates for both hypermutability and disease association.
Keywords: codon substitution, CpG, natural selection, selection mutation balance
Introduction
Distinguishing phenotypically influential from nonfunctional genetic variants remains a major challenge in human genetics. The increasing ease of obtaining whole-genome sequences emphasizes the importance of distinguishing genetic variants likely to contribute to disease risk which can in part be addressed by prior identification of candidate positions for disease causation (Cooper et al. 2010). One approach to this problem is to exploit comparative genomics information. It has been argued that genetic variants at a position conserved in related species are strong candidates for phenotypic effect in humans (Miller and Kumar 2001; Kumar et al. 2009). Here, we advance this approach, showing that consideration of differences in mutation pressure can enhance the discrimination of disease/nondisease genetic variants. In particular, we exploit knowledge of the hypermutability of cytosine within the CpG dinucleotide (Sved and Bird 1990) for analyses of exonic sequences, demonstrating CpG codons are subjected to stronger purifying natural selection. We further identify the genes containing these codons and predict specific functionally important residues.
Classic population genetics theory establishes the equilibrium frequency of a sequence state is governed by the relationship between mutagenicity and functional significance. To illustrate this, consider a genetic locus with two alleles A and a. If the mutation rate of is μ and of is ν, the equilibrium frequencies are for allele A and for allele a (Sved and Bird 1990). When the two alleles exert different effects on phenotype, the equilibrium frequencies depend on the mode of natural selection. If allele a is recessive, strongly deleterious and present at a low frequency (so we ignore ν); the relative fitnesses of each genotype AA, Aa, and aa are 1, 1, and 1 − s, respectively, where s is the selection coefficient against genotype aa. The equilibrium frequency of allele a is then approximately 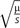 (Hartl and Clark 2007). In this simple scenario, either higher mutability (large μ) or less disadvantage (small s) of allele a will increase the equilibrium frequency of a. These within-population processes should also manifest in sequence divergence between species such that the probability of a sequence state not being substituted depends on the mutation rate and functional significance. Hence, conservation of hypermutable bases at a sequence position implies very large s (strong purifying selection) and thus functional importance.
(Hartl and Clark 2007). In this simple scenario, either higher mutability (large μ) or less disadvantage (small s) of allele a will increase the equilibrium frequency of a. These within-population processes should also manifest in sequence divergence between species such that the probability of a sequence state not being substituted depends on the mutation rate and functional significance. Hence, conservation of hypermutable bases at a sequence position implies very large s (strong purifying selection) and thus functional importance.
The hypermutability of CpG dinucleotides in vertebrates (Coulondre et al. 1978; Cooper and Youssoufian 1988) suggests a distinctive functional profile for CpG-containing codons. CpG sites exhibit higher mutability than other dinucleotides due to the addition of a methyl group to cytosine, resulting in 5-methyl-cytosine (5mC). This mutation pressure is applicable to CpG-containing codons because exons are generally methylated (Tornaletti and Pfeifer 1995; Rabinowicz et al. 2003). Opposition to the loss of exonic CpG arises because CpG-containing codons encode a collection of amino acids (alanine, arginine, proline, serine, and threonine) with different physico-chemical properties that can be the target of natural selection.
To illustrate the difference in CpG equilibrium frequencies expected under different modes of selection, consider the impact on a species’ genome when the species moves from a state of no DNA methylation to one of methylation at CpG dinucleotides. Given the CpG → TpG/CpA mutation rate is about one order of magnitude higher than the reverse (Duncan and Miller 1980; Bulmer 1986; Sved and Bird 1990), 1) neutrally evolving codons will move toward a lower frequency of CpG; 2) CpG’s at functionally significant codons will be maintained by substantial purifying selection against amino acid changes. A direct consequence of this process is that conserved CpG dinucleotides have an increased likelihood of functional significance compared with other dinucleotides. This conjecture has an important implication for finding positions where amino acid polymorphisms affect protein function and are associated with disease.
Previous examinations of the interaction between CpG mutability and natural selection within protein-codingsequences have flaws. One approach was to apply codon substitution models as these allowed formal hypothesis testing under the phylogeny-based maximum likelihood framework. Extending Goldman and Yang’s model (Goldman and Yang 1994), Huttley (2004) introduced CpG context-dependent substitution parameters to assess relative substitution rate and selective strength at CpG’s compared with other types of nucleotide substitutions. CpG-containing codons were estimated to exhibit strikingly higher substitution rates and a lower ratio of nonsynonymous to synonymous substitution rates in the BRCA1 gene. However, we have recently demonstrated that the model of Goldman and Yang is unsuitable for measuring context-dependent processes (Lindsay et al. 2008; Yap et al. 2010). Moreover, a confounding influence arising from different selective constraints affecting the amino acids encoded by CpG was not explicitly addressed. Misawa and Kikuno (2009) and Schmidt et al. (2008) both compared mutation probabilities arising from CpG and non-CpG content for the same amino acid exchanges using a parsimony-based method. Misawa and Kikuno address the issue of amino-acid properties by utilizing Grantham’s distances (Grantham 1974) but they did not develop significance testing for individual genes or residues. Schmidt et al. (2008) found a substantially elevated CpG nonsynonymous transition substitution rate by comparing the same amino acid exchanges with or without CpG context and reduced fixation probability for CpG nonsynonymous transitions by comparing the ratios of transition rates within a CpG context to that of outside a CpG context between nonsynonymous and other substitutions. The analyses had a number of limitations. There were no formal tests for statistical significance and background selective constraints affecting CpG-encoded amino acids were not adjusted for. Furthermore, since closely related primates were used, concatenating substitutions from a large number of genes was required. Using a CpG transition rate derived from whole genomes is also not reasonable given substitution rate varies substantially across mammalian genomes (Wolfe et al. 1989; Matassi et al. 1999; Lercher et al. 2001; Smith et al. 2002; Ellegren et al. 2003; Arndt et al. 2005). Finally, the critical step of identifying the individual proteins and CpG codons most plausibly subjected to strong negative selection has not yet been done.
