

Abstract
Clofazimine is a lipophilic antibiotic with an extremely long pharmacokinetic half-life associated with the appearance of crystal-like drug inclusions, in vivo. Here, we studied how clofazimine accumulates inside cells in the presence of supersaturating, extracellular concentrations of the drug (in the range of physiological drug concentrations). Based on a combination of molecular imaging, biochemical analysis and electron microscopy techniques, clofazimine mass increased inside cells in vitro, over a period of several days, with discrete clofazimine inclusions forming in the cytoplasm. These inclusions grew in size, number and density, as long as the drug-containing medium was replenished. With Raman confocal microscopy, clofazimine’s spectral signature in these inclusions resembled that of amorphous clofazimine precipitates and was unlike that of clofazimine crystals. Additional experiments revealed that clofazimine first accumulated in mitochondria, with ensuing changes in mitochondrial structure and function. In turn, the degenerating organelles coalesced, fused with each other and condensed to form prominent drug-membrane aggregates (dubbed autophagosome-like drug inclusions or “aldis”). Like clofazimine, it is possible that intracellular drug-membrane aggregate formation is a common phenomenon underlying the reported phenotypic effects of many other small molecule drugs.
Keywords: Drug sequestration, autophagosome-like drug inclusion, drug-membrane aggregates, clofazimine, confocal Raman imaging, phospholipidosis, intracellular crystals
Introduction
Intracellular drug targeting can be useful to minimize drug toxicity and maximize efficacy. Nevertheless, the sequestration of drugs within intracellular organelles can also affect pharmacokinetics and pharmacodynamics after systemic delivery1–3. Among several known organelle sequestration mechanisms, ion-trapping of drugs within acidic compartments, i.e. lysosomes, has been most extensively studied4, 5. Protonation of weakly basic molecules leads to increased solubility and decreased membrane permeability, and promotes retention within lysosomes and endosomes. In the case of lipophilic cations, the electrical potential present across the inner membrane of mitochondria drives accumulation of membrane permeant lipophilic cations into this organelle 1, 3, 6, 7. Beyond transmembrane pH gradients (i.e. lysosomotropism) and electrical potentials (i.e. mitochondriotropism), interactions of chemical agents with intracellular membranes could directly promote their accumulation. To study this phenomenon, the FDA approved antibacterial drug clofazimine was selected as a model compound. Clofazimine is highly lipophilic and possesses and unusually long pharmacokinetic half-life of up to 70 days 8 associated with extensive accumulation of the drug in the body9.
Although clofazimine’s low solubility has been suspected to be responsible for its atypical pharmacokinetics, the mechanism by which drug accumulates intracellularly is not known8–12. To study this, we decided to pursue a cell-based approach to determine whether clofazimine forms intracellular precipitates or crystal-like deposits in Madin Darby Canine Kidney (MDCK) cells, in vitro. MDCK cells were selected because they are considered a robust in vitro assay system for drug transport studies. While these cells originated from the epithelia of the distal renal tubules, they are also used as a model to study the passive diffusive component of intestinal drug absorption2 and to study drug bioaccumulation associated with ion-trapping13 and phospholipidosis3, 14. Taking advantage of clofazimine’s red color, the drug’s absorbance, fluorescence, and biodistribution properties could be readily measured. In addition, Raman spectral imaging and electron microscopic examination were performed to reveal the mechanism of clofazimine’s intracellular accumulation: the formation of organelle-derived, intracellular drug-membrane aggregates.
Materials and Methods
Chemicals
Reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise indicated.
Drug mass measurement
MDCK (CCL-34 ™ ATCC, Manassas, VA) cells were maintained with DMEM plus 10 % FBS (Gibco® 10082), 10 % non-essential amino acid (Gibco® 11140 from Invitrogen, Carlsbad, CA), and with 10 % penicillin/streptomycin (Gibco® 15140) in a 37°C 5% CO2 incubator. For experiments, MDCK cells were incubated with clofazimine solutions: clofazimine-containing medium was made by diluting 10 mM clofazimine stock (in DMSO) into DMEM plus 5 % FBS immediately before adding to cells (clofazimine added to cell culture medium without FBS formed amorphous precipitates, but in 5% FBS no visible precipitates formed due to drug binding to serum proteins). For clofazimine mass measurements, cells were detached using Trypsin-EDTA (Gibco® 15050), resuspended in 0.1 M citric acid/0.1 M trisodium citrate buffer (pH 5) and counted with hemocytometer. Equal number and volume of cells were transferred to 96-well plates and dissolved with 100 μl ATCC detergent (American Type Culture Collection, 30–1010K). The clofazimine absorbance at 495 nm was measured with a plate reader (Synergy-2, Biotek Instruments, Winooski, VT) and mass was calculated with the aid of a standard curve generated separately.
