

Abstract
Synapsin III was discovered in 1998, more than two decades after the first two synapsins (synapsins I and II) were identified. Although the biology of synapsin III is not as well understood as synapsins I and II, this gene is emerging as an important factor in the regulation of the early stages of neurodevelopment and dopaminergic neurotransmission, and in certain neuropsychiatric illnesses. Molecular genetic and clinical studies of synapsin III have determined that its neurodevelopmental effects are exerted at the levels of neurogenesis and axonogenesis. In vitro voltammetry studies have shown that synapsin III can control dopamine release in the striatum. Since dopaminergic dysfunction is implicated in many neuropsychiatric conditions, one may anticipate that polymorphisms in synapsin III can exert pervasive effects, especially since it is localized to extrasynaptic sites. Indeed, mutations in this gene have been identified in individuals diagnosed with schizophrenia, bipolar disorder and multiple sclerosis. These and other findings indicate that the roles of synapsin III differ significantly from those of synapsins I and II. Here, we focus on the unique roles of the newest synapsin, and where relevant, compare and contrast these with the actions of synapsins I and II.
Keywords: phosphoprotein, neurogenesis, axon formation, dopamine, schizophrenia, polymorphism
1. Introduction
Synapsins are a family of three neuron-specific genes – designated synapsins I, II and III– which encode phosphoproteins that play crucial roles in the regulation of neurotransmission and neurodevelopment [1, 2]. Synapsins I and II were identified in the late 1970s [3, 4], and have since been studied intensively as regulators of synaptic function. Synapsin III, which is the subject of this review, was discovered in 1998 as a result of early work on the human genome project [5, 6]. The human synapsin III gene was identified via homologous sequences on chromosome 22 that were different from synapsin I (localized to chromosome X) or synapsin II (localized to chromosome 3). Synapsin III shares structural and functional properties with synapsins I and II, but also possesses unique features as described below.
The human synapsin III gene spans an unusually large region within the genome (494,000 bases) compared to synapsin I (48,000 bases) or synapsin II (43,500 bases). Despite this difference, the gene possesses a conserved intron-exon structure very similar to those of synapsins I and II [6]. The full-length synapsin III protein (isoform IIIa) exhibits protein homology with the other two synapsins: they share conserved domains A, C and E. Domain B is not conserved among the synapsins, and synapsin III possesses a unique region termed domain J (Fig. 1). Like the other two synapsins, synapsin III is also an avid substrate for several protein kinases [7].
Figure 1.
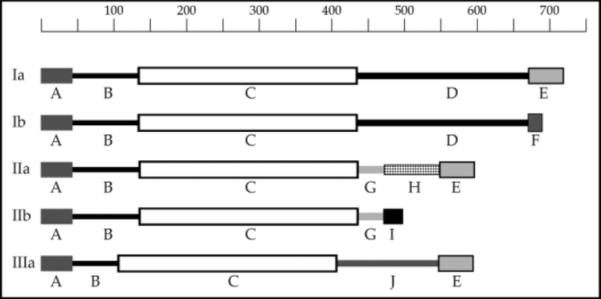
Protein Domain Structure of the Synapsins. Domains are schematically represented and indicated by letters A–J. The length of the polypeptide chains is shown at the top in number of amino acid residues.
Although the synapsins share similar domain structures and sequence homologies, synapsin III has a unique transcriptional profile with respect to alternative splicing [8]. Six different mRNAs (IIIa to IIIf) are transcribed from the synapsin III gene, whereas only two spliced isoforms are known to arise from the synapsin I (Ia and Ib) and II (IIa and IIb) genes. The synapsin III mRNAs exhibit differential tissue- and developmental stage-specific expression. Three of the neuronal transcripts are detected in fetal and to a lesser extent adult brain (IIIa–IIIc), whereas one (IIId) is detected only in fetal brain. Additionally, two transcripts (IIIe and IIIf) are detected only in nonneuronal tissues. Parenthetically, a putative second promoter, which is contained within an intron in the synapsin III gene locus, appears to generate the nonneuronal synapsin IIIe and IIIf transcripts [8]. This level of transcriptional complexity is far greater than that described previously for the synapsin I and II genes, and suggests that synapsin III may have functions distinct from those ascribed to synapsins I and II [8].
Structural studies also imply a distinct role for synapsin III. For instance, the C domains of synapsins are structurally homologous to a family of ATP-utilizing enzymes [9]. Consistent with this observation, synapsins were shown to bind to ATP with a high affinity and to ADP with a low affinity, suggesting that synapsins might also be ATP-utilizing enzymes [5, 9, 10]. Curiously, calcium inhibited the binding of ATP to synapsin III, activated the binding of ATP to synapsin I, but did not affect the binding of ATP to synapsin II [5]. This intriguing data suggests a biological rationale for the existence of three synapsins, but to date, the significance of these findings have yet to be determined.
The expression profile of synapsin III also suggests a function distinct from the other two synapsins. In the adult brain, synapsin III expression is much lower than that of synapsins I or II [6], and it is developmentally regulated in a manner that differs from that of the other two synapsins [11]. Although synapsin III, like synapsins I and II, is localized to the synapse [6], the synapsin III protein is primarily localized to regions outside of the synapse in the adult brain [11]. Thus, the subcellular localization of synapsin III includes extrasynaptic sites, such as the cell body and growth cones [11], whereas synapsins I and II are almost exclusively localized at synaptic sites [11]. These findings suggested that unlike synapsins I and II, synapsin III does not play a major role in synaptic activity, but rather, in early neural development. Despite these dissimilarities, synapsin III does not function in isolation – all synapsins form hetero-and homodimers, and therefore the function of synapsin III is entwined with the other synapsins [12]. Here, we present our current understanding of the newest member of the synapsin gene family, synapsin III, with regards to its relationship with the other synapsins, its role in neurodevelopment and neurotransmission, and its potential relevance to human illness.
2. Neurotransmission
2.1 Kinetics
A role for synapsins in the regulation of neurotransmitter release was first reported more than two decades ago [13]. The creation of mice bearing homozygous deletions of individual synapsins of all three synapsin genes (triple knockout or TKO) has proven to be invaluable tools in deciphering the precise mechanisms by which synapsins exert their physiological actions.
Initial experiments with mice bearing a homozygous deletion of the synapsin III gene (synapsin III knockout mice) revealed subtle changes in neurotransmission. Superficially, synapsin III knockout mice do not display neurological abnormalities such as seizures or motor disturbances [14]. This contrasts strongly with synapsin I and II knockout mice, which exhibit seizures after 1–2 months of age [15, 16]. In both synapsin I and II knockout mice, there is depletion in the reserve pool of synaptic vesicles [15–17], which results in a rapid decrease of neurotransmission after repeated stimulation. This presumably accounts for the seizure phenotype in synapsin I and II knockout mice. However, in mice lacking synapsin III, the size of the recycling pool is actually increased, but release kinetics is slower [14]. Consequently, the number of vesicles released per action potential is similar between synapsin III knockout and wild-type mice. Consistent with this observation, the density and distribution of synaptic vesicles in synapsin III knockout mice does not differ from those of the wild-type controls [14].
While there were no major changes in the reserve pool of synaptic vesicles, a significant reduction in inhibitory postsynaptic currents was observed in synapsin III-deficient neurons [14]. Synaptic depression was also substantially reduced at strong synapses. In this context, synapsin III appears to act as a negative regulator while synapsins I and II act as positive regulators of neurotransmission.