Despite differences between the forward and reverse nucleotide mutation rates operating in primate genomes, the general time reversible (hereafter GTR, [Lanave et al. 1984]) variant of the conditional nucleotide frequency (hereafter CNF) codon substitution model successfully distinguished neutral mutation processes from selection in primates (Yap et al. 2010). In this paper, we modify the codon model approach of Huttley (2004) to use this CNF codon model form (Yap et al. 2010) to improve the estimation of the mode of natural selection affecting CpG-containing codons. The objectives were to assess the extent of elevated CpG mutation properties within coding sequences and whether CpG-encoded amino acids were subjected to greater purifying selection than an appropriately defined background rate. Using yeast, whose genomes are putatively methylation free (Proffitt et al. 1984), as a naturally occurring biological control, we show that codon substitution models can formally establish specific context-dependent mutation profiles and distinguish unique selective constraints on a subset of defined amino acids. We further present evidence of stronger selective constraints on CpG codons by examining disease-causing genes.
Materials and Methods
Statistical Models of Codon Evolution
We employ the CNF matrix form to model codon evolution (Yap et al. 2010). Several continuous-time Markov process models have been defined for codon-based analyses and their instantaneous rate matrices, conventionally denoted Q, all share the following general definition for substitutions between codons i and j.
 |
(1) |
where r(i, j) is the product of rate parameters affecting exchanges between the codons and πx is an equilibrium frequency. For instance, in a commonly employed codon model form, r(i, j) includes combinations of the parameters ω and κ for: synonymous transversions, r(i, j) = 1; synonymous transitions, r(i, j) = κ; nonsynonymous transversions, r(i, j) = ω; and nonsynonymous transitions, r(i, j) = κ·ω. The competing model forms differ in their definition of πx. The (Muse and Gaut 1994, MG94) model uses the frequency of the nucleotide in codon j that differs from codon i, with the result that the equilibrium codon frequencies are the product of nucleotide frequencies (normalized for the omission of stop codons). This multiplicative feature of the MG94 model is unlikely to be satisfied in coding sequences and has been shown to bias parameter estimates (Yap et al. 2010). πx in the Goldman and Yang (1994, GY94) model is the frequency of codon j, so the equilibrium codon frequencies readily match those observed, but this formulation confounds the single nucleotide substitution event with the frequency of other sequence states. This confounding has the undesired effect of causing the GY94 model to show context-dependent effects when they do not exist (Lindsay et al. 2008). Therefore, parameter estimates in both the MG94 and GY94 models can be biased by sequence composition. πx in the CNF model, by contrast, is the frequency of the nucleotide in codon j that differs from codon i, conditional on the other two nucleotides in codon j, which can be expressed as:
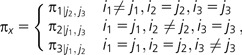 |
(2) |
where i1, i2, i3, and j1, j2, j3 represent the nucleotide states at the three codon positions in codon i and j, respectively. The merit of the CNF model is that it nests the independent substitution process but also allows equilibrium codon frequencies to be nonmultiplicative (for a complete explanation, see Yap et al. 2010).
A parameterization of the CNF form that included the most general reversible nucleotide process (Lanave et al. 1984) has been established as being most suitable for the estimation of evolutionary rates within codons (Yap et al. 2010). A striking feature of the mammal genomes is that the type profile and total rate of neutral nucleotide substitutions varies between regions and that this can confound estimation of context-dependent rates (Yap et al. 2010). Robust estimation of context-dependent rates was achieved by including parameters from the most GTR nucleotide substitution model Yap et al. (2010). This GTR model employs six separate parameters r(A, C), r(A, G), r(A, T), r(C, G), r(C, T), r(G, T) to represent nucleotide exchanges within codon models (Pond and Muse 2005; Yap et al. 2010). We omit r(G, T) (thus constraining it to equal one) to calibrate the matrix and scale Q so that time is measured as the expected number of substitutions per codon (Yap et al. 2010). Returning to the definition of r(i, j) in equation (1), in the interests of simplifying the notation for the codon models employed here, we use rGTR to represent the nucleotide GTR component, for example, r(i, j) = rGTR(i, j) is a synonymous rate and r(i, j) = rGTR(i, j)·ω a nonsynonymous rate. Finally, all models employed satisfy the condition of reversibility (by constraining r(i, j) = r(j, i)) and stationarity (π does not change). This completes the definition of the baseline codon model, which we will subsequently refer to as CNFGTR.
The substitution dynamics of CpG-containing codons are measured by adding parameters to the CNFGTR baseline. How comparisons among these model forms are used to make inference about biological process is described in the next section. Here, we define the parameters used to assess mutagenic propensities and then to measure selective influences. We measure a common CpG mutation effect as
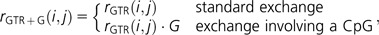 |
a CpG transition effect as
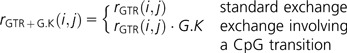 |
(3) |
and both a common and transition-specific CpG effects as
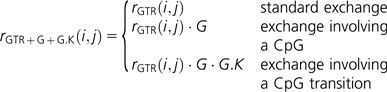 |
How these parameters are interpreted is discussed in the next section.
The influence of natural selection on CpG transitions was measured by a parameter corresponding to the intersection of nonsynonymous and CpG transition substitutions. The ω parameter represents the common influence of natural selection on the rate of substitution for all amino acids. The amino acid exchanges, resulting from CpG transitions, may also result from non-CpG events (table 1). For instance, substitutions between alanine and valine can arise from CpG transitions (e.g., GCG ↔ GTG) or not (e.g., GCA ↔ GTA). The rate of replacements between the 12 methylation-affected amino acids (hereafter MAA, table 1) are likely to differ from the “average” amino acid exchange due to the distinctive changes in physico-chemical properties. To assess whether the positions at which the MAAs are CpG encoded evolve differently from other MAA positions, we introduce a parameter, α, to account for the substitution rate common to the MAA amino acid substitutions listed in table 1.
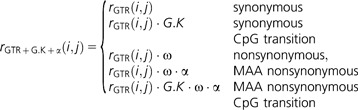 |
Table 1.
MAA Substitutions
| Sub type | aa sub. | Codon sub. |
| CpG transitions | A ↔ V | GCG ↔ GTG |
| C ↔ R | TGC ↔ CGC, TGT ↔ CGT | |
| H ↔ R | CAC ↔ CGC, CAT ↔ CGT | |
| L ↔ P | CTG ↔ CCG | |
| L ↔ S | TTG ↔ TCG | |
| M ↔ T | ATG ↔ ACG | |
| Q ↔ R | CAA ↔ CGA, CAG ↔ CGG | |
| R ↔ W | CGG ↔ TGG | |
| A ↔ V | GCA ↔ GTA, GCC ↔ GTC, GCT ↔ GTT | |
| Non-CpG counterparts | L ↔ P | CTA ↔ CCA, CTC ↔ CCC, CTT ↔ CCT |
| L ↔ S | TTA ↔ TCA | |
| R ↔ W | AGG ↔ TGG |
Note.—The MAAs are defined by amino acid substitutions resulting from CpG transitions. The non-CpG counterparts are other codon substitution events that give rise to the same amino acid substitution. Sub. type, substitution type; aa sub., amino acid substitution; codon sub., codon substitutions.
