

Abstract
The poly(A)-binding protein (PABP) recognizes the 3′ mRNA poly(A) tail and plays an essential role in eukaryotic translation initiation and mRNA stabilization/degradation. PABP is a modular protein, with four N-terminal RNA-binding domains and an extensive C terminus. The C-terminal region of PABP is essential for normal growth in yeast and has been implicated in mediating PABP homo-oligomerization and protein–protein interactions. A small, proteolytically stable, highly conserved domain has been identified within this C-terminal segment. Remarkably, this domain is also present in the hyperplastic discs protein (HYD) family of ubiquitin ligases. To better understand the function of this conserved region, an x-ray structure of the PABP-like segment of the human HYD protein has been determined at 1.04-Å resolution. The conserved domain adopts a novel fold resembling a right-handed supercoil of four α-helices. Sequence profile searches and comparative protein structure modeling identified a small ORF from the Arabidopsis thaliana genome that encodes a structurally similar but distantly related PABP/HYD domain. Phylogenetic analysis of the experimentally determined (HYD) and homology modeled (PABP) protein surfaces revealed a conserved feature that may be responsible for binding to a PABP interacting protein, Paip1, and other shared interaction partners.
Eukaryotic mRNA translation initiation is an intricate, exquisitely controlled process involving assembly of a large multiprotein-RNA complex that directs the ribosome to the initiation codon. Like transcription initiation, translation initiation represents a critical, rate-limiting step at which eukaryotic gene expression is regulated in response to developmental/environmental signals (1). Eukaryotic mRNAs (excluding organellar mRNAs) are distinguished by the presence of a 5′ 7-methyl-G cap structure and a 3′ poly(A) tail that synergize in stimulating translation (reviewed in refs. 2 and 3).
In the most general case, protein synthesis is stimulated by protein–protein interactions that serve to connect the 5′ 7-methyl-G cap to the poly(A) tail found at the 3′ ends of most cellular mRNAs. Protein synthesis begins with cap recognition by eukaryotic initiation factor 4E (eIF4E or cap-binding protein; reviewed in ref. 4). eIF4E is a component of the eIF4F complex, which also includes eIF4G, a bridging protein, and eIF4A, an ATP-dependent RNA helicase. After cap recognition by eIF4E, eIF4G directs the 40S ribosomal subunit (plus methionyl tRNA and various accessory factors) to the appropriate initiation codon, where it is joined by the 60S ribosomal subunit to form the 80S ribosomal initiation complex.
Ribosomal recruitment and translation initiation have been shown to be stimulated by the poly(A) tail and its associated protein, the poly(A)-binding protein (PABP) (5–7). This stimulation is effected by a direct interaction between eIF4G and PABP, giving rise to a closed-loop model for translation initiation (reviewed in ref. 2). mRNA circularization has been demonstrated in vitro by using recombinant yeast proteins (8) and is thought to increase the efficiency of translation by promoting reinitiation of terminating ribosomes on the same mRNA. Consequently, capped, polyadenylated mRNAs are translated more efficiently than those lacking one or both terminal modifications (reviewed in ref. 3). In higher eukaryotes, PABP also seems to stimulate translation initiation indirectly with the aid of a PABP interacting protein (Paip1) (9). Paip1 interacts with eIF4A, and overexpression of Paip1 increases the rate of translation initiation, suggesting that there at least two ways to close the loop between the 5′ and 3′ ends of mRNAs.
Interactions between the 5′ and 3′ ends of mRNA also influence mRNA stability. The poly(A) tail stabilizes most mRNAs via association with the poly(A)-binding protein (reviewed in refs. 10 and 11). In yeast, PABP contributes to mRNA stability by inhibiting removal of the 7-methyl-G cap, because this decapping step is a prerequisite to the degradation of a number of mRNAs (12, 13). In higher eukaryotes, PABP and Paip1 are part of a multiprotein complex that stabilizes the c-fos mRNA, presumably via mRNA circularization (14). Given its critical functions in promoting translation initiation and mRNA stabilization, it is not surprising that the poly(A)-binding protein gene is essential in yeast (15).