LC/MS analysis of intracellular clofazimine
Analysis of clofazimine’s chemical stability was performed for samples treated with drug-containing media (10 μM or 0 μM), and for pure drug (1 μg/ml). For extraction, samples were resuspended in methanol to dissolve and extract the drug, and then centrifuged at 15,000g for 10 minutes to remove insoluble components. Analysis was performed with HPLC (Agilent1200, Zorbax RX-C18 column) coupled to a mass spectrometer (Applied Biosystems QTRAP 3200) eluted with H2O:acetonitrile gradient (Analyst1.4, Applied Biosystems). Full scanning of total ion current (TIC) from 200 to 800 of the reference drug sample revealed intact clofazimine 473.2 and two ion transition fragments of 431.2 and 429.2 (m/z). For treated cells, multiple reaction monitoring (MRM) was performed, scanning for 44 common metabolic transformations of the intact molecule and the two fragments of 431.2 and 429.2 m/z.
Light and fluorescence microscopy
Cells on cover glasses were cultured with clofaziminemedium for the indicated times. For cellular lipid staining, 1 μg/ml Nile red was added for 2 hours. MTR staining was carried out with or without CFZ in media with 150 nM of MTR (from 10 mM DMSO stock solution) incubated for 45 minutes. After incubation, cover glasses were washed twice with DPBS and inverted onto glass slides for visualization on Olympus 51X upright epifluorescence/polarization microscope equipped 100× objective (1.40 NA, PlanApo oil emersion), cross polarizers, U-MWIBA3 (eGFP) for green U-MWG2 (rhodamine) filter cube for red channel, and an Olympus DP-70 color camera. Images were acquired using DP controller 3.1.1.267 under same exposure settings.
Transmission Electron Microscopy
Samples were processed as previously described 15 and imaged with a Philips CM-100 at magnifications from ×2,600 to ×130,000. Objects from pictures of cells taken during triplicate experiments were counted and scored for each category (with 6 to 10 individual cells analyzed per sample).
Mitochondrial respiration measurement
Mitochondrial respiration was measured using Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience Inc., North Billerica, MA) equipped with optical sensors that measure oxygen and protons in the extracellular media16. Briefly, 24 hours before the measurement, 50,000 cells were seeded onto wells of XF24 cell culture microplates (V7, Seahorse Bioscience) with 150 μl DMEM, and additional 100 μl was added after 4 hours when cells attached firmly. After 24 hours, cells were washed and preincubated with bicarbonate-free media (DMEM base XF Assay Medium, Seahorse Bioscience 100965) in 37 °C incubator without CO2 control. Injection cartridge (Seahorse Bioscience 100840) was pre-incubated with calibration media 24 hour-prior to the assay and four different injection ports were loaded with following compounds containing 20 μM BSA with 10 fold concentrate of the drugs in media: CFZ (100 μM, port A), oligomycin (10 μM, port B), FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone, 30 μM, port C), and Antimycin A (15 μg/ml, port D). After automated calibration of the sensors, at pre-determined time points, O2 level was measured and OCR (oxygen consumption rate) was calculated from the depleting oxygen level and data were analyzed using algorithm described by the manufacturer17. For plotting drug effects, OCR measurements for each well were subtracted by the internal, baseline OCR obtained for that same well, based on the measurement made before injecting any drug (arbitrarily set to the 27th minute read after cells equilibrate in the instrument).