Finally, the mode of natural selection affecting nonsynonymous CpG transitions is measured using an analogous definition to that of Huttley (2004). The parameter G.K.ω represents the rate of nonsynonymous CpG transitions relative to the background rate.
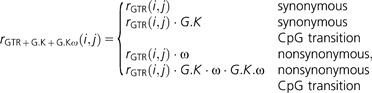 |
Distinguishing between the α and G.K.ω terms is done by defining an additional model that combines the above two definitions. In that model, the G.K.ω parameter is included in cases where both G.K and α are present.
Hypothesis Testing
Likelihood ratio tests (LRTs) between nested hypotheses were used to compare the models. The test statistic, computed as LR = 2(ln Lalt − ln Lnull), is asymptotically distributed χ2 with degrees of freedom (hereafter, df) equal to the difference in number of free parameters between the null and alternate hypotheses. Employing the standard assumptions of time reversibility, stationarity, and homogeneity (between codon positions and between branches), we determined the set of parameter values in the evolutionary model that maximized the likelihood for each multiple alignment using the method of Felsenstein (1981). The interpretation of the LRT statistics and the associated parameter maximum-likelihood estimates (MLEs) derives from the nested relationship between competing models. We illustrate this with reference to the CNFGTR + G.K model (eq. 3). By setting G.K = 1, the rates defined in equation (3) reduce to CNFGTR rates. The interpretation of parameter MLEs therefore hinges on their position relative to 1 and on the other parameters in the model. For instance, values for G.K that are > 1 from the CNFGTR + G.K model would indicate CpG transition substitutions occur at a higher rate than the baseline substitution rate. We further emphasize here that these parameters are being estimated across all aligned codon positions.
Given the strength of prior evidence supporting CpG transitions as the dominant mutation process (Coulondre et al. 1978; Cooper and Youssoufian 1988; Sved and Bird 1990; Krawczak et al. 1998; Sommer et al. 2001), we evaluated the contribution of CpG transitions to codon evolution by comparing the CNFGTR null hypothesis against the CNFGTR + G.K alternate. As these two models differ in just one parameter (G.K), the probability of the observed, or larger, LRT statistic occurring by chance is determined from the distribution. If the G.K term proved significant, we then evaluated support for a distinct CpG transversion rate by comparing the CNFGTR + G.K null hypothesis against the CNFGTR + G.K + G alternate, again comparing the LRT test statistic to the distribution. The sequential Bonferroni correction (Holm 1979) was applied to adjust for multiple tests.
Evaluation of selection effects on CpG substitutions were also established through hierarchical hypothesis tests. First, we are interested in whether CpG-containing codons evolve at a rate that cannot be accounted for by the physico-chemical properties of the amino acids alone. We address this potential confounding by explicitly representing the physico-chemical properties of the amino acids with the parameter α (as discussed above). We evaluated whether a distinctive nonsynonymous CpG rate existed by first comparing the CNFGTR + G.K null hypothesis against the CNFGTR + G.K + α alternate followed by comparing CNFGTR + G.K + α against CNFGTR + G.K + α + G.K.ω. We separately employed a more direct evaluation of the G.K.ω parameter both to evaluate the broad support for distinctive nonsynonymous CpG substitution rate and for predicting functionally important CpG sites; comparing the CNFGTR + G.K null against the CNFGTR + G.K + G.K.ω alternate hypothesis.
Model Fitting
The different substitution models were implemented using standard features of PyCogent 1.5.0.dev (Knight et al. 2007). Numerical optimization of functions was done using the Powell local optimizer using a maximum of five restarts, exit tolerance of 10−8 and a maximum of 103 function evaluations. All functions were checked to ensure that they had not exited due to exceeding the maximum evaluations limit as this implies the function was not maximized (this condition did not occur). Substitution models were fit to each alignment in a manner that ensured the alternate model likelihood always improved over the null. The resulting MLEs for model parameters were used as starting values for the alternate model. For example, for each alignment, the CNFGTR model was fit and the resulting MLEs used to then fit CNFGTR + G.K, with resulting MLEs used to then fit CNFGTR + G.K + G. For all models, the equilibrium codon frequencies (π) were estimated as the average codon frequency across the observed sequences.
Predicting CpG-Encoded Amino acids Subjected to Strong Purifying Selection
For genes where there was support for strong purifying selection operating against CpG transitions (), it is of interest to identify the codons involved. One potential approach to this classification problem is to use a mixture model in which the alternate hypothesis defines two site classes: 1) codons at which CpG substitutions are unrestricted and 2) codons where CpG substitutions are selected against. There are a number of challenges in using such a mixture model approach. A key limitation is that the increased number of free parameters over the null reduces statistical power, an effect exacerbated by the typically low frequency of CpG dinucleotides. These models are also well known to be difficult to fit numerically (e.g., see Wong et al. 2004), a challenge again exacerbated by the low frequency of CpG’s. Hence, we considered mixture models impractical for the current case. As an alternative, we developed an approach that requires no additional model parameters to be specified. The LRT statistics described above can be decomposed into the contributions from individual codon positions. Codons whose evolution is markedly more consistent with the alternate (compared with the null) hypothesis will exhibit large positive position-wise LRT statistics (hereafter LRTP). Conversely, codons whose evolution strongly contradicts the alternate hypothesis will exhibit negative LRTP with large absolute magnitude. To assess the LRTP that lay beyond that expected when the null hypothesis is correct, we determined the 1st and 99th quantiles of LRTp under the null from simulated alignments. We used the LRT(5) statistic for this purpose since using parametrically richer null models (e.g., CNFGTR + G.K + α) can identify changes at non-CpG-encoded positions as influential due to their support for elevation of rates at non-CpG positions which is not of interest here.
Data Sampling
Sampling Protein-Coding Sequences
Ensembl release 56 (Hubbard et al. 2009) was queried for nuclear-encoded primate protein-coding genes with one-to-one orthologs in the primate species Homo sapiens, Pan troglodytes, Macaca mulatta, and Pongo pygmaeus. We excluded genes if the (Ensembl defined) canonical transcript for any one of the species was <900 nucleotides in length or the gene could not be translated using the standard genetic code.