2.2 Specificity for Dopamine Release
Investigations of the role for synapsins in neurotransmission revealed that synapsin I is primarily responsible for maintaining the pool of GABAergic vesicles [18, 19] while synapsin IIa maintains glutamatergic synaptic vesicles [20]. Nevertheless, synapsins do not appear to be involved in the release of serotonin in the substantia nigra [21] or acetylcholine at neuromuscular junctions [22].
Recently, it was demonstrated that synapsin III is specifically involved in the release of dopamine in the striatum [21]. Initially, this observation was made using substantia nigra pars reticulata slices prepared from TKO mice. Release of dopamine in response to electrical stimulation was approximately doubled, both in vivo and in striatal slices from TKO mice, compared to wild-type controls [21]. A similar magnitude of dopamine release was also observed in slices derived from synapsin III knockout mice, suggesting that synapsin III is primarily responsible for the regulation of dopamine release in the striatum. It should be emphasized that dopamine is a neurotransmitter that is clinically important in many neuropsychiatric disorders, and this finding has potentially significant implications for synapsin III in brain disorders, as described below.
3. Neurodevelopment
3.1 Neurogenesis
Synapsin III protein is enriched in young neuronal precursor cells of the hippocampal dentate gyrus [23], a region of the brain where neurogenesis is known to persist well into adulthood (reviewed in [24]). In synapsin III knockout mice [14], neurogenesis was markedly altered, suggesting a direct link between synapsin III and neurogenesis. Since neurogenesis consists of a number of stages of development, proliferation, survival, and differentiation of neural progenitor cells were systematically quantitated in the hippocampal dentate gyrus of adult synapsin III knockout and wild-type mice [25]. A 30% decrease in proliferation and a 55% increase in survival of neural progenitor cells were observed in synapsin III knockout mice. No difference in the volume of the dentate gyrus was noted between synapsin III knockout and wild-type mice, suggesting that the decrease in proliferation was compensated by the increased survival of neural progenitor cells [25]. A 6% increase in the number of neural progenitor cells that differentiated into neurons was also observed. Immunocytochemistry of the adult hippocampal dentate gyrus revealed that synapsin III co-localizes with markers of neural progenitor cell development (i.e. Nestin, PSA-NCAM, NeuN, and Tuj1), but synapsin III immunoreactivity did not co-localize with markers of mitosis (i.e. Ki67 and PCNA) (Fig. 2). These results suggest a complex role for synapsin III during this stage of neurodevelopment, because deletion of synapsin III affects each step during the process of neurogenesis in the hippocampal dentate gyrus.
Figure 2.
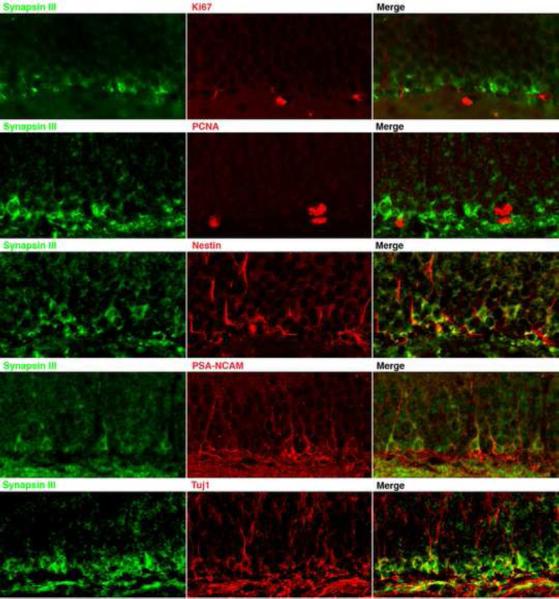
Synapsin III Co-localizes with Markers of Early Neuronal Development. Sections from the hippocampal dentate gyrus from a wild-type adult mouse were stained with antibodies specific to synapsin III and to the indicated proteins; images were obtained using confocal microscopy. Synapsin III does not co-localize with the mitotic markers Ki67 and PCNA, but it does co-localize with markers of early neuronal development that include Nestin, PSA-NCAM, and Tuj1.
As will be discussed later, there is increasing evidence that adult neurogenesis is highly relevant to psychiatric illness. For instance neurogenesis in the hippocampal dentate gyrus is associated with facilitated learning and memory [26], is disrupted by depression and stress [27, 28], but is stimulated by some antidepressants [29, 30], lithium [31, 32] and certain antipsychotic drugs [33–38].
3.2 Axonogenesis
To determine if synapsin III has a role in the morphological development of neurons, the hippocampal culture system, in which morphological stages of neuronal development are well-established [39, 40], was employed [11]. These experiments demonstrated that synapsin III protein is expressed at an earlier developmental time-frame than synapsins I and II [11]. Remarkably, immunohistochemical experiments revealed that, in contrast to synapsins I and II, synapsin III failed to co-localize at synaptic sites with synaptic markers (e.g. synaptophysin). In contrast, synapsin III was concentrated in all cell bodies and most growth cones, suggesting a prominent role in axon growth. In support of this notion, depletion of synapsin III by either antisense oligonucleotides [11] or genetic ablation [14] led to hypertrophied growth cones and stunted axons. Depleting synapsin III after axons had formed did not appear to have subsequent effects on neuronal maturation [11]. The results indicate a distinct role for synapsin III in axonogenesis.
Significantly, specific stages of neurodevelopment are not affected by depletion of individual synapsin genes. For instance, depletion of synapsin III has no effect on synapse formation or maintenance, processes that are regulated both by synapsin I [41] and synapsin II [42]. Conversely, the lack of synapsin I and II immunoreactivity in neural progenitor cells [23] suggests that these synapsins are not involved in neurogenesis. These observations indicate that synapsin III plays a much earlier role in neurodevelopment, which contrasts with the later roles of synapsins I and II in the formation and maintenance of synapses.
4. Neuropsychiatric Disorders
4.1 Importance of Neurotransmission
Since the advent of psychopharmacology in the 1950s and 1960s, it has been recognized that specific neurotransmitters play an integral role in the pathophysiology of various neuropsychiatric disorders. It is well established that most antidepressants in current use enhance serotonin and/or noradrenaline accumulation at the synapse [43]. Typical antipsychotic drugs are all selective antagonists of the dopamine D2 receptor, and their potency at this receptor is directly correlated with clinical efficacy [44]. Conversely, reagents that increase synaptic dopamine (e.g. antagonists of the dopamine transporter such as cocaine and amphetamines) exacerbate psychosis [45]. These observations led to the formulation of the dopamine hypothesis of schizophrenia [46]. While the mechanism of action of lithium in the treatment of bipolar disorder remains poorly understood, most of the newer treatments for this disorder are anticonvulsants, suggesting a generalized dysfunction of neurotransmission in this condition [47]. Since synapsin III is involved in dopamine release, its dysfunction fits well with a potential role in psychosis.
Although dysfunctions in specific neurotransmitters such as the serotonin, noradrenaline and dopamine systems are often viewed as potential players in susceptibility to mental illness, the etiology of psychiatric disorders remains poorly understood. Indeed, many investigators have raised the possibility that abnormal neurotransmission may not be the primary locus of dysfunction in mental disorders. As will be discussed in the next section, growing evidence suggests that neurodevelopmental abnormalities may underlie the pathogenesis of some psychiatric disorders.