Confocal Raman microscopy
Cover glasses were inverted on glass slides, sealed and frozen in liquid N2 and kept at <−20 °C until using a WITec alpha300R instrument equipped with CCD camera, UHTS300 Raman spectrometer and with 10 mW power 532 nm doubled Nd:YAG laser analyzed (WITec GmbH©, Germany). For analysis of clofazimine signal, a 2nd order polynomial was fitted to the raw data for background fluorescence subtraction, and the remaining signal was median-2 filtered (WITec project 2.00 software).
Lipid analysis
Cells were prepared on 10 cm-diameter dish and incubated with media containing either 10 μM or 0 μM of clofazimine for 36 hours. Cells were trypsinized, counted and kept frozen until analysis at the Lipidomics lab at Michigan Metabolomics and Obesity Center. Lipids were extracted using methanol:chloroform (2:1) solution and isolated with thin layer chromatography (TLC Silica gel 60, E.Merck). Chloroform:methanol:acetic acid:water (100:40:12:4) was used as mobile phase and TLC bands were scraped off and weighed for neutral lipid and phospholipid species, while phospholipid species (lyso phosphatidylcholine, phosphatidic acid, phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl serine, phosphatidyl inositol, sphingomyelin, cardiolipin) were quantified according to the inorganic phosphorous assay18. For each lipid species, reference standards were purchased from Sigma. The experiments were independently repeated three times and the results analyzed with the Student’s T-test.
Mitochondria isolation and clofazimine partitioning
Mitochondria were isolated using a commercial kit (MITOISO1, Sigma) from approximately 108 cells and homogenized with rotating motor (200 rpm, >50 times) in 4 – 5 ml volume of Extraction Buffer A (E2778 Sigma) followed by centrifugation at 1,000g for 5 min, and then at 11,000g for 10 min. After the enrichment procedure, a small brown-white color pellet comprised of enriched mitochondria was verified using the JC-1 accumulation assay, following the kit’s instructions. To measure CFZ binding to isolated mitochondria, 5 μM CFZ solution was made in Storage Buffer (S9689 Sigma), which contains ATP/ADP and DTT in potassium buffer to sustain the mitochondrial respiration, then added to the isolated mitochondria. The mix was vortexed until homogenous, then centrifuged at 11,000g for 10 min. Clofazimine that was bound to the pellet, that remained dissolved in the liquid, or that was absorbed to the sides of the tube were extracted with MeOH. MeOH extracts were cleared by after centrifugation at 11,000g for 10 min. The mass of clofazimine in each fraction was determined from the concentration of clofazimine in the extracts, using the absorbance plate reader, with the aid of a standard curve. The mass of clofazimine remaining in the supernatants was determined by adding the mass of clofazimine that adsorbed to the sides of the centrifuge tube to that which remained dissolved in the liquid.
Image processing
For display, images were digitally enhanced and overlaid using Photoshop®. For control vs. experimental comparisons within the same figure, contrast and brightness settings were similarly adjusted.
Results
1. Biochemical analysis and transmitted light microscopy reveal intracellular clofazimine inclusion formation
Clofazimine accumulated intracellularly upon incubation with cells (Figure 1A). Based on the calculated single cell volume of 1.6 pL5, cellular concentration approximated 4 mM. However, concentration in the extracellular medium was 10 μM, mostly in protein-bound form (SI. 1), which is within range of serum concentrations in vivo19. We observed a rapid increase of cellassociated drug mass within minutes after the start of incubation, followed by a continuous, gradual increase that continued for as long as the cells were maintained in the clofazimine containing medium (Figure 1B). To determine if intracellular clofazimine was metabolized, we performed LC/MS chemical analysis of intracellular clofazimine in cells incubated with or without 10 μM clofazimine in culture medium for 90 hours. Intracellular clofazimine was present intact with no major metabolites detected (SI. 2).
Figure 1.

Clofazimine accumulation kinetics. A) Time course analysis of intracellular clofazimine accumulation. (error bars represent the s.e.m., N=3). B) Red intracellular drug inclusions grow in size. Brightfield images were acquired during 0 to 120 hour incubation in 10 μM clofazimine containing 5% FBS-DMEM. Lines were manually drawn to indicate individual cell boundaries. N: nucleus. C) Intrinsic fluorescence of CFZ and Nile Red (NR) staining of control and drug-treated cells. Cells were incubated with CFZ containing DMEM for 24 hours and imaged with the standard eGFP (green) and rhodamine (red) fluorescence filter set. Arrows point to large clofazimine inclusions lacking green fluorescence (whereas small inclusions are visibly fluorescent). Nile Red staining (NR) of CFZ treated vs. untreated samples were acquired and displayed under the same exposure settings, showing significantly greater NR fluorescence in association with clofazimine inclusions.