All available PABP sequences (over 30 to date) include four conserved RNA-binding domains known as RNA-recognition motifs (RRMs), arranged in tandem, followed by a C-terminal region of variable length (Fig. 1; refs. 16–18). In vivo studies in yeast indicate that all four RRMs alone are not sufficient for normal growth (19), underscoring the functional importance of the C-terminal portion of PABP.
Figure 1.

Domain organization of PABP and HYD protein families. (A) Sequence alignment of the conserved C-terminal domain of PABP (Homo sapiens, Xenopus laevis, Drosophila melanogaster, Caenorhabditis elegans, Schizosaccharomyces pombe, Saccharomyces cerevisiae, Nicotiana tabacum), the orthologous region of the three HYD proteins (H. sapiens, Rat norvegicus, D. melanogaster), and a small ORF from the Arabidopsis thaliana genome. Sequence conservation among family members is color-coded by a yellow-green continuum; pale yellow represents low homology, dark green represents identity. (B) The four RRM domains of PABP (Top) are depicted by red boxes and are connected by interdomain linkers depicted as lines. The conserved C-terminal portion of the protein is shown as a blue ellipse. The location of the common coxsackie and poliovirus 2A protease site is indicated by the blue arrow. The HYD protein's domain organization (Middle) depicts the PABP-like region as a blue ellipse, and the HECT domain as a yellow hexagon. The PABP-like region of the A. thaliana small ORF (Bottom) is also represented as a blue ellipse.
This C-terminal region (266 aa in human PABP) has been attributed numerous biochemical functions, including homo-oligomerization (20) and protein–protein interactions. Amino acid sequence alignments demonstrate the presence of a highly conserved 60-residue segment within the PABP C terminus (Fig. 1). Although the biological significance of this highly conserved portion of PABP is not known, deletion of this conserved C-terminal motif (PABP-C) in yeast containing a PABP-MS2 fusion protein instead of wild-type PABP is lethal (13). Further support for the importance of the PABP C-terminal motif arises from the fact that the 2A proteases of both coxsackievirus and poliovirus cleave PABP between RRM4 and the C-terminal motif (Fig. 1B), thereby diminishing translation initiation in vitro (21, 22).
A Ψ blast (23) search demonstrated that the conserved C-terminal motif of PABP occurs in a second family of proteins implicated in development [named HYD for hyperplastic discs in Drosophila melanogaster (24), EDD in human (25), and 100-kDa protein in rat (26)]. The human homologue of HYD and human PABP are 56% identical over this region.
We have determined the x-ray structure of the PABP-like region of the human HYD protein (HYD-P) at atomic resolution. The structure reveals a domain with four α-helices wound in a loose right-handed supercoil. Comparative protein structure modeling was used to derive high-quality model structures for four HYD homologues and 30 PABP C-terminal domain orthologs, plus another structurally similar ortholog encoded by the Arabidopsis thaliana genome. Analysis of the molecular surfaces of HYD and PABP family members revealed a phylogenetically conserved surface feature that may be responsible for protein–protein interactions. Finally, in vitro binding assays demonstrated that both the PABP-C and the orthologous HYD-P domains can bind Paip1, suggesting that the two domains may share one or more interaction partners.
Materials and Methods
Protein Preparation and Crystallization.
cDNA corresponding to residues 533–633 of human PABP was inserted into the pet28a (Novagen) expression vector. The resulting N-terminal hexa-histidine fusion protein was expressed in Escherichia coli [BL21 (DE3)] and purified to homogeneity by Ni2+ ion-affinity and size-exclusion chromatographies. The purified protein was treated with trypsin and Glu-C proteases at mass ratios of protease to protein of 1:100, and mass spectrometry was used to identify cleavage at Glu-554, Lys-617, and Glu-618 (data not shown). Crystallization trials with PABP(533–619) and PABP(553–619) yielded small, poorly diffracting crystals.