4.2 Importance of Neurodevelopment
There is increasing evidence that neurodevelopmental abnormalities may underlie major psychiatric disorders such as schizophrenia [48, 49], bipolar disorder [50] and depression [51]. The evidence is strongest for schizophrenia, where data from childhood and adolescent observations [52, 53], postmortem analyses (for review see [54]), and neuroimaging studies [55] suggest this progressive brain disorder originates early in life.
At a cellular level, postmortem analyses of brains from individuals with schizophrenia have demonstrated the existence of a malformed cytoarchitecture [56], dendritic abnormalities [57], decreased dendritic spine densities [58], reduced neuropil [59] and cell body sizes [60], and decreased expression of synaptic proteins [61] and mRNA [62], suggesting disrupted synaptic connections in this disorder. Although there is a wide range of cellular abnormalities, they encompass the types of abnormalities that have been observed in synapsin III knockout mice, including aberrations in axon outgrowth.
Recently, abnormalities of neurogenesis in the adult have been investigated as a potential major contributor to mental illness (for review see [63]). The existence of adult neurogenesis was debated for many years after it was first described [64]. It is now accepted that limited neurogenesis occurs in all adult mammalian brains, primarily at two sites: the subgranular zone of the hippocampal dentate gyrus [64, 65], and the subventricular zone which gives rise to neurons in the rostral migratory stream and olfactory bulb [66–68]. Studies have shown that stem cells responsible for adult neurogenesis are functional and contribute to cognition [26, 69, 70]. The enrichment of synapsin III in neural progenitor cells in precisely these same regions, and its effects on various neurogenic stages and behavior when depleted, implies that synapsin III exerts a functional role in neurogenesis, which may have broad implications for neuropsychiatric disorders [25].
4.3 Behavioral Studies
Although psychiatric disorders are difficult to study in rodents, over the years a number of different behavioral tests have been developed that may have face, predictive, and construct validity. One such neurophysiological correlate is prepulse inhibition (PPI), a phenomenon in which a weak stimulus (the prepulse) preceding a stronger stimulus inhibits a startle response. Patients with schizophrenia and some other neuropsychiatric disorders have impaired PPI (reviewed in [71]). Impaired PPI in these patient populations is thought to reflect dysfunctional sensorimotor gating mechanisms, and similar deficits in PPI are produced in rats by pharmacological or developmental manipulations, and in mice by pharmacological and genetic manipulations.
Cognitive impairments have also been reported in major depression [72], bipolar disorder [73] and schizophrenia [74]. Domains of cognition that are disrupted significantly in schizophrenia include attention, executive function, verbal and visual-spatial working memory [74]. Impairments of working and semantic memory are primarily due to dysfunction of the frontal cortex, temporal cortex and hippocampus.
A neurophysiological screen was initially conducted to exclude gross deficits in the synapsin III knockout mice [75]. No abnormalities were detected in anxiety- or depressive-like responses, or in gross sensory or motor function; however, synapsin III knockout mice increased their exploratory activities when placed into a novel environment [76]. Hippocampal function was examined using the Morris water maze [77], novel object recognition [78] and social transmission of food preference tests [79, 80]. Synapsin III knockout mice did not display abnormalities in the Morris water maze test, but showed deficits in the novel object recognition and social transmission of food preference tests. Both the object recognition and social transmission tasks require an intact hippocampus, and successful performance on the tests relies upon somewhat different brain circuits and areas [79, 81–83]. Hence, synapsin III may play a selective role in learning and memory functions where spatial learning and memory are largely intact, while processes underlying explicit memory are affected [76]. Synapsin III knockout mice were also examined in tests of conditioned fear, a type of emotional memory that requires an intact hippocampus and amygdala, the latter is a brain region implicated in paranoia [84, 85], a form of psychosis seen in subtypes of schizophrenia. Synapsin III knockout mice displayed abnormalities in both contextual and cued fear conditioning [76] suggesting that depletion of synapsin III has widespread behavioral consequences involving connections between the amygdala, hippocampus and frontal cortex. The amygdala deficits in the synapsin III knockouts were further supported by abnormalities in fear-potentiated startle.
Sensorimotor gating was also evaluated in synapsin III knockout mice using auditory PPI, and by challenging these mice with drugs that are typically used to treat individuals with schizophrenia. Synapsin III knockout mice are deficient in PPI and fail to show prepulse-dependency of the response (Fig. 3). Clozapine, an atypical antipsychotic used in the treatment of schizophrenia, restores the PPI performance of synapsin III knockout mice to that of the vehicle-treated wild-type controls (Fig. 3).
Figure 3.
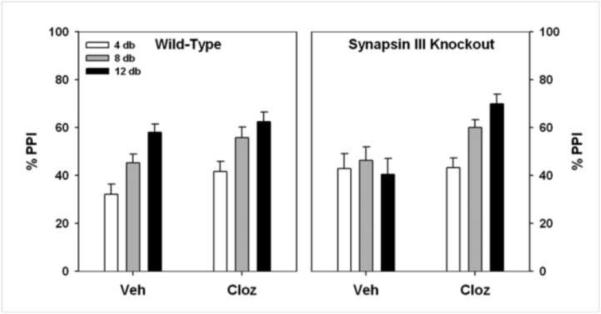
PPI is Abnormal in Synapsin III Knockout Mice, and is Rescued by an Atypical Antipsychotic Drug. Wild-type and synapsin III knockout mice were injected with vehicle saline (Veh) or 1 mg/kg clozapine (intraperitoneal). Wild-type mice show normal responses in PPI: increasing PPI with increasing prepulse intensity, which is unaffected by clozapine. By contrast, synapsin III knockout mice have an abnormal response to PPI (vehicle injection), which is restored when they are treated with clozapine.
Taken together, synapsin III knockout mice demonstrate deficits in cognition, emotional processing and sensorimotor gating. Similar deficits have also been described in patients with psychosis. Thus, synapsin III knockout mice may serve as a useful model system for further investigations of mechanisms underlying this symptom.
4.4 Post-Mortem Studies
Several synaptic proteins are decreased in dorsolateral prefrontal cortex and hippocampus of post-mortem brain, two regions that are implicated in brain disorders (for review see [61]). These findings are consistent with the hypothesis that neural mis-connectivity contributes to schizophrenia and other neuropsychiatric disorders [86].
The hippocampus is considered to be one region of the brain where a convergence of imaging, postmortem, and functional studies has revealed subtle changes in schizophrenia (for review, see [54]) and bipolar disorder (for reviews see [87, 88]). Synapsin III levels were decreased by 75% in postmortem hippocampi of individuals with bipolar disorder, and decreased by more than 50% decreased in individuals with schizophrenia [89].
Another region implicated in both schizophrenia [90] and bipolar disorder [91] is the dorsolateral prefrontal cortex. At the mRNA level, synapsin II is one of the most consistently down-regulated genes in the prefrontal cortex of individuals with schizophrenia [62]. In agreement with this observation, synapsin III protein levels were also significantly decreased in the prefrontal cortex of individuals with schizophrenia [92].
Curiously, synapsin I levels are not significantly different between schizophrenia and controls in the hippocampus [89] or prefrontal cortex [93], indicating a potential disparity in the involvement of specific synapsins in mental disorders. This result is relevant because synapsins are thought to function together as heterodimers [12]. If confirmed, the involvement of synapsins II and III but not synapsin I in specific mental disorders may be an important clue to investigating molecular mechanisms underlying these conditions.