We used a parallel analysis of yeast genes as a natural biological control because budding yeast does not methylate its DNA (Proffitt et al. 1984). Yeast nuclear protein-coding genes were sampled from four closely related species, namely Saccharomyces cerevisiae, S. paradoxus, S. mikatae, and S. bayanus (fig. 1). Orthologous yeast sequences were downloaded from the Saccharomyces Genome Database. Each downloaded file is an alignment from the four yeast species corresponding to one S. cerevisiae open reading frame and its flanking sequences provided by Kellis et al. (2003). In the file, coding sequences were distinguished by uppercase letters; whereas noncoding sequences, including intergenic and intronic sequences, were recorded in lowercase. To be consistent with the workflow of primates, the yeast-coding sequences were extracted and aligned using the PyCogent codon aligner (see below). The criteria used for filtering primate coding sequences were also applied.
FIG. 1.

Distributions of CpG substitution parameter MLEs from primate and yeast. is the MLE for G.K from the CNFGTR + G.K model where LRT(1) was nominally significant (P < 0.05). is the MLE for G from the CNFGTR + G.K + G model where both LRT(1) and LRT(2) were nominally significant (P < 0.05). Frequency is the number of significant alignments whose MLE lay within the indicated bin. The vertical dotted blue line corresponds to the Parameter having no effect. MLE estimates > 15 were collapsed into the 15+ bin.
Human SNPs
Allelic nuclear single nucleotide polymorphisms (SNPs) in sampled protein-coding sequences were obtained from the Ensembl variation database (Hubbard et al. 2009) using PyCogent (Knight et al. 2007). This database includes SNPs from National Center for Biotechnology Information (NCBI) dbSNP and other sources, such as the supporting databases for the Affy GeneChip 100 k Array. Both validated (genotyped for a certain number of individuals within a population) and nonvalidated SNPs were considered. We used the more recent release 57 rather than Ensembl release 56 as we identified a substantial number of errors in flanking sequences obtained from the latter release compared with the NCBI records (result not shown). Each SNP was classified by its effect on the corresponding codon as synonymous or nonsynonymous. The 5’ and 3’ flanking sequences of a SNP were used to determine whether it was within a CpG dinucleotide. For example, an A/G SNP including flanking sequence of 5’-C [A/G] G-3’ is a CpG allele, whereas 5’-T [A/G] G-3’ is not.
Sequence Alignment
Orthologous codon sequences were aligned using the built-in progressive Hidden Markov Model alignment algorithm of PyCogent (Knight et al. 2007). This algorithm is motivated by the approach developed by Loytynoja and Goldman (2005), which has been argued to provide a better solution than traditional alignment algorithms that typically overmatch sequences by underestimation of insertions (Loytynoja and Goldman 2005). In our case, we used the CNFHKY codon substitution model (Yap et al. 2010) because it preserves the sequence reading frame and distinguishes transition from transversion and nonsynonymous from synonymous substitutions with just two exchangeability parameters. In our case, the alignment method used pairwise distances estimated using the provided codon model to estimate a phylogenetic tree using the Neighbor Joining algorithm. This tree, along with the median MLEs for ω and κ from the pairwise estimates were used in the progressive alignment step.
Quantifying the Role of Conserved Exonic CpG in Human Disease
The NCBI Online Mendelian Inheritance in Man (OMIM) database was used to query disease-associated genes. OMIM catalogues human genes and genetic disorders with evidence from the literature (Antonarakis and McKusick 2000). Each gene record provides information like gene description, gene function, and gene structure. Some genes have allelic variants which, according to the OMIM curation process (Antonarakis and McKusick 2000), is evidence a gene has been associated with human disease. Although not all allelic variants in OMIM cause disease, most alleles are related to pathological disorders (Antonarakis and McKusick 2000). Accordingly, we used genes with allelic variants to represent disease-associated genes.
Gene OMIM accession numbers and associated symbols were downloaded on 29 July, 2009. OMIM gene symbols were then used to query the Ensembl database. If not found, alternative symbols from the OMIM gene table were employed. This resulted in 11,129 human nuclear protein-coding genes recorded in both the Ensembl and OMIM databases. Among these genes, 7,629 were included in the sampled primate alignments with 1,563 genes classified as disease associated.
Source Code Availability
All scripts and data employed for this study are available on request from the authors.
Results
Elevated CpG Transition Rate
Our analyses of primate genes provided the expected support for elevated CpG transition rates and modest support for a CpG transversion effect. Although a substantial 66% of primate genes showed nominal (P < 0.05) significance, the proportion of genes significant for LRT(1) after correcting for multiple tests dropped substantially to 17% (table 2). We suggest this reflects relatively low statistical power in the primate data set due to a low of divergence and typically reduced frequencies of CpG-containing dinucleotides. The number of alignments exhibiting a common CpG effect (G), when only genes significant after the test correction were considered, was <1%, indicating only modest support for a distinct CpG transversion rate affecting primate genes.
Table 2.
LRTs for Distinctive Mutation and Selection Influences Affecting CpG-Containing Codons
| Clade | LRT (#) | Ho | +Param | % (P < 0.05) | % (Bonf P < 0.05) |
| Primate | 1 | CNFGTR | G.K | 66.33 | 17.36 |
| 2 | CNFGTR + G.K | G | 17.63 | 0.82 | |
| 3 | CNFGTR + G.K | α | 22.57 | 0.75 | |
| 4 | CNFGTR + G.K + α | G.K.ω | 11.02 | 0.07 | |
| 5 | CNFGTR + G.K | G.K.ω | 22.50 | 1.03 | |
| Yeast | 1 | CNFGTR | G.K | 15.59 | 0.00 |
| 2 | CNFGTR + G.K | G | 9.20 | 0.00 | |
| 3 | CNFGTR + G.K | α | 38.29 | 1.11 | |
| 4 | CNFGTR + G.K + α | G.K.ω | 11.37 | 0.00 | |
| 5 | CNFGTR + G.K | G.K.ω | 18.12 | 0.16 |
Note.—The total number of alignments was 12,092 for primate and 2,533 for yeast. Ho, null hypothesis; +Param, parameter added to null to make the alternate hypothesis; % (P < 0.05), percent of total alignments significant at the nominal 0.05 level; % (Bonf P < 0.05), percentage of alignments significant after adjustment for multiple tests. If the model was the first in a series, the adjustment was the total number of alignments, if they were second in a series, the adjustment was for the corrected significant number from the earlier result. For example, the correction for primate LRT(1) was 12,092 but for LRT(2), it was the number of LRT(1) significant after correcting for multiple tests, 2,099.