A human cDNA KIAA0896 (residues 1568–2798 of the human HYD homologue) was obtained from the Kazusa DNA Research Institute (Chiba, Japan). Residues 2391–2455 of HYD were expressed in E. coli by using the same strategy as for PABP-C. Ni2 + ion-affinity and gel filtration chromatography yielded purified selenomethionine (Se-Met) HYD-P. Matrix-assisted laser desorption mass spectrometry confirmed that the protein was purified to homogeneity (observed mass = 7,979 ± 7; predicted mass = 7,979.7). Se-Met HYD-P was dialyzed against storage buffer (100 mM NaCl/20 mM Hepes, pH 7.25) and concentrated to 14 mg/ml. Crystallization trials yielded Se-Met HYD-P crystals via hanging-drop diffusion at 4°C against 34% 1,4-dioxane (unbuffered). Pyramidal crystals in the primitive orthorhombic space group p212121 with one molecule/asymmetric unit (unit cell: a = 32.96 Å, b = 38.12 Å c = 51.08 Å; Matthews coefficient = 2.0 Å3/Da) appeared overnight. Crystal cryoprotection was achieved by adding ethylene glycol to a final concentration of 17% (vol/vol).
HYD-P Structure Determination.
All diffraction data were collected at Beamline X9B at the Brookhaven National Laboratory National Synchrotron Light Source and Sector 19 at the Advanced Photon Source under standard cryogenic conditions. A Multiwavelength Anomalous Diffraction (MAD) experiment was conducted by recording diffraction data at three x-ray wavelengths (Se absorption edge inflection point, peak, and high-energy remote) with one high-quality crystal. Data were processed by using denzo/scalepack (27). All four possible Se sites were located with SnB (28). Experimental MAD phases were estimated at 1.7 Å resolution by using mlphare (29). After density modification, the resulting phases were input into arp/warp (30) for automated structure determination. Residues 2392–2452 of HYD were built without manual intervention. Iterative rounds of model adjustment/refinement were performed with o (31) and cns (32), followed by shelx (33). The current refinement model consists of residues 2392–2452 and 112 water molecules, giving a working R factor of 14.3% and a free R value of 17.1% (Table 1). procheck (34) showed no unfavorable (φ,ψ) combinations; main-chain and side-chain stereochemical parameters were better than average (overall G value = 0.4)
Table 1.
Refinement statistics for native data, |F| > 4σ|F|
| Resolution, Å | 20.0–1.04 |
| Completeness, % | 85.5 |
| R factor | 0.143 |
| Free R factor | 0.171 |
| rmsd, | |
| Bond lengths, Å | 0.0108 |
| Bond angles, ° | 2.06 |
Root-mean-square deviation (rmsd) bond lengths and rmsd bond angles are the respective root-mean-square deviations from ideal values. rmsd Free R factor was calculated with 5% of data omitted from the structure refinement.
Paip1 Binding Analysis.
A fragment of the gene encoding human Paip1 (corresponding to amino acids 113–480) was subcloned into pGEX6P1 (Amersham Pharmacia). A glutathione S-transferase (GST)-Paip1(113–480) fusion and GST alone were expressed in E. coli, purified by glutathione-Sepharose chromatography, and dialyzed against binding buffer (100 mM NaCl/20 mM Hepes, pH 7.3/1 mM DTT/0.1% Triton X-100). His-tagged HYD-P(2391–2455) and PABP-C(541–619) were prepared as described above. GST (30 μg) and GST-Paip1 (42 μg) were each immobilized on 40 μl of glutathione-Sepharose resin (Amersham Pharmacia), and the unbound protein was washed away. To 20 μl of both the GST-resin and GST-Paip1-resin mixture were added 7 μg of PABP-C or 6 μg of HYD-P. Binding reactions were diluted with 100 μl of binding buffer and incubated at 22°C for 30 min. After washing with 100 μl of binding buffer three times, the resin was harvested by centrifugation and bound proteins were detected by SDS/PAGE electrophoresis.
Results
Crystallization and Structure Determination.
Limited proteolysis combined with mass spectrometry (35) identified a proteolytically stable domain within the C-terminal region of human PABP (Fig. 1A). Crystallization trials with human PABP(553–619) proved unsuccessful, yielding small, poorly diffracting crystals. In parallel, the orthologous region of the human HYD protein was subcloned, expressed, and purified to homogeneity. HYD(2391–2455) yielded high-quality crystals with one protomer/asymmetric unit. The structure of the HYD-P domain was determined via MAD (36) at 1.7 Å resolution. The current refinement model has an R factor of 14.3% and a free R value of 17.1% (37) at 1.04 Å resolution, with excellent stereochemistry. (See Table 2 and Materials and Methods for a complete description of structure determination.)