4.5 Genetic Studies
Early twin and familial studies strongly support the hypothesis that genetics plays a strong role in the susceptibility to schizophrenia [94]. Subsequent studies have shown that genetics also plays a significant role in a variety of other major psychiatric disorders (for review, see [95]), including bipolar disorder, major depression, anxiety disorders and addiction. However, genetic studies have shown that for virtually all these conditions, inheritance of the disorder is complex, and that susceptibility is influenced by environmental factors that have yet to be determined. Thus, in a recent opinion, “high heritability has not … translated into a satisfying search for genetic lesions” [96].
One potential explanation for the difficulty in identifying susceptibility genes for mental disorders is that multiple genes of weak effect may work in concert with environmental factors and/or among themselves to produce susceptibility to major psychiatric illness [95]. The majority of psychiatric genetic studies are not designed to detect such genes of weak effect. Consequently, a recent strategy has emerged in psychiatric genetics where investigators are now seeking rare variants in mental disorders to better understand a biological basis of these conditions [97, 98].
In this regard, synapsin III is located on human chromosome 22q12.3 [6]. Several studies have consistently demonstrated evidence for a susceptibility gene located on chromosome 22q12.3 for schizophrenia [99–101] and bipolar disorder [102, 103]. Indeed, a number of chromosomal loci linked to bipolar disorder are identical to those linked to schizophrenia, raising the possibility of genetic overlap between the two disorders [104–107].
Since the identification of synapsin III, several studies have conducted genetic associations between polymorphisms in the synapsin III gene and schizophrenia [108–113]. In most studies, the polymorphic variations employed were located in introns, and would likely be nonfunctional. In the next section, we describe potentially functional polymorphisms in synapsin III: (1) S470N, which abolishes a functional mitogen-activated protein kinase (MAPK) site in the synapsin III protein that may affect neurotrophic signaling; and (2) synapsin III promoter mutations, of which one has been shown to alter DNA-protein interactions at a transcription factor binding site.
4.6 Functional Polymorphisms in the Synapsin III Gene
In a search for potentially functional polymorphisms in the synapsin III gene, exons from this gene were sequenced from probands derived from families that displayed linkage to schizophrenia at chromosome 22q12–13 [7]. Two polymorphisms residing on exon 12 were identified: one polymorphism affected the third base of the codon for Leu469, which was silent (L469L), and the other mutation consisted of amino acid 470, that was changed from Ser to Asn (S470N). In this study sample, all individuals that possessed S470N also inherited L469L [7].
Using association analyses, the S470N polymorphism was found to be more frequent in individuals with schizophrenia than in controls [7]. A subsequent follow-up study also found that S470N appeared more frequently in schizophrenia in a Caucasian population [114], but the sample was too small to draw firm conclusions. Curiously, these investigations found a 50-fold increase in the frequency of both the S470N and L469L polymorphisms in the African population. Parenthetically, in this population, both polymorphisms were found more frequently in schizophrenia, with L469L being statistically significant and S470N exhibiting a trend towards significance. S470N was also found more frequently in individuals with schizophrenia in another independent Caucasian population, but the frequency of the polymorphism was too rare to draw conclusions [115].
It is highly unlikely that a single gene can act alone to confer susceptibility to schizophrenia or to other major mental disorders. Moreover, there are very few studies that analyze multiple genes at a time. However, in recent Bayesian analyses of single nucleotide polymorphisms, 30 candidate genes for schizophrenia were studied [115]. These analyses revealed that S470N was one out of six polymorphisms that were considered risk factors for schizophrenia when considered in combination with other mutations [115].
Subsequent studies revealed that S470N might affect neurodevelopment. Synapsin III is predominantly expressed during mouse neonatal development [7], which corresponds to the peak of neurogenesis during brain cortical development [116]. Synapsin III is also a substrate for abundant brain proline-directed kinases, such as cyclin-dependent protein kinase 5 (cdk5) and mitogen-activated protein kinase (MAPK). Ser-470 is an evolutionarily conserved site for the MAPK known as Erk1/2, a downstream effector of neurotrophin action [7]. Further studies have revealed that Ser-470 is phosphorylated by MAPK almost exclusively during neonatal development, and is stimulated by the neurotrophins, NT-3 [7] and BDNF [unpublished observation]. The degree of phosphorylation at Ser-470 parallels the levels of phospho-Erk1/2, suggesting that Ser-470 on synapsin III is a physiological substrate for MAPK and a downstream target of neurotrophins. These observations raise the possibility that S470N could affect neurotrophic signaling.
Taken together, these findings are consistent with the accumulating evidence of a neurodevelopmental defect in schizophrenia. In particular, abnormalities in neurotrophic signaling have been implicated in this disorder. Studies have demonstrated aberrant levels of BDNF [117, 118], NT-3 [119], and their respective cognate receptors, trkB [117, 118, 120] and trkC [120, 121] in this condition. Moreover, there are selective changes in the expression levels of downstream activated MAPK signaling molecules that were observed in postmortem brain from individuals with schizophrenia [122].
Two single nucleotide polymorphisms (SNPs) in the synapsin III promoter have also been investigated by a number of research groups: rs133946 (position -631) and rs133945 (position -196). The SNP rs133945 is potentially functional, because an ATTT motif is located at position -196 that resembles the octamer sequence recognized by members of the POU family of transcription factors [123]. Indeed, the G-allele of this SNP has consistently resulted in greater binding to brain proteins than the A-allele, suggesting that this polymorphism has an effect on DNA-protein complexes [123]. The functional relevance of rs133946 has not been investigated.
Interestingly, no striking differences in the frequencies of these SNPs were observed in schizophrenia [113, 123] or bipolar disorder [123], although there were trends towards significance in some ethnic samples, suggesting that the studies were underpowered. However, there were significant differences in protein-DNA interactions using protein extracts from postmortem brains of individuals with bipolar disorder and synthetic DNA corresponding to alleles of rs133945 [123]. These results suggest that additional variables (e.g.. composition of protein extracts) should be considered when investigating the functional relevance of a polymorphism.
Both rs133946 and rs133945 have also been investigated in the context of multiple sclerosis. An initial report in an Italian cohort showed that the C631/A196 haplotype seems to confer a significant protection against this neurological condition [124]. A subsequent study in a German cohort has failed to find any association between these polymorphisms and multiple sclerosis [125]. However, a recent study in a Spanish Basque cohort has also reported that C631/A196 may be protective factors against multiple sclerosis [126].
5. Conclusions
Synapsin III is emerging as an important regulator of early neurodevelopmental processes as well as dopaminergic neurotransmission. Synapsin III appears to play significant roles during neurogenesis and axon formation, while synapsins I and II are primarily involved in the formation and maintenance of synapses. Synapsin III is involved in slow synaptic transmission, particularly dopamine release [21], whereas synapsins I and II are involved in fast neurotransmitter release (i.e. glutamate and GABA [127]).