The distribution of parameter MLEs from nominally significant alignments were consistent with the elevation of both CpG transition and transversion substitutions. The distribution of from LRT(3) (fig. 1) with nearly all MLEs > 1 supports CpG transitions having an elevated rate of substitution over other substitution types, consistent with biochemical evidence of the hypermutability of CpG putatively arising from 5mC. We considered from only those genes where both LRT(1) and LRT(2) were nominally significant. The distribution of these MLEs were also typically > 1, again supporting an elevated CpG transversion rate at these genes in primates. It is noteworthy that a moderate number of were < 1, indicating significant suppression of CpG substitutions for a number of genes. These observations highlight the fact both the and remain potentially confounded by the influence of natural selection in the CNFGTR + G.K and CNFGTR + G.K + G models. Suppression of the general CpG substitution rate may reflect the operation of strong purifying natural selection at those genes. Consistent with this interpretation, a substantial fraction (44/131) of the genes with that were nominally significant for both LRT(1) and LRT(2) were also nominally significant for LRT(5) with .
The primate results starkly contrast with those from our parallel analyses on the yeast negative control data. Focusing on the % of genes significant after multiple test correction, we did not observe a single yeast gene at which G.K was significant for LRT(1). This result is consistent with the absence of a distinctive CpG transition rate in yeast. We point out the absence of a significant G.K effect in yeast was despite the yeast data having considerably greater statistical power than the primate data to detect such effects (see supplementary material, Supplementary Material online). Despite this lack of support for distinctive mutagenic processes operating on CpG in yeast, we present the distributions of and from nominally significant yeast genes as a point of comparison with the results from primates (fig. 1).
Evidence for a Distinctive CpG Nonsynonymous Substitution Rate
In protein-coding sequences, the context dependence of mutagenic processes are entangled with the selective constraints operating on the sequences. As a consequence, it was necessary to distinguish the contribution to CpG substitution rate arising from mutation and natural selection. One way in which natural selection may confound estimates of CpG mutation processes or selection profiles specific to CpG-containing codon positions is if the amino acid exchanges that characterize CpG transition events themselves have distinctive rates due to the changes in physico-chemical properties. We specifically evaluated this possibility through defining the α rate parameter which assigned a distinctive nonsynonymous substitution rate common to all MAA exchanges. The role of selection operating on positions whose encoding involved CpG dinucleotides, a subset of the MAA exchanges, was represented by the G.K.ω parameter.
The analyses of primate alignments indicate that a distinct nonsynonymous substitution rate exists at CpG-encoded codons that is not attributable to just selection on amino acid properties. Significant support for distinctive selection affecting MAA was evident at only a small number of genes (table 2 LRT(3)). Of those genes with a significant LRT(3) (for α) after correcting for multiple tests, a subset were also significant (after correcting for multiple tests) for LRT(4). The latter result confirms that the encoding of MAA does contribute to their distinctive evolutionary process in primates. The distributions of parameter MLEs determined from primate alignments (fig. 2) were dominantly < 1, indicating the rate of nonsynonymous CpG transition substitutions is suppressed relative to the common nonsynonymous substitution rate (ω). In contrast, the analyses of yeast alignments suggested the MAA exchanges are not affected by their encoding. A larger percentage of yeast genes were significant for α, LRT(3), supporting a distinctive evolutionary MAA substitution rate (table 2 LRT(3)). After correcting for multiple tests, no yeast genes were significant for G.K.ω, (table 2, LRT(4)). The latter result is consistent with the absence of elevated CpG transition rate in this clade.
Fig. 2.

Selection affecting CpG-containing codons in primates. MLEs were from CNFGTR + G.K + G.K.ω + α for genes with nominally significant LRT(5) (P < 0.05).
The combination of results from primates, which methylate their DNA, and from yeast, which do not methylate their DNA, endorse the role of 5mC in affecting distinctive position-specific selection at CpG-containing primate codons.
Genes with Significant Suppression of Nonsynonymous CpG Changes Are Enriched in OMIM Disease-Causing Genes
The suppression of nonsynonymous CpG changes in primate suggests functional significance at such CpG positions, which increases the likelihood of disorders if mutations occur. This hypothesis was tested from the frequencies of disease association in CpG effect and non-CpG effect genes. CpG effect genes were those where the overall rate of CpG transitions was elevated (significant LRT(1) with > 1) but the rate of nonsynonymous CpG transitions was suppressed (significant LRT(5) with < 1). Among sampled OMIM genes, 1,361 genes are CpG effect genes with 321 genes being disease associated. Accordingly, the sampled OMIM genes were classified into four categories (table 3). Using a Fisher’s Exact test, the genes whose CpG codons exhibited higher mutation rate and stronger purifying selection were significantly enriched for association with human disease.
Table 3.
Genes Identified with Suppression of Nonsynonymous CpG Substitutions Were Enriched in OMIM
| Nondisease Associationa | Disease Associationa | Pb | |
| Non-CpG effectc | 5,026 | 1,242 | |
| CpG effectc | 1,040 | 321 | 1.14 × 10−3 |
Disease association status was defined according to presence/absence of an allelic variant in the OMIM record.
The probability from a Fisher’s Exact test that frequency of CpG effect genes in the disease association class is the same, or lower, than the frequency in the nondisease class.
Both LRT(3) and LRT(5) were nominally significant, Ĝ.K from LRT(3) was > 1 and ω from LRT(5) was < 1.
Classification of Constrained CpG Positions
We developed a novel strategy for the identification of exonic CpG positions likely to encode important functional properties. Our approach uses the position-wise likelihood ratio statistic (LRTP) from LRT(5) (see Materials and Methods). The position-specific statistics are computationally efficient, straightforward to obtain from maximized likelihood functions and allow a relatively straightforward interpretation of the resulting statistic. We use these statistics to identify codon positions that contributed substantially to the significant LRT(5) result. Prior to discussion of the genome-wide distribution of selectively constrained CpG positions, we illustrate its application to the F8 gene for which disease-associated CpG polymorphism have been reported (Krawczak et al. 1998; Antonarakis et al. 2000).