Table 2.
Statistics of the crystallographic analysis
| Data set | Resolution Å | Reflections (measured/unique) | Completeness % (overall/outer shell) | Rsym % (overall/outer shell) | Phasing power Iso (acentric/centric) | Rcullis Ano/Iso |
|---|---|---|---|---|---|---|
| MAD Structure determination (4 Se sites) | ||||||
| λ1 = (0.97926 Å) | 30.0–1.60 | 104,037/15,968 | 97.0/77.2 | 3.2/8.4 | — | 0.24/— |
| λ2 = (0.97878 Å) | 30.0–1.60 | 65,171/15,920 | 96.6/75.8 | 3.1/7.6 | 3.09/2.05 | 0.23/0.40 |
| λ3 = (0.96112 Å) | 30.0–1.60 | 53,600/16,106 | 97.7/80.8 | 2.7/5.6 | 4.62/3.39 | 0.29/0.30 |
| Native data collection | ||||||
| λ = (1.1000 Å) | 20.0–1.04 | 460,042/31,619 | 99.7/98.0 | 5.9/33.6 | — | — |
Rsym = Σ|I − 〈I〉|/∑I, where I = observed intensity, 〈I〉 = average intensity obtained from multiple observations of symmetry related reflections. Phasing power = root-mean-square deviation (rmsd) (|FH|/E), |FH| = heavy atom structure factor amplitude and E = residual lack of closure. MAD figure of merit (28-1.7 Å) 0.92.
HYD-P Is a Right-Handed α-Helical Supercoil.
The three-dimensional structure of the PABP-C-like domain of HYD is illustrated in Fig. 2A. The polypeptide chain folds into a compact domain with four α-helices (H1-H4) wound in a loose right-handed supercoil with dimensions of 31 Å (length) × 25 Å (height) × 19 Å (width) and 11 Å radius. The interhelical angles are 144° (H1-H2), 136° (H2-H3), and 118° (H3-H4), and the average interhelical distance parallel to the supercoil axis is 11 Å. Structure-based sequence alignments of various HYD and PABP homologues show significant pairwise identities (44%–96%) and demonstrate that the hydrophobic core (Fig. 3) is conserved, whereas insertions map to a surface loop (H1-H2). We conclude, therefore, that the phylogenetically conserved regions of all known members of the HYD and PABP families share the same three-dimensional structure illustrated in Fig. 2 (38). To the best of our knowledge, the structure of HYD-P represents an additional protein fold. A search with the DALI server (39) revealed a maximum Z score of 4.8, obtained with a fragment of an unrelated α-helical protein (RNA polymerase primary sigma factor, PDB accession code 1sig). There is no evidence of HYD-P oligomers within our crystals, and HYD-P(2391–2455) is monomeric in aqueous solution (data not shown).
Figure 2.