These functions of synapsin III are particularly relevant to psychiatric disorders that encompass psychosis, a symptom shared by schizophrenia and bipolar disorder. Schizophrenia is currently believed to have a neurodevelopmental basis [96], and recent experiments posit neurogenesis, which is affected by synapsin III [25], as a stage that is affected in this condition [63]. A role for synapsin III in psychiatric disorders is also supported by postmortem studies [89, 92]. Additionally, support also derives from its role in dopaminergic transmission [21], which is believed to be the site of action of antipsychotic drugs, as well as a behavioral phenotype in synapsin III knockout mice that is consistent with psychosis [76]. Functional polymorphisms in the synapsin III gene may be relevant to schizophrenia, bipolar disorder and possibly multiple sclerosis (Section 5.6.2). The discovery of the synapsin III polymorphism, S470N, is particularly instructive, as it could affect neurotrophic signaling. Taken together, these investigations into synapsin III have provided new insights into mechanisms of neural plasticity as they may relate to a variety of neuropsychiatric disorders.
Here are five bullet points highlighting the core findings of our article:
We compare the actions of Synapsin III with those of Synapsins I and II.
Synapsin III is mainly involved in the regulation of neurogenesis and axonogenesis.
By contrast, Synapsins I and II are involved in synapse formation and maintenance.
Synapsin III, but not I or II, is implicated in dopamine release in the striatum.
Human genetic and animal studies support a role for synapsin III in psychosis.
Acknowledgements
Supported by grants NIH MH070898 and NS047209.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–5. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- [2].Kao HT, Porton B, Hilfiker S, Stefani G, Pieribone VA, DeSalle R, et al. Molecular evolution of the synapsin gene family. J Exp Zool. 1999;285:360–77. [PubMed] [Google Scholar]
- [3].Forn J, Greengard P. Depolarizing agents and cyclic nucleotides regulate the phosphorylation of specific neuronal proteins in rat cerebral cortex slices. Proc Natl Acad Sci U S A. 1978;75:5195–9. doi: 10.1073/pnas.75.10.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ueda T, Greengard P. Adenosine 3':5'-monophosphate-regulated phosphoprotein system of neuronal membranes. I. Solubilization, purification, and some properties of an endogenous phosphoprotein. J Biol Chem. 1977;252:5155–63. [PubMed] [Google Scholar]
- [5].Hosaka M, Sudhof TC. Synapsin III, a novel synapsin with an unusual regulation by Ca2+ J Biol Chem. 1998;273:13371–4. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- [6].Kao HT, Porton B, Czernik AJ, Feng J, Yiu G, Haring M, et al. A third member of the synapsin gene family. Proc Natl Acad Sci U S A. 1998;95:4667–72. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Porton B, Ferreira A, DeLisi LE, Kao HT. A rare polymorphism affects a mitogen-activated protein kinase site in synapsin III: possible relationship to schizophrenia. Biol Psychiatry. 2004;55:118–25. doi: 10.1016/j.biopsych.2003.07.002. [DOI] [PubMed] [Google Scholar]
- [8].Porton B, Kao HT, Greengard P. Characterization of transcripts from the synapsin III gene locus. J Neurochem. 1999;73:2266–71. doi: 10.1046/j.1471-4159.1999.0732266.x. [DOI] [PubMed] [Google Scholar]
- [9].Esser L, Wang CR, Hosaka M, Smagula CS, Sudhof TC, Deisenhofer J. Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO J. 1998;17:977–84. doi: 10.1093/emboj/17.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hosaka M, Sudhof TC. Synapsins I and II are ATP-binding proteins with differential Ca2+ regulation. J Biol Chem. 1998;273:1425–9. doi: 10.1074/jbc.273.3.1425. [DOI] [PubMed] [Google Scholar]
- [11].Ferreira A, Kao HT, Feng J, Rapoport M, Greengard P. Synapsin III: developmental expression, subcellular localization, and role in axon formation. J Neurosci. 2000;20:3736–44. doi: 10.1523/JNEUROSCI.20-10-03736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hosaka M, Sudhof TC. Homo- and heterodimerization of synapsins. J Biol Chem. 1999;274:16747–53. doi: 10.1074/jbc.274.24.16747. [DOI] [PubMed] [Google Scholar]
- [13].Greengard P, Browning MD, McGuinness TL, Llinas R. Synapsin I, a phosphoprotein associated with synaptic vesicles: possible role in regulation of neurotransmitter release. Adv Exp Med Biol. 1987;221:135–53. doi: 10.1007/978-1-4684-7618-7_11. [DOI] [PubMed] [Google Scholar]
- [14].Feng J, Chi P, Blanpied TA, Xu Y, Magarinos AM, Ferreira A, et al. Regulation of neurotransmitter release by synapsin III. J Neurosci. 2002;22:4372–80. doi: 10.1523/JNEUROSCI.22-11-04372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, et al. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–93. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- [16].Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9235–9. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ryan TA, Li L, Chin LS, Greengard P, Smith SJ. Synaptic vesicle recycling in synapsin I knock-out mice. J Cell Biol. 1996;134:1219–27. doi: 10.1083/jcb.134.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baldelli P, Fassio A, Valtorta F, Benfenati F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci. 2007;27:13520–31. doi: 10.1523/JNEUROSCI.3151-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, et al. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004;24:11368–80. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gitler D, Cheng Q, Greengard P, Augustine GJ. Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci. 2008;28:10835–43. doi: 10.1523/JNEUROSCI.0924-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kile BM, Guillot TS, Venton BJ, Wetsel WC, Augustine GJ, Wightman RM. Synapsins differentially control dopamine and serotonin release. J Neurosci. 2010;30:9762–70. doi: 10.1523/JNEUROSCI.2071-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gaffield MA, Betz WJ. Synaptic vesicle mobility in mouse motor nerve terminals with and without synapsin. J Neurosci. 2007;27:13691–700. doi: 10.1523/JNEUROSCI.3910-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pieribone VA, Porton B, Rendon B, Feng J, Greengard P, Kao HT. Expression of synapsin III in nerve terminals and neurogenic regions of the adult brain. J Comp Neurol. 2002;454:105–14. doi: 10.1002/cne.10417. [DOI] [PubMed] [Google Scholar]
- [24].Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- [25].Kao HT, Li P, Chao HM, Janoschka S, Pham K, Feng J, et al. Early involvement of synapsin III in neural progenitor cell development in the adult hippocampus. J Comp Neurol. 2008;507:1860–70. doi: 10.1002/cne.21643. [DOI] [PubMed] [Google Scholar]