The distribution of LRTp for F8 (fig. 3) discriminates functionally constrained from neutral candidates by strong positive and negative LRTp, respectively. LRT(5) compares the null model of CNFGTR + G.K. against the alternate of CNFGTR + G.K + G.K.ω. As the distribution of from these tests in primates were predominantly < 1, a large LRTp statistic indicates suppression of nonsynonymous CpG substitutions compared with an unrestricted occurrence of synonymous CpG substitutions. Furthermore, codon positions that provide support for the alternate hypothesis should exhibit a positive LRTP. Codon positions that do not support the alternate hypothesis will be more consistent with the null and are expected to exhibit negative LRTP. The “significance” of an individual LRTP was evaluated by comparison to a distribution of LRTp produced from a parametric bootstrap procedure (see Materials and Methods), and the 1st and 99th quantiles for the null distributions LRTP statistics are shown as the red lines above and below LRTP = 0. As expected, for F8, the largest LRTP fell on codon positions at which a synonymous substitution had occurred within the CpG (indicated by star symbol, fig. 3). Only one codon with a synonymous CpG substitutions exhibited LRTP < 0 (fig. 3). Conserved codons for which no substitution was recorded also exhibited a positive LRTP but these were typically smaller in magnitude and a small number of codons did not follow this pattern. The smaller magnitude LRTP for conserved CpG derives from the fact that such a state has two possible “causes”: no mutation occurred and thus there has been no opportunity for natural selection or strong purifying selection. In either case, the absence of change at an otherwise hypermutable base contributes to the < 1. The large positive LRTP at synonymous CpG sites also reflects their contribution to as such changes strongly suggest these dinucleotides have been methylated, have mutated, and that natural selection has prevented nonsynonymous changes from being fixed. Also illustrated in figure 3 is that not all CpG-containing codons fit these basic patterns. The cause for LRTP < 0 are the values of other parameters in the model: the CNFs, the rGTR, ω, and G.K terms. As r terms are affected by other parameters in the model, inclusion of G.K.ω is most noticeably going to affect ω, G.K, and the r(A, G), r(C, T) parameters, potentially decreasing the accuracy of rate estimates for other transition substitutions.
Fig. 3.

Identification of phenotypically influential CpG-containing codons in F8. Horizontal red lines—are the 1st and 99th quantiles of LRTp from data simulated under the null hypothesis; yellow triangle—codons with conserved CpG; magenta star—codons with synonymous CpG substitutions; green diamond—codons with nonsynonymous CpG substitutions.
Based on the detailed inspection of LRTP from F8 and other genes, we defined an ordinal ranking of exonic CpG with respect to their potential impact on human phenotype (table 4). A number of factors limit the value of applying the current statistical modeling framework to quantifying the potential phenotypic impact of CpG mutations. First, the statistical power to detect suppression of nonsynonymous CpG substitutions differs between genes. These differences can arise from low divergence rates between species, low CpG frequency, and/or a short gene length. Each of these will have the effect of reducing the number of detectable events from comparisons between the primate species. Second, the statistical modeling framework has the limitation of not counting events occurring within CpG that span codon boundaries. For these reasons, we devised an ordinal ranking scheme to provide a relative indicator of the potential for mutations within a human CpG to contribute to disease. A higher rank indicates a CpG that has likely been historically subjected to strong purifying selection and thus a genetic variant at the CpG is considered more likely to be deleterious. CpG within genes showing a nominally significant LRT(5) were given stronger weight than other genes because this test directly assesses the existence of distinctive selection opposing CpG variation. Genes that fail this test, however, should not be entirely discounted as the failure may reflect low statistical power as discussed above. Within a gene, any CpG that has undergone a synonymous substitution is weighted more strongly than a CpG which has not. Obviously in the latter case, the potential for such positions to exhibit genetic variation depends on whether a CpG remains within humans. Conserved CpG exonic positions should be ranked highly but less than those exhibiting synonymous change. Codons exhibiting nonsynonymous variation should be given a low ranking, especially if changes occurred in multiple lineages. The results of this classification are available as a supplementary data file (Supplementary Material online) for display as a custom track on the UCSC genome browser.
Table 4.
Ranking of Exonic CpG for Potential Deleterious Impact of Genetic Variation
| Rank | Criteria | Numbera |
| 1 | No CpG in humans | 185,919 |
| 2 | CpG in humans but nonsynonymous changes in other species or nonsynonymous in humans at a codon position not within the CpG | 222,582 |
| 3 | CpG in humans with nonsynonymous SNP, CpG is minor allele | 5,630 |
| 4 | Conserved CpG | 359,388 |
| 5 | CpG in humans with synonymous change in human or other species or with SNP, CpG is major allele | 123,896 |
| 6 | Conserved CpG within gene showing a nominally significant LRT(5) | 102,738 |
| 7 | CpG in humans with synonymous change in human or other species within gene showing a nominally significant LRT(5) or with human SNP where CpG is major allele | 42,057 |
Note.—From 14,348 genes, there were 1,042,210 exonic CpG. Number is the total of CpG at each rank.
CpG within humans satisfying criteria.
Discussion
Our analyses firmly support a 5mC-derived shift in mutation-selection balance in primate protein-coding genes indicating that CpG-containing codons play a disproportionate role in human disease. The elevation of CpG transitions was identified as a general feature affecting primate protein-coding gene evolution. Moreover, strong purifying natural selection specifically targeting CpG-containing codons was also evident for a substantial fraction of human genes. We further established that genes subjected to significant purifying selection opposing CpG variation are enriched for disease association. The robustness of these results was endorsed by the absence of equivalent signals in our parallel analyses of the yeast clade which, since it lacks 5mC, served as our biological control.
After correcting for multiple tests, our analyses of the yeast data did not provide support for a distinctive mutagenic influence associated with CpG dinucleotides (table 2 LRT(1),LRT(2)). For genomes free of CpG methylation with no impact from other evolutionary forces on CpG sites, these dinucleotides should evolve at the same rate as that of the background. We therefore expected few, if any, yeast genes to display statistical support for the term. Consistent with this, the proportion of yeast genes significant after multiple test correction was 0 (table 2 LRT(1),LRT(2)). Coupled with the greater statistical power of yeast data (see supplementary material, Supplementary Material online), these results confirm no mutagenic distinction of the CpG dinucleotide in yeast. The absence of support for a distinctive role for CpG dinucleotides in our negative biological control affirms the suitability of the statistical modeling framework for evaluating methylation-associated phenomena in the primate clade where DNA methylation does occur.