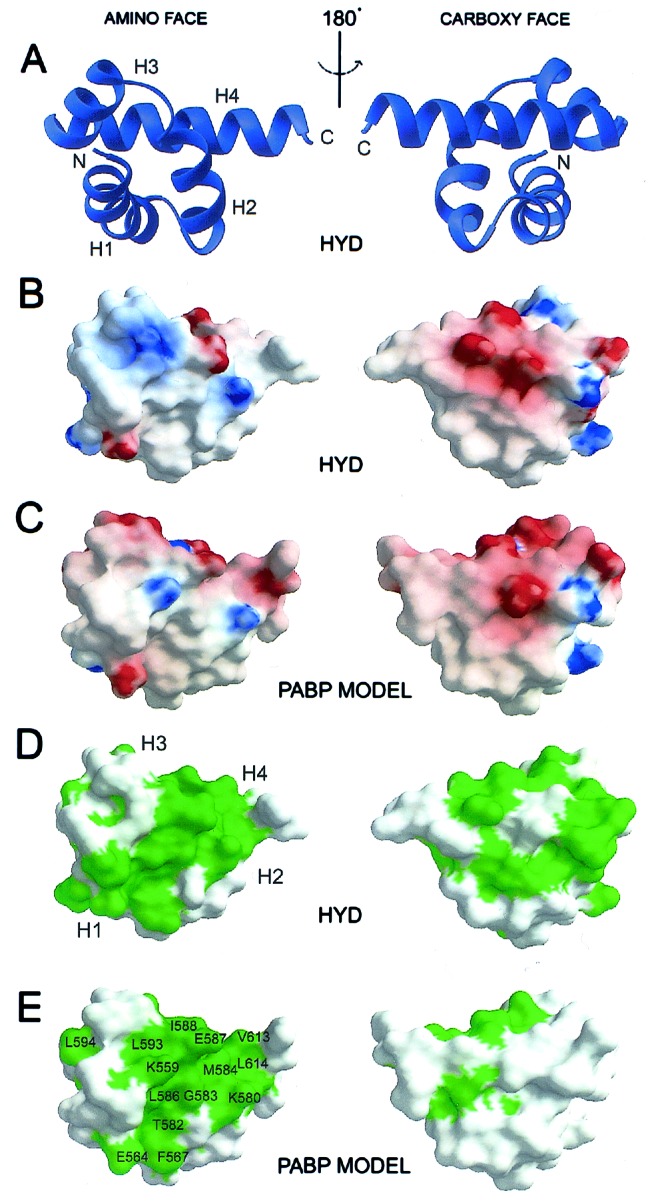
Structure and surface properties of the amino (Left) and carboxyl (Right) faces of HYD-P and homology model of PABP-C. (A) ribbons (51) drawing of the HYD-P domain, where the four α-helices are labeled H1, H2, H3, and H4, as are the N and C termini. (B and C) grasp (52) representations of the chemical properties of the solvent-accessible molecular surface of HYD-P (B) and PABP-C (C) calculated by using a water probe (radius = 1.4 Å.). The surface electrostatic potential is color-coded red and blue, representing electrostatic potentials <−10 to >+10 kBT, where kB is the Boltzmann constant and T is the temperature. The calculations were performed with an ionic strength of 0 and dielectric constants of 80 and 2 for solvent and protein, respectively (53). (D and E) Depiction of the molecular surfaces of HYD-P (D) and PABP-C (E), color-coded green for phylogenetic conservation. Conserved amino-face residues of PABP-C are identified by a one-letter code.
Figure 3.

The hydrophobic core of HYD-P. ribbons (51) stereodrawing of residues that constitute the hydrophobic core of the HYD-P domain. α-helices are labeled H1, H3, and H4 (see Fig. 2). Amino acids, labeled in gold, are identified by a one-letter code.
Comparative Protein Structure Modeling.
All 30 extant PABP orthologs and four other known HYD proteins were modeled by using modeller (40) with the HYD-P crystal structure as template. Comparative protein-structure modeling involves the alignment of the modeling template with the sequence of a homologue for which no structural information is available. Satisfaction of spatial restraints obtained from the alignment is used to construct a three-dimensional structural model of the homologue. A quantitative energetic analysis of the model can then be performed to analyze its quality. Model scores in excess of 0.7 (on a scale of 0–1) connote useful models. All calculated PABP and HYD models surpassed this threshold. In addition to modeling known PABP and HYD proteins, we constructed a high-quality model for an orthologous 103 residue protein (42% identity with HYD-P), encoded by a small ORF (GI = 9229338) within the A. thaliana genome (Fig. 1).
HYD-P and PABP Share a Conserved Hydrophobic Surface Feature.
The solvent-accessible molecular surface of the human HYD illustrated in Fig. 2 B–E, with similar views of the surface of our homology model of the conserved C-terminal domain of human PABP. The electrostatic properties of the two molecular surfaces are remarkably similar. The two N-terminal surfaces (amino face—Fig. 2 B and C, Left) are largely hydrophobic with two basic patches, whereas the C-terminal surfaces (carboxyl face—Fig. 2 B and C, Right) are acidic, basic, and hydrophobic. Fig. 2 D and E, Left demonstrate that the amino faces of HYD-P and PABP-C are conserved within their respective families. Further analyses revealed that the solvent-accessible residues on the green color-coded N-terminal surfaces of α-helices H1, H2, and H4 are also conserved between the HYD and PABP protein families (Fig. 1A). This finding suggests that the two proteins share at least one common interaction partner. The limited phylogenetic conservation of the C-terminal faces of the two proteins suggests that this region is probably not involved in common biochemical functions.