- [26].Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–6. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- [27].Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci. 1997;17:2492–8. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pham K, Nacher J, Hof PR, McEwen BS. Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci. 2003;17:879–86. doi: 10.1046/j.1460-9568.2003.02513.x. [DOI] [PubMed] [Google Scholar]
- [29].Malberg JE, Duman RS. Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology. 2003;28:1562–71. doi: 10.1038/sj.npp.1300234. [DOI] [PubMed] [Google Scholar]
- [30].Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–10. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. Enhancement of hippocampal neurogenesis by lithium. J Neurochem. 2000;75:1729–34. doi: 10.1046/j.1471-4159.2000.0751729.x. [DOI] [PubMed] [Google Scholar]
- [32].Hanson ND, Nemeroff CB, Owens MJ. Lithium, but not fluoxetine or the corticotropin-releasing factor receptor 1 receptor antagonist r121919, increases cell proliferation in the adult dentate gyrus. J Pharmacol Exp Ther. 2011;337:180–6. doi: 10.1124/jpet.110.175372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wakade CG, Mahadik SP, Waller JL, Chiu FC. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69:72–9. doi: 10.1002/jnr.10281. [DOI] [PubMed] [Google Scholar]
- [34].Halim ND, Weickert CS, McClintock BW, Weinberger DR, Lipska BK. Effects of chronic haloperidol and clozapine treatment on neurogenesis in the adult rat hippocampus. Neuropsychopharmacology. 2004;29:1063–9. doi: 10.1038/sj.npp.1300422. [DOI] [PubMed] [Google Scholar]
- [35].Keilhoff G, Grecksch G, Bernstein HG, Roskoden T, Becker A. Risperidone and haloperidol promote survival of stem cells in the rat hippocampus. Eur Arch Psychiatry Clin Neurosci. 2010;260:151–62. doi: 10.1007/s00406-009-0033-1. [DOI] [PubMed] [Google Scholar]
- [36].Kodama M, Fujioka T, Duman RS. Chronic olanzapine or fluoxetine administration increases cell proliferation in hippocampus and prefrontal cortex of adult rat. Biol Psychiatry. 2004;56:570–80. doi: 10.1016/j.biopsych.2004.07.008. [DOI] [PubMed] [Google Scholar]
- [37].Nasrallah HA, Hopkins T, Pixley SK. Differential effects of antipsychotic and antidepressant drugs on neurogenic regions in rats. Brain Res. 2010;1354:23–9. doi: 10.1016/j.brainres.2010.07.075. [DOI] [PubMed] [Google Scholar]
- [38].Wang HD, Dunnavant FD, Jarman T, Deutch AY. Effects of antipsychotic drugs on neurogenesis in the forebrain of the adult rat. Neuropsychopharmacology. 2004;29:1230–8. doi: 10.1038/sj.npp.1300449. [DOI] [PubMed] [Google Scholar]
- [39].Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–42. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- [40].Banker GA, Cowan WM. Further observations on hippocampal neurons in dispersed cell culture. J Comp Neurol. 1979;187:469–93. doi: 10.1002/cne.901870302. [DOI] [PubMed] [Google Scholar]
- [41].Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9230–4. doi: 10.1073/pnas.92.20.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ferreira A, Han HQ, Greengard P, Kosik KS. Suppression of synapsin II inhibits the formation and maintenance of synapses in hippocampal culture. Proc Natl Acad Sci U S A. 1995;92:9225–9. doi: 10.1073/pnas.92.20.9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Feighner JP. Mechanism of action of antidepressant medications. J Clin Psychiatry. 1999;60(Suppl 4):4–11. discussion 2-3. [PubMed] [Google Scholar]
- [44].Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261:717–9. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- [45].Lieberman JA, Kinon BJ, Loebel AD. Dopaminergic mechanisms in idiopathic and drug-induced psychoses. Schizophr Bull. 1990;16:97–110. doi: 10.1093/schbul/16.1.97. [DOI] [PubMed] [Google Scholar]
- [46].Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1988;1:179–86. doi: 10.1016/0893-133x(88)90012-7. [DOI] [PubMed] [Google Scholar]
- [47].Post RM, Weiss SR, Chuang DM. Mechanisms of action of anticonvulsants in affective disorders: comparisons with lithium. J Clin Psychopharmacol. 1992;12:23S–35S. doi: 10.1097/00004714-199202001-00005. [DOI] [PubMed] [Google Scholar]
- [48].Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–34. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- [49].Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–9. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- [50].Blumberg HP, Kaufman J, Martin A, Charney DS, Krystal JH, Peterson BS. Significance of adolescent neurodevelopment for the neural circuitry of bipolar disorder. Ann N Y Acad Sci. 2004;1021:376–83. doi: 10.1196/annals.1308.048. [DOI] [PubMed] [Google Scholar]
- [51].Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, et al. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–9. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 1982;17:319–34. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- [53].Walker E, Green M. Soft signs of neurological dysfunction in schizophrenia: an investigation of lateral performance. Biol Psychiatry. 1982;17:381–6. [PubMed] [Google Scholar]
- [54].Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122(Pt 4):593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- [55].DeLisi LE, Sakuma M, Tew W, Kushner M, Hoff AL, Grimson R. Schizophrenia as a chronic active brain process: a study of progressive brain structural change subsequent to the onset of schizophrenia. Psychiatry Res. 1997;74:129–40. doi: 10.1016/s0925-4927(97)00012-7. [DOI] [PubMed] [Google Scholar]
- [56].Arnold SE, Hyman BT, Van Hoesen GW, Damasio AR. Some cytoarchitectural abnormalities of the entorhinal cortex in schizophrenia. Arch Gen Psychiatry. 1991;48:625–32. doi: 10.1001/archpsyc.1991.01810310043008. [DOI] [PubMed] [Google Scholar]
- [57].Rosoklija G, Toomayan G, Ellis SP, Keilp J, Mann JJ, Latov N, et al. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch Gen Psychiatry. 2000;57:349–56. doi: 10.1001/archpsyc.57.4.349. [DOI] [PubMed] [Google Scholar]
- [58].Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- [59].Rajkowska G, Selemon LD, Goldman-Rakic PS. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry. 1998;55:215–24. doi: 10.1001/archpsyc.55.3.215. [DOI] [PubMed] [Google Scholar]
- [60].Benes FM, Sorensen I, Bird ED. Reduced neuronal size in posterior hippocampus of schizophrenic patients. Schizophr Bull. 1991;17:597–608. doi: 10.1093/schbul/17.4.597. [DOI] [PubMed] [Google Scholar]
- [61].Kao H-T, Porton B. Synaptic Vesicle Associated Proteins and Schizophrenia. In: Lajtha A, Javitt DC, Kantrowitz J, editors. Handbook of Neurochemistry and MolecularNeurobiology Schizophrenia. Springer; New York: 2009. pp. 267–84. [Google Scholar]
- [62].Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- [63].Eisch AJ, Cameron HA, Encinas JM, Meltzer LA, Ming GL, Overstreet-Wadiche LS. Adult neurogenesis, mental health, and mental illness: hope or hype? The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:11785–91. doi: 10.1523/JNEUROSCI.3798-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–35. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- [65].Kaplan MS, Hinds JW. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science. 1977;197:1092–4. doi: 10.1126/science.887941. [DOI] [PubMed] [Google Scholar]