Our analyses of yeast identified distinctive evolutionary rates for the MAA. That was predominantly less than 1, indicates that the amino acid exchanges involving the MAA are generally less permissive than the common nonsynonymous substitution rate (represented by ω) affecting all amino acids. Importantly, the α parameter conferred the largest improvement to model fit in yeast for any parameter (table 2 LRT(3)). After the multiple test correction, none of the genes significant for α were also significant for G.K.ω. As our hypothesized suppression of CpG substitutions by natural selection will also lead to parameter estimates for G.K.ω < 1, a common MAA effect on nonsynonymous CpG substitution rates is potentially confounding and needs to be considered in analyses of genomes where methylation occurs.
Results from our analyses of primate genes were strikingly different to those from yeast, supporting a systemic influence of hypermutable CpG across primate genes. That transitions are the major methylation-induced mutations was confirmed by a substantially elevated rate of CpG transitions over the background for the majority of primate genes. As spontaneous deamination of 5mC results in T, CpG transitions can simply arise either from unrepaired T/G mismatches being incorporated during DNA replication or “incorrect” repair on the opposite strand, which produces a T/A pair from the original C/G pair. Both situations create permanent transition mutations in the cell. Thus, the observed dominant transition effect on CpGs is concordant with the expected methylation-derived mutations.
Although far fewer primate genes showed support for an elevated CpG transversion rate, the number of genes significant after correcting for multiple tests was still substantial (table 2 LRT(2)) and suggests either confounding with selective influences or additional mutagenic outcomes other than just 5mC deamination. One possible mutagenic cause is base misincorporation during DNA repair. Compared with DNA replication, DNA repair processes tend to be error prone, possibly due to the use of low-fidelity DNA polymerases (Johnson et al. 2000). Thus, the high 5mC deamination rate may be accompanied with a high probability of complete replacement during DNA repair and lead to a high CpG transversion rate. The other possibility is that CpG sites are DNA damage hotspots irrespective of methylation status. For instance, CpG has been identified as a preferred target of oxidative damage (Radford and Lobachevsky 2008). Oxidative reactions are one of the major mechanisms of DNA damage and some of these predominantly produce transversions, for example, 8-OH-dG (Cheng et al. 1992). Thus, an elevated CpG transversion rate may reflect oxidative mechanisms.
Our analyses of primate genes support the hypothesis that CpG-encoded MAA are more likely to affect trait evolution than their non-CpG counterparts (table 1). The fraction of primate genes exhibiting significant support for strong purifying selection operating on CpG-encoded amino acids was approximately an order of magnitude larger than that evident in yeast. Like the case for yeast, α was predominantly less than 1 in primates, suggesting that the amino acid exchanges involved in MAA are generally less permissive than other amino acids. Even after taking account of the common selective constraints against the MAA modeled by α, CpG codons were further distinguished from non-CpG codons by stronger purifying selection (fig. 2). The decreased rate of CpG nonsynonymous exchanges is consistent with our hypothesis that the CpG-encoded MAA occupy functionally significant positions. This conjecture was supported by the significant enrichment of CpG-effected genes with diseases (table 3).
Motivated by these results, we devised an ordinal ranking system (table 4) for classifying CpG dinucleotides within the human genome. An ordinal approach was required because the codon substitution model classes treat neighboring codons as independent and thus CpG dinucleotides that span codon boundaries (e.g., NNC GNN) are not considered. Furthermore, a number of genes were excluded from our substitution modeling analyses due to incomplete data in the other species or they were too short. In addition to reducing statistical power to detect CpG effects, this model property precludes formal estimation of nucleotide position-wise statistics (e.g., LRTp). The question of how to model transition probabilities on neighboring codons within a CpG and non-CpG dinucleotide needs further examination.
Application of the scoring system identified 1,042,210 CpG’s within the human genome and identified 144,795 CpG with the highest risk (ranks 6 and 7). We emphasize that the SNP-based ranking system only utilizes information regarding CpG-related processes. As a result, SNPs that do not result from CpG decay can receive a low rank and yet may cause disease. Such is the case for rs28935207 within the F8 gene. We also note that while the ranking system drew on whether a gene returned a significant result for the LRT(5), this test is conservative because it is affected by both the length of the gene and the divergence between orthologs. The number of genes showing a significant CpG effect was limited by statistical power in primates. Because the sampled yeast genes diverge further than primate genes, the statistical power is greater for the yeast data than that for primates (see supplementary material, Supplementary Material online). It was clear that when the CpG transition rate is 2-fold higher than the background, it has a 98% chance of being detected in yeast but only an ∼60% chance in primates. In addition, sequential LRTs further reduce the number of significant genes at later hypothesis tests. A compensation for the lack of statistical power in primates is the alignment length. With more columns to be counted, larger genes are more likely to be significant in LRTs. This was evident in that CpG-affected genes with (or without) disease association were generally larger than overall OMIM allelic genes (data not shown). This limitation will result in a failure to detect some smaller genes with a significant CpG effect. Despite these limitations, however, our results indicate the ordinal classification system, which is applied independent of alignment properties, is robust. Examination of the relationship between the highest ranked CpG dinucleotides and disease association was also strikingly significant (P < 1 × 10−15, see supplementary material, Supplementary Material online).
Our estimate of the magnitude of influence exerted by CpG-related substitutions on nonsynonymous variation in primate genomes is likely to be an underestimate. The statistical power to detect an elevated CpG transition rate was much greater in yeast than primates (supplementary material, Supplementary Material online). In light of this, our primate analyses likely exhibit a high Type II error rate. Consequently, many of the genes that were nominally significant (table 2, LRT(1), LRT(5)) but not significant after the multiple test correction, may be truly CpG effect genes. Resolving the classification of individual genes may be possible with the increasing availability of other primate genomes. An increase in number of taxa would refine both the classification of CpG effect genes and also the assignment of ranks to individual human CpG dinucleotides. For the analyses of nonprimate lineages, comparable evaluations of statistical power to those we undertook (supplementary material, Supplementary Material online) will be required.
As the CpG dinucleotide is overrepresented in human SNPs, genetic variants within this dinucleotide are expected to be abundant in genome resequencing studies. By integrating the evolutionary history at CpG positions, we have endeavored to improve the prediction of functionally significant genetic variants. Our results specifically identify a large number of genomic positions at which genetic variants are candidates for disease association studies. Utilization of this classification should improve the statistical power of disease association studies.
Web Resources
PyCogent, for genome analysis, http://pycogent.sourceforge.net/.
The Ensembl genome browser, http://www.ensembl.org.
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/.
Alternative symbols from the OMIM gene table, http://www.ncbi.nlm.nih.gov/Omim/Index/genetable.html.