Within the first 12 hours of clofazimine accumulation, small and scattered clofazimine inclusions in the cytoplasm formed nanometer-sized bright red speckles, visible with transmitted light illumination. After 24 hours or longer, inclusions grew in size, ranging from 1 to 2 μm in length, and assumed irregular shapes and heterogeneous red color. They continued growing in size for as long as the extracellular medium was replenished with fresh, clofazimine containing medium (Figure 1B, 120 hours). The size, shape and color of the inclusions were consistent with previous report in mouse lung macrophages10. Intracellular clofazimine accumulation was reversible. After clofazimine-treated cells were transferred to drug-free medium, the efflux half-life was between 2 and 3 hours, with some red inclusions remaining visible even after 6 hours of efflux. At these concentrations, clofazimine incubation inhibited cell growth and intracellular drug mass accumulation was proportional to the drug concentration in the medium (SI. 3).
2. Fluorescence microscopy yields evidence that large clofazimine inclusions form from condensation and aggregation of smaller vesicles
In buffer, dilute clofazimine solutions (< 10 μM) fluoresce under 450 nm illumination (528 nm emission), but more concentrated solutions result in fluorescence quenching (SI. 4). With epifluorescence microscopy, we found small intracellular inclusions were fluorescent at those wavelengths (460 – 495 nm excitation, 515 – 550 nm emission), but that the larger, redder inclusions apparent at later time points did not fluoresce (arrow, Fig 1C). We inferred that clofazimine first accumulated in the small fluorescent vesicles, and then became increasingly concentrated in the larger, red (but nonfluorescent) inclusions.
The larger clofazimine inclusions were brightly stained with Nile Red (NR), a highly lipophilic, neutral fluorophore that is commonly used as cellular lipid stain. This suggested that the larger clofazimine inclusions were enriched in a hydrophobic component. Without clofazimine, cells showed significantly lower NR staining which bleached very quickly, without evidence of organelle-associated NR staining. However, measurement of total cellular lipid, neutral lipid and various phospholipids did not reveal statistically significant alterations in lipid mass or relative contents after clofazimine treatment (data not shown). Thus, while the growth of clofazimine inclusions was associated with increased NR staining and photostability, there were no measurable changes in lipid profiles.
3. Chemical imaging yields evidence of clofazimine aggregates but no evidence of intracellular crystals
To investigate whether intracellular clofazimine inclusions were formed by crystallization of clofazimine, cells were analyzed with polarization microscopy. Birefringence from intracellular clofazimine inclusions was not detectable (SI. 5). With confocal Raman microscopy20, 21, clofazimine-treated cells exhibited a fluorescence signal (Figure 2A) superimposed on a drug-specific vibrational Raman signal (Figure 2B). Clofazimine fluorescence (Figure 2C) and Raman signals (Figure 2D) was localized almost exclusively to the cytoplasm, mostly in discrete spots in the perinuclear region corresponding to the red clofazimine inclusions observed by transmitted light microscopy (Figure 1).
Figure 2.

Confocal Raman imaging of clofazimine-treated cells. A) Polynomial fitting of a representative, raw spectral scan acquired from a clofazimine treated cell. B) Background subtracted Raman vibrational spectra from A. C) The spectral intensity map of a selected fluorescence spectral region (2199 – 2296 cm−1), highlighted in blue in panel A. D) The spectral map of the selected, background subtracted, Raman spectral region (1100 – 1600 cm−1), highlighted in blue in panel B. E) spectra of various regions of in D (1: cytosol, 2: perinuclear region, 3: center of the nucleus, 4: perinuclear region; 5: center of the nucleus; 6–8: various punctate cytoplasmic foci. F) Representative, fluorescence-subtracted Raman spectra of untreated MDCK cells (a); clofazimine precipitates in different buffer solutions (b – g); dry clofazimine crystals (h); and, drug-treated cells (i). The intensity is in arbitrary units, normalized to the same scale. 1436 and 1461 cm−1 (*) are vibrational peaks that are prominent in crystalline clofazimine but almost absent the amorphous clofazimine (bold line).