- [66].Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11:173–89. doi: 10.1016/0896-6273(93)90281-u. [DOI] [PubMed] [Google Scholar]
- [67].Lois C, Alvarez-Buylla A. Long-distance neuronal migration in the adult mammalian brain. Science. 1994;264:1145–8. doi: 10.1126/science.8178174. [DOI] [PubMed] [Google Scholar]
- [68].Lois C, Garcia-Verdugo JM, Alvarez-Buylla A. Chain migration of neuronal precursors. Science. 1996;271:978–81. doi: 10.1126/science.271.5251.978. [DOI] [PubMed] [Google Scholar]
- [69].Song HJ, Stevens CF, Gage FH. Neural stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nat Neurosci. 2002;5:438–45. doi: 10.1038/nn844. [DOI] [PubMed] [Google Scholar]
- [70].van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–4. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–54. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- [72].Marazziti D, Consoli G, Picchetti M, Carlini M, Faravelli L. Cognitive impairment in major depression. Eur J Pharmacol. 2010;626:83–6. doi: 10.1016/j.ejphar.2009.08.046. [DOI] [PubMed] [Google Scholar]
- [73].Osuji IJ, Cullum CM. Cognition in bipolar disorder. Psychiatr Clin North Am. 2005;28:427–41. doi: 10.1016/j.psc.2005.02.005. [DOI] [PubMed] [Google Scholar]
- [74].MacDonald AW, 3rd, Carter CS. Cognitive experimental approaches to investigating impaired cognition in schizophrenia: a paradigm shift. J Clin Exp Neuropsychol. 2002;24:873–82. doi: 10.1076/jcen.24.7.873.8386. [DOI] [PubMed] [Google Scholar]
- [75].Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- [76].Porton B, Rodriguiz RM, Phillips LE, Gilbert JWt, Feng J, Greengard P, et al. Mice lacking synapsin III show abnormalities in explicit memory and conditioned fear. Genes Brain Behav. 2010;9:257–68. doi: 10.1111/j.1601-183X.2009.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–6. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- [78].Dere E, Huston JP, De Souza Silva MA. The pharmacology, neuroanatomy and neurogenetics of one-trial object recognition in rodents. Neurosci Biobehav Rev. 2007;31:673–704. doi: 10.1016/j.neubiorev.2007.01.005. [DOI] [PubMed] [Google Scholar]
- [79].Holmes A, Wrenn CC, Harris AP, Thayer KE, Crawley JN. Behavioral profiles of inbred strains on novel olfactory, spatial and emotional tests for reference memory in mice. Genes Brain Behav. 2002;1:55–69. doi: 10.1046/j.1601-1848.2001.00005.x. [DOI] [PubMed] [Google Scholar]
- [80].Ross RS, Eichenbaum H. Dynamics of hippocampal and cortical activation during consolidation of a nonspatial memory. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:4852–9. doi: 10.1523/JNEUROSCI.0659-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bunsey M, Eichenbaum H. Critical role of the parahippocampal region for paired-associate learning in rats. Behav Neurosci. 1993;107:740–7. doi: 10.1037//0735-7044.107.5.740. [DOI] [PubMed] [Google Scholar]
- [82].Winocur G, McDonald RM, Moscovitch M. Anterograde and retrograde amnesia in rats with large hippocampal lesions. Hippocampus. 2001;11:18–26. doi: 10.1002/1098-1063(2001)11:1<18::AID-HIPO1016>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- [83].Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111:104–13. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- [84].Tebartz Van Elst L, Baeumer D, Lemieux L, Woermann FG, Koepp M, Krishnamoorthy S, et al. Amygdala pathology in psychosis of epilepsy: A magnetic resonance imaging study in patients with temporal lobe epilepsy. Brain. 2002;125:140–9. doi: 10.1093/brain/awf008. [DOI] [PubMed] [Google Scholar]
- [85].Williams LM, Das P, Liddell BJ, Olivieri G, Peduto AS, David AS, et al. Fronto-limbic and autonomic disjunctions to negative emotion distinguish schizophrenia subtypes. Psychiatry Res. 2007;155:29–44. doi: 10.1016/j.pscychresns.2006.12.018. [DOI] [PubMed] [Google Scholar]
- [86].Hyman S. Mental illness: genetically complex disorders of neural circuitry and neural communication. Neuron. 2000;28:321–3. doi: 10.1016/s0896-6273(00)00110-0. [DOI] [PubMed] [Google Scholar]
- [87].Frey BN, Andreazza AC, Nery FG, Martins MR, Quevedo J, Soares JC, et al. The role of hippocampus in the pathophysiology of bipolar disorder. Behav Pharmacol. 2007;18:419–30. doi: 10.1097/FBP.0b013e3282df3cde. [DOI] [PubMed] [Google Scholar]
- [88].Ng WX, Lau IY, Graham S, Sim K. Neurobiological evidence for thalamic, hippocampal and related glutamatergic abnormalities in bipolar disorder: a review and synthesis. Neurosci Biobehav Rev. 2009;33:336–54. doi: 10.1016/j.neubiorev.2008.10.001. [DOI] [PubMed] [Google Scholar]
- [89].Vawter MP, Thatcher L, Usen N, Hyde TM, Kleinman JE, Freed WJ. Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:571–8. doi: 10.1038/sj.mp.4001158. [DOI] [PubMed] [Google Scholar]
- [90].Volk DW, Lewis DA. Prefrontal cortical circuits in schizophrenia. Curr Top Behav Neurosci. 2010;4:485–508. doi: 10.1007/7854_2010_44. [DOI] [PubMed] [Google Scholar]
- [91].Cerullo MA, Adler CM, Delbello MP, Strakowski SM. The functional neuroanatomy of bipolar disorder. Int Rev Psychiatry. 2009;21:314–22. doi: 10.1080/09540260902962107. [DOI] [PubMed] [Google Scholar]
- [92].Porton B, Wetsel WC. Reduction of synapsin III in the prefrontal cortex of individuals with schizophrenia. Schizophr Res. 2007;94:366–70. doi: 10.1016/j.schres.2007.04.016. [DOI] [PubMed] [Google Scholar]
- [93].Albert KA, Hemmings HC, Jr., Adamo AI, Potkin SG, Akbarian S, Sandman CA, et al. Evidence for decreased DARPP-32 in the prefrontal cortex of patients with schizophrenia. Arch Gen Psychiatry. 2002;59:705–12. doi: 10.1001/archpsyc.59.8.705. [DOI] [PubMed] [Google Scholar]
- [94].Kety SS, Rosenthal D, Wender PH, Schulsinger F. Studies based on a total sample of adopted individuals and their relatives: why they were necessary, what they demonstrated and failed to demonstrate. Schizophr Bull. 1976;2:413–28. doi: 10.1093/schbul/2.3.413. [DOI] [PubMed] [Google Scholar]
- [95].Abdolmaleky HM, Thiagalingam S, Wilcox M. Genetics and epigenetics in major psychiatric disorders: dilemmas, achievements, applications, and future scope. Am J Pharmacogenomics. 2005;5:149–60. doi: 10.2165/00129785-200505030-00002. [DOI] [PubMed] [Google Scholar]
- [96].Insel TR. Rethinking schizophrenia. Nature. 2010;468:187–93. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]
- [97].Joober R, Boksa P. A new wave in the genetics of psychiatric disorders: the copy number variant tsunami. J Psychiatry Neurosci. 2009;34:55–9. [PMC free article] [PubMed] [Google Scholar]
- [98].McClellan J, King MC. Genomic analysis of mental illness: a changing landscape. JAMA. 2010;303:2523–4. doi: 10.1001/jama.2010.869. [DOI] [PubMed] [Google Scholar]