The PABP-C and HYD-P Domains Both Interact with Paip1.
Given that human Paip1 interacts with Xenopus laevis PABP somewhere within the C-terminal 237 residues (41), we tested Paip1 for binding to PABP-C and HYD-P. Fig. 4 demonstrates that Paip1 interacts with both protein domains in vitro. Presumably the conserved amino faces of the structurally similar domains contribute, at least in part, to the observed interactions. The relative stoichiometry of the binding reactions was difficult to assess because PABP-C and HYD-P are not well stained by Coomassie blue in SDS/PAGE gels. We did not observe any binding of Paip1 to two unrelated eukaryotic translation initiation factors, eIF5 or eIF6 (data not shown).
Figure 4.

The PABP-C and HYD-P domains bind to Paip1 in vitro. Coomassie blue-stained SDS/PAGE gel of GST-affinity assays shows binding of Paip1 to both HYD-P (lane 2) and PABP-C (lane 3) with appropriate negative controls (lanes 1 and 4).
Discussion
The multidomain, modular nature of PABP is essential to its many biochemical functions. The four N-terminal RNA recognition motifs (RRMs) serve primarily to tether multiple PABP molecules to the 5′ poly(A) tail of mRNAs specificity (20). Additionally, the N-terminal two RRMs mediate interaction with eIF4G (19, 42) and, partly, with Paip1 (41). The C terminus of PABP has emerged as an important protein–protein interaction domain that recruits other proteins to the poly(A) tail. These interaction partners include mediators of translation termination (eRF3) (43), translation enhancement (Paip1) (9), mRNA stabilization (hnRNPE) (44), polyadenylation (45), and viral replication (potyvirus RNA polymerase) (46). We have identified a conserved domain within this C terminus that is sufficient for interaction with Paip1. This domain has also been shown to be sufficient for binding to the potyvirus RNA polymerase (46) and may serve to assist in binding to one or more of the PABP-binding partners described above. With comparative protein structure modeling, we have identified a conserved surface feature that may be responsible, at least in part, for these interactions. Mutational analysis will further elucidate the importance of this conserved feature to PABP function.
It is remarkable that this surface feature is also present in the PABP-like domain of the HYD family of proteins. Moreover, we have shown that the structurally similar HYD-P domain can bind Paip1 and may well interact with other binding partners of PABP. HYD proteins are ubiquitin ligases with an active C-terminal HECT (homologous to E6-AP C terminus) ubiquitin ligase domain. Ubiquitination of proteins generally requires three proteins: the ubiquitin activating enzyme (E1), the ubiquitin-carrier or conjugating enzyme (E2), and the ubiquitin ligase (E3), which transfers (or assists the transfer of) activated ubiquitin to the protein substrate (reviewed in refs. 47 and 48). Ubiquitinated substrate proteins are then targeted for degradation by the 26S proteasome. Specificity in the ubiquitination reaction is determined by the E3 ubiquitin ligase (reviewed in ref. 49). Specific protein degradation through ubiquitination is used extensively in biological systems to regulate such cellular processes as signal transduction, cell-cycle progression, and development (reviewed in ref. 50).
We propose that the PABP-like domain of HYD contributes to the specificity of the ubiquitin ligase activity of the HECT domain. The HYD family of proteins could bind to and ubiquitinate proteins such as Paip1 that also are able to interact with the C-terminal domain of PABP. Specific ubiquitination and degradation of these substrates would, therefore, serve as a means of regulating PABP function. We suggest that protein synthesis is regulated by targeted destruction of proteins that recognize both PABP and HYD.