The multiple sequence alignments from the Saccharomyces Genome Database, ftp://genome-ftp.stanford.edu/pub/yeast/sequence/fungal_genomes/Multiple_species_align/other/MIT_Spar_Sbay_Smik_Scer.
Supplementary Material
Supplementary material is available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Dr V. B. Yap for comments on an earlier draft, Dr James Cai for assistance in obtaining the yeast data and acknowledge funding to G.A.H. from the National Health and Medical Research Council of Australia.
References
- Antonarakis SE, Krawczak M, Cooper DN. Disease-causing mutations in the human genome. Eur J Pediatr. 2000;159(Suppl 3):S173–S178. doi: 10.1007/pl00014395. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, McKusick VA. OMIM passes the 1,000-disease-gene mark. Nat Genet. 2000;25(1):11. doi: 10.1038/75497. [DOI] [PubMed] [Google Scholar]
- Arndt PF, Hwa T, Petrov DA. Substantial regional variation in substitution rates in the human genome: importance of GC content, gene density, and telomere-specific effects. J Mol Evol. 2005;60:748–763. doi: 10.1007/s00239-004-0222-5. [DOI] [PubMed] [Google Scholar]
- Bulmer M. Neighboring base effects on substitution rates in pseudogenes. Mol Biol Evol. 1986;3(4):322–329. doi: 10.1093/oxfordjournals.molbev.a040401. [DOI] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G—T and A—C substitutions. J Biol Chem. 1992;267(1):166–172. [PubMed] [Google Scholar]
- Cooper DN, et al. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat. 2010;31(6):631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78(2):151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274(5673):775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287(5782):560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Smith NGC, Webster MT. Mutation rate variation in the mammalian genome. Curr Opin Genet Dev. 2003;13(6):562–568. doi: 10.1016/j.gde.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17(6):368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11(5):725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. Principles of population genetics. Sunderland (MA): Sinauer Associates; 2007. [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Statist. 1979;6(2):65–70. [Google Scholar]
- Hubbard TJP, et al. Ensembl 2009. Nucleic Acids Res. 2009;37(Database issue):D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttley GA. Modeling the impact of DNA methylation on the evolution of BRCA1 in mammals. Mol Biol Evol. 2004;21(9):1760–1768. doi: 10.1093/molbev/msh187. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase eta. J Biol Chem. 2000;275(11):7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423(6937):241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Knight R, et al. PyCogent: a toolkit for making sense from sequence. Genome Biol. 2007;8(8):R171. doi: 10.1186/gb-2007-8-8-r171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M, Ball EV, Cooper DN. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet. 1998;63:474–488. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, et al. Positional conservation and amino acids shape the correct diagnosis and population frequencies of benign and damaging personal amino acid mutations. Genome Res. 2009;19(9):1562–1569. doi: 10.1101/gr.091991.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanave C, Preparata G, Saccone C, Serio G. A new method for calculating evolutionary substitution rates. J Mol Evol. 1984;20(1):86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Williams EJ, Hurst LD. Local similarity in evolutionary rates extends over whole chromosomes in human-rodent and mouse-rat comparisons: implications for understanding the mechanistic basis of the male mutation bias. Mol Biol Evol. 2001;18(11):2032–2039. doi: 10.1093/oxfordjournals.molbev.a003744. [DOI] [PubMed] [Google Scholar]
- Lindsay H, Yap VB, Ying H, Huttley GA. Pitfalls of the most commonly used models of context dependent substitution. Biol Direct. 2008;3:52. doi: 10.1186/1745-6150-3-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 2005;102(0027–8424):10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matassi G, Sharp PM, Gautier C. Chromosomal location effects on gene sequence evolution in mammals. Curr Biol. 1999;12(9):786–791. doi: 10.1016/s0960-9822(99)80361-3. [DOI] [PubMed] [Google Scholar]
- Miller MP, Kumar S. Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet. 2001;10(21):2319–2328. doi: 10.1093/hmg/10.21.2319. [DOI] [PubMed] [Google Scholar]
- Misawa K, Kikuno RF. Evaluation of the effect of CpG hypermutability on human codon substitution. Gene. 2009;431(1–2):18–22. doi: 10.1016/j.gene.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11(5):715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Pond SK, Muse SV. Site-to-site variation of synonymous substitution rates. Mol Biol Evol. 2005;22(12):2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Proffitt JH, Davie JR, Swinton D, Hattman S. 5-methylcytosine is not detectable in Saccharomyces cerevisiae DNA. Mol Cell Biol. 1984;4(5):985–988. doi: 10.1128/mcb.4.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowicz PD, et al. Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res. 2003;13(12):2658–2664. doi: 10.1101/gr.1784803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford IR, Lobachevsky PN. Clustered DNA lesion sites as a source of mutations during human colorectal tumourigenesis. Mutat Res. 2008;646(1–2):60–68. doi: 10.1016/j.mrfmmm.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Schmidt S, et al. Hypermutable non-synonymous sites are under stronger negative selection. PLoS Genet. 2008;4(11):e1000281. doi: 10.1371/journal.pgen.1000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NGC, Webster MT, Ellegren H. Deterministic mutation rate variation in the human genome. Genome Res. 2002;12(9):1350–1356. doi: 10.1101/gr.220502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer SS, Scaringe WA, Hill KA. Human germline mutation in the factor ix gene. Mutat Res. 2001;487(1–2):1–17. doi: 10.1016/s0921-8777(01)00108-2. [DOI] [PubMed] [Google Scholar]
- Sved J, Bird A. The expected equilibrium of the cpg dinucleotide in vertebrate genomes under a mutation model. Proc Natl Acad Sci U S A. 1990;87:4692–4696. doi: 10.1073/pnas.87.12.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S, Pfeifer GP. Complete and tissue-independent methylation of cpg sites in the p53 gene: implications for mutations in human cancers. Oncogene. 1995;10(8):1493–1499. [PubMed] [Google Scholar]
- Wolfe KH, Sharp PM, Li WH. Mutation rates differ among regions of the mammalian genome. Nature. 1989;337(6204):283–285. doi: 10.1038/337283a0. [DOI] [PubMed] [Google Scholar]
- Wong WSW, Yang Z, Goldman N, Nielsen R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168(2):1041–1051. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap VB, Lindsay H, Easteal S, Huttley G. Estimates of the effect of natural selection on protein coding content. Mol Biol Evol. 2010;27(3):726–734. doi: 10.1093/molbev/msp232. [DOI] [PMC free article] [PubMed] [Google Scholar]