- [99].Gill M, Vallada H, Collier D, Sham P, Holmans P, Murray R, et al. A combined analysis of D22S278 marker alleles in affected sib-pairs: support for a susceptibility locus for schizophrenia at chromosome 22q12. Schizophrenia Collaborative Linkage Group (Chromosome 22) Am J Med Genet. 1996;67:40–5. doi: 10.1002/(SICI)1096-8628(19960216)67:1<40::AID-AJMG6>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [100].Liu YL, Fann CS, Liu CM, Chen WJ, Wu JY, Hung SI, et al. RASD2, MYH9, and CACNG2 genes at chromosome 22q12 associated with the subgroup of schizophrenia with non-deficit in sustained attention and executive function. Biol Psychiatry. 2008;64:789–96. doi: 10.1016/j.biopsych.2008.04.035. [DOI] [PubMed] [Google Scholar]
- [101].Potash JB, Buervenich S, Cox NJ, Zandi PP, Akula N, Steele J, et al. Gene-based SNP mapping of a psychotic bipolar affective disorder linkage region on 22q12.3: association with HMG2L1 and TOM1. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:59–67. doi: 10.1002/ajmg.b.30574. [DOI] [PubMed] [Google Scholar]
- [102].Kelsoe JR, Spence MA, Loetscher E, Foguet M, Sadovnick AD, Remick RA, et al. A genome survey indicates a possible susceptibility locus for bipolar disorder on chromosome 22. Proc Natl Acad Sci U S A. 2001;98:585–90. doi: 10.1073/pnas.011358498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Grover D, Verma R, Goes FS, Mahon PL, Gershon ES, McMahon FJ, et al. Family-based association of YWHAH in psychotic bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:977–83. doi: 10.1002/ajmg.b.30927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Badner JA, Gershon ES. Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:405–11. doi: 10.1038/sj.mp.4001012. [DOI] [PubMed] [Google Scholar]
- [105].Berrettini W. Bipolar disorder and schizophrenia: convergent molecular data. Neuromolecular Med. 2004;5:109–17. doi: 10.1385/NMM:5:1:109. [DOI] [PubMed] [Google Scholar]
- [106].Craddock N, O'Donovan MC, Owen MJ. Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr Bull. 2006;32:9–16. doi: 10.1093/schbul/sbj033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Wildenauer DB, Schwab SG, Maier W, Detera-Wadleigh SD. Do schizophrenia and affective disorder share susceptibility genes? Schizophr Res. 1999;39:107–11. doi: 10.1016/s0920-9964(99)00108-5. discussion 60. [DOI] [PubMed] [Google Scholar]
- [108].Chen Q, Che R, Wang X, O'Neill FA, Walsh D, Tang W, et al. Association and expression study of synapsin III and schizophrenia. Neurosci Lett. 2009;465:248–51. doi: 10.1016/j.neulet.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ohmori O, Shinkai T, Hori H, Kojima H, Nakamura J. Synapsin III gene polymorphisms and schizophrenia. Neurosci Lett. 2000;279:125–7. doi: 10.1016/s0304-3940(99)00970-2. [DOI] [PubMed] [Google Scholar]
- [110].Tsai MT, Hung CC, Tsai CY, Liu MY, Su YC, Chen YH, et al. Mutation analysis of synapsin III gene in schizophrenia. Am J Med Genet. 2002;114:79–83. doi: 10.1002/ajmg.10116. [DOI] [PubMed] [Google Scholar]
- [111].Ohtsuki T, Ichiki R, Toru M, Arinami T. Mutational analysis of the synapsin III gene on chromosome 22q12-q13 in schizophrenia. Psychiatry Res. 2000;94:1–7. doi: 10.1016/s0165-1781(00)00123-2. [DOI] [PubMed] [Google Scholar]
- [112].Stober G, Meyer J, Nanda I, Wienker TF, Saar K, Knapp M, et al. Linkage and family-based association study of schizophrenia and the synapsin III locus that maps to chromosome 22q13. Am J Med Genet. 2000;96:392–7. doi: 10.1002/1096-8628(20000612)96:3<392::aid-ajmg29>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- [113].Imai K, Harada S, Kawanishi Y, Tachikawa H, Okubo T, Suzuki T. Polymorphisms in the promoter and coding regions of the synapsin III gene. A lack of association with schizophrenia. Neuropsychobiology. 2001;43:237–41. doi: 10.1159/000054896. [DOI] [PubMed] [Google Scholar]
- [114].Lachman HM, Stopkova P, Rafael MA, Saito T. Association of schizophrenia in African Americans to polymorphism in synapsin III gene. Psychiatr Genet. 2005;15:127–32. doi: 10.1097/00041444-200506000-00009. [DOI] [PubMed] [Google Scholar]
- [115].Hall H, Lawyer G, Sillen A, Jonsson EG, Agartz I, Terenius L, et al. Potential genetic variants in schizophrenia: a Bayesian analysis. World J Biol Psychiatry. 2007;8:12–22. doi: 10.1080/15622970600892004. [DOI] [PubMed] [Google Scholar]
- [116].McConnell SK. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761–8. doi: 10.1016/0896-6273(95)90168-x. [DOI] [PubMed] [Google Scholar]
- [117].Thompson Ray M, Weickert CS, Wyatt E, Webster MJ. Decreased BDNF, trkB-TK+ and GAD(67) mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J Psychiatry Neurosci. 2011;36:195–203. doi: 10.1503/jpn.100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Takahashi M, Shirakawa O, Toyooka K, Kitamura N, Hashimoto T, Maeda K, et al. Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol Psychiatry. 2000;5:293–300. doi: 10.1038/sj.mp.4000718. [DOI] [PubMed] [Google Scholar]
- [119].Durany N, Michel T, Zochling R, Boissl KW, Cruz-Sanchez FF, Riederer P, et al. Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr Res. 2001;52:79–86. doi: 10.1016/s0920-9964(00)00084-0. [DOI] [PubMed] [Google Scholar]
- [120].Weickert CS, Ligons DL, Romanczyk T, Ungaro G, Hyde TM, Herman MM, et al. Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2005;10:637–50. doi: 10.1038/sj.mp.4001678. [DOI] [PubMed] [Google Scholar]
- [121].Schramm M, Falkai P, Feldmann N, Knable MB, Bayer TA. Reduced tyrosine kinase receptor C mRNA levels in the frontal cortex of patients with schizophrenia. Neurosci Lett. 1998;257:65–8. doi: 10.1016/s0304-3940(98)00807-6. [DOI] [PubMed] [Google Scholar]
- [122].Kyosseva SV. Differential expression of mitogen-activated protein kinases and immediate early genes fos and jun in thalamus in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:997–1006. doi: 10.1016/j.pnpbp.2004.05.017. [DOI] [PubMed] [Google Scholar]
- [123].Lachman HM, Stopkova P, Papolos DF, Pedrosa E, Margolis B, Aghalar MR, et al. Analysis of synapsin III-196 promoter mutation in schizophrenia and bipolar disorder. Neuropsychobiology. 2006;53:57–62. doi: 10.1159/000091720. [DOI] [PubMed] [Google Scholar]
- [124].Liguori M, Cittadella R, Manna I, Valentino P, La Russa A, Serra P, et al. Association between Synapsin III gene promoter polymorphisms and multiple sclerosis. J Neurol. 2004;251:165–70. doi: 10.1007/s00415-004-0293-7. [DOI] [PubMed] [Google Scholar]
- [125].Akkad DA, Godde R, Epplen JT. No association between synapsin III gene promoter polymorphisms and multiple sclerosis in German patients. J Neurol. 2006;253:1365–6. doi: 10.1007/s00415-006-0214-z. [DOI] [PubMed] [Google Scholar]
- [126].Otaegui D, Zuriarrain O, Castillo-Trivino T, Aransay A, Ruiz-Martinez J, Olaskoaga J, et al. Association between synapsin III gene promoter SNPs and multiple sclerosis in Basque patients. Mult Scler. 2009;15:124–8. doi: 10.1177/1352458508096682. [DOI] [PubMed] [Google Scholar]
- [127].Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–30. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]