Acknowledgments
We thank Drs. Z. Dauter and K. Rajashankar (at the National Synchrotron Light Source) for their help with Beamline X9B. We thank Drs. Andrejz Joachimiak and Stephen Ginell and the SBC staff (at the Advanced Photon Source) for their help with the ID19 Beamline. We thank Drs. J. E. Darnell, J. B. Bonanno, A. W. Craig, C. Groft, A. Kahwajian, K. Kamada, C. Kielkopf, J. Kuriyan, J. Marcotrigiano, S. K. Nair, and G. A. Petsko for many useful discussions, and Ms. T. Niven for editorial assistance. S.K.B. is an Investigator in the Howard Hughes Medical Institute, and N.S. is a Howard Hughes Medical Institute International Scholar. This work was supported in part by The Rockefeller University (S.K.B.) and the Medical Research Council of Canada (N.S.). R.C.D. was supported by the Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-Ph.D. Program and the Andrew W. Mellon Foundation.
Abbreviations
- eIF4E
eukaryotic initiation factor 4E
- PABP
poly(A)-binding protein
- Paip1
poly(A)-binding protein interacting protein 1
- PABP-C
PABP C-terminal domain
- HYD
hyperplastic discs protein
- HYD-P
PABP-like domain of Homo sapiens hyperplastic discs protein
- GST
glutathione S-transferase
Note Added in Proof.
Recently, we identified a 12-aa motif within Paip1 (127) that is both necessary and sufficient for binding to the PABP-C and HYD-P domains (R.C.D., N.S., S.K.B., unpublished data). Amino acid sequence searches have revealed that this motif also occurs in a number of other proteins, including Paip2, eRF3, TOB, ataxin2, deubiquitinating enzyme (GI 1136437), mei2188, map205, and blackjack.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1I2T).
See commentary on page 4288.
References
- 1.Mathews M B, Sonenberg N, Hershey J W B. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 1–32. [Google Scholar]
- 2.Gingras A-C, Raught B, Sonenberg N. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 3.Sachs A. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 447–467. [Google Scholar]
- 4.Hershey J W B, Merrick W C. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 33–88. [Google Scholar]
- 5.Tarun S Z, Jr, Sachs A B. EMBO J. 1996;15:7168–7177. [PMC free article] [PubMed] [Google Scholar]
- 6.Tarun S Z, Wells S E, Deardorff V A, Sachs A. Proc Natl Acad Sci USA. 1997;94:9046–9051. doi: 10.1073/pnas.94.17.9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michel Y M, Poncet D, Piron M, Kean K M, Borman A M. J Biol Chem. 2000;275:32268–32276. doi: 10.1074/jbc.M004304200. [DOI] [PubMed] [Google Scholar]
- 8.Wells S E, Hillner P E, Vale R D, Sachs A B. Mol Cell. 1998;2:135–140. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- 9.Craig A W, Haghighat A, Yu A T, Sonenberg N. Nature (London) 1998;392:520–523. doi: 10.1038/33198. [DOI] [PubMed] [Google Scholar]
- 10.Jacobson A. In: Translational Control. Hershey J W B, Mathews M B, Sonenberg N, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 451–480. [Google Scholar]
- 11.Schwartz D C, Parker R. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 807–826. [Google Scholar]
- 12.Caponigro G, Parker R. Genes Dev. 1995;9:2421–2432. doi: 10.1101/gad.9.19.2421. [DOI] [PubMed] [Google Scholar]
- 13.Coller J M, Gray N K, Wickens M P. Genes Dev. 1998;12:3226–3235. doi: 10.1101/gad.12.20.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grosset C, Chyi-Ying A C, Xu N, Sonenberg N, Jacqemin-Sablon H, Shyu A-B. Cell. 2000;103:29–40. doi: 10.1016/s0092-8674(00)00102-1. [DOI] [PubMed] [Google Scholar]
- 15.Sachs A B, Davis R W, Kornberg R D. Mol Cell Biol. 1987;7:3268–3276. doi: 10.1128/mcb.7.9.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sachs A B, Bond M W, Kornberg R D. Cell. 1986;45:827–835. doi: 10.1016/0092-8674(86)90557-x. [DOI] [PubMed] [Google Scholar]
- 17.Adam S A, Nakagawa T, Swanson M S, Woodruff T K, Dreyfuss G. Mol Cell Biol. 1986;6:2932–2943. doi: 10.1128/mcb.6.8.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deo R C, Bonanno J B, Sonenberg N, Burley S K. Cell. 1999;98:835–845. doi: 10.1016/s0092-8674(00)81517-2. [DOI] [PubMed] [Google Scholar]
- 19.Kessler S H, Sachs A B. Mol Cell Biol. 1998;18:51–57. doi: 10.1128/mcb.18.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhn U, Pieler T. J Mol Biol. 1996;256:20–30. doi: 10.1006/jmbi.1996.0065. [DOI] [PubMed] [Google Scholar]
- 21.Joachims M, Van Breugel P C, Lloyd R E. J Virol. 1999;73:718–727. doi: 10.1128/jvi.73.1.718-727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerekatte V, Keiper B D, Badorff C, Cai A, Knowlton K U, Rhoads R E. J Virol. 1999;73:709–717. doi: 10.1128/jvi.73.1.709-717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschul S F, Madden T L, Schaffer A A, Zhang J Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansfield E, Hersperger E, Biggs J, Shearn A. Dev Biol. 1994;165:507–526. doi: 10.1006/dbio.1994.1271. [DOI] [PubMed] [Google Scholar]
- 25.Callaghan M J, Russell A J, Woollatt E, Sutherland G R, Sutherland R L, Watts C K. Oncogene. 1998;17:3479–3491. doi: 10.1038/sj.onc.1202249. [DOI] [PubMed] [Google Scholar]
- 26.Muller D, Rehbein M, Baumeister H, Richter D. Nucleic Acids Res. 1992;20:1471–1475. doi: 10.1093/nar/20.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Weeks C, Miller R. J Appl Crystallogr. 1999;32:120–124. [Google Scholar]
- 29.Dodson E J, Winn M, Ralph A. Methods Enzymol. 1997;277:620–633. doi: 10.1016/s0076-6879(97)77034-4. [DOI] [PubMed] [Google Scholar]
- 30.Lamzin V S, Wilson K S. Acta Crystallogr D. 1993;49:129–149. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 31.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 32.Brünger A, Adams P D, Clore G M, Gros P, Grosse-Kuntsleve R W, Jiang J-S, Kuszewski J, Nilges M, Pannu N S, Read R J. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Sheldrick G M, Schneider T R. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 34.Laskowski R J, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–290. [Google Scholar]
- 35.Cohen S L, Ferre-D'Amare A R, Burley S K, Chait B T. Protein Sci. 1995;4:1088–1099. doi: 10.1002/pro.5560040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendrickson W. Science. 1991;254:51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- 37.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 38.Sander C, Schneider R. Proteins. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- 39.Holm L, Sander C. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 40.Sali A, Blundell T L. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 41.Gray N K, Coller J M, Dickson K S, Wickens M. EMBO J. 2000;19:4723–4733. doi: 10.1093/emboj/19.17.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imataka H, Gradi A, Sonenberg N. EMBO J. 1998;17:7480–7489. doi: 10.1093/emboj/17.24.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoshino S, Imai M, Kobayashi T, Uchida N, Katada T. J Biol Chem. 1999;274:16677–16680. doi: 10.1074/jbc.274.24.16677. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Day N, Trifillis P, Kiledjian M. Mol Cell Biol. 1999;19:4552–4560. doi: 10.1128/mcb.19.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mangus D A, Amrani N, Jacobson A. Mol Cell Biol. 1998;18:7383–7396. doi: 10.1128/mcb.18.12.7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Ullah Z, Grumet R. Virology. 2000;275:433–443. doi: 10.1006/viro.2000.0509. [DOI] [PubMed] [Google Scholar]
- 47.Hershko A, Ciechanover A, Varshavsky A. Nat Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- 48.Hochstrasser M. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 49.Hershko A, Ciechanover A. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz A L, Ciechanover A. Annu Rev Med. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- 51.Carson M. J Appl Crystallogr. 1991;24:958–961. [Google Scholar]
- 52.Nicholls A, Sharp K, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 53.Gilson M, Sharp K, Honig B. J Comput Chem. 1988;9:327–335. [Google Scholar]