Clofazimine’s vibrational signal (Figure 2D, in the region between 1100 and 1600 cm−1) overlapped with the overall fluorescence signal. After subtracting the fluorescence signal, different parts of the cells exhibited similar Raman spectra, only varying in intensity (Figure 2E). The strongest vibrational signals came from the punctate foci in the perinuclear region (Figure 2E 2, 4, 6, 7, and 8). In contrast, regions inside the nucleus and at the cell periphery lacked Raman signal (Figure 2E 1, 3, and 5). Again, the specific Raman chemical signal of clofazimine corresponded to the distribution of red inclusions observed with transmitted light microscopy (Figure 1).
As a reference, the vibrational Raman spectra of untreated cells (negative control; Figure 2Fa) and the vibrational spectra of crystalline and amorphous clofazimine precipitates in various buffers (positive controls; Figure 2Fb – h) were compared to the Raman spectra of clofazimine-treated cells. The vibrational spectra acquired from the punctate foci of clofazimine-treated cells (Figure 2Fi) resembled that of positive clofazimine controls (2Fb – h), with signature peaks at 1167, 1255, 1353, and several other peaks between 1406 and 1565 cm−1. In buffer (2Fb – e), peaks at 1298 and 1493 cm−1 decreased in intensity, whereas the peak at 1565 cm−1 only appeared in association with crystalline clofazimine in pH 5 and 7, indicating vibrational signals of clofazimine are sensitive to the local microenvironment.
Most importantly, the reference spectrum of amorphous clofazimine at pH 5 (Figure 2Fc) resembled that of amorphous clofazimine at pH 3, whereas the spectrum of crystalline in pH 5 (Figure 2Fd) resembled that of crystalline clofazimine at pH 7 (SI. 6). Remarkably, the spectra from drug-treated cells (Figure 2Fi) clearly lacked the peaks of 1436 (*) and 1461 (*) cm−1 which were the prominent, signature peaks of crystalline clofazimine. These peaks were absent in amorphous clofazimine aggregates (Figures 2Fc and f, bold). Thus, chemical imaging of intracellular drug inclusions also did not yield evidence for the presence of clofazimine crystals.
4. Transmission electron microscopy reveals an intracellular drug-membrane aggregation pathway, leading to aldi formation without intracellular crystals
With TEM, the cytoplasm of control cells appeared normal in morphology (Figure 3A and SI. 7a – c). In comparison, incubation with clofazimine-containing medium for 24 hours induced many dense, osmiophilic multivesicular bodies (MVB) and many other darkly stained, osmiophilic vesicles filled with granular material (Figure 3B and SI. 7g – i). Upon prolonged exposure (10 μM clofazimine for >72 hours), the bulk of cellular membrane content became reorganized into morphologically-distinct and atypical inclusions, many possessing multilamellar membrane features (Figure 3C and SI. 7g – l). These inclusions were highly complex and heterogeneous in their internal structure, resembling autophagosomes22. The number, size, location and appearance of these heterogeneous, membranous inclusions corresponded in size, number and distribution to the red clofazimine inclusions observed under transmitted light microscopy (Figures 3D – F and SI. 7j – p). Hence, we inferred these structures corresponded to clofazimine-membrane aggregates, and labeled them autophagosome-like drug inclusion, aldi (SI. 7m – p).
Figure 3.

TEM images of control cells (A, vehicle-only with 5% FBS-DMEM after 24 hr treatment); and cells incubated with 10 μM clofazimine for 24 hr (B) or 87 hr (C). D, E, and F are zoom-in of boxed regions in A, B, and C, respectively. Cytoplasmic objects were categorized into autophagosome-like drug inclusions (aldis) if they did not show characteristic features of the typical organelles, while exhibiting lipid-rich (lamellar or vesicular) internal structures stained with osmium tetroxide.
Cytoplasmic object counts from TEM sections of cells exposed to clofazimine containing cell culture media for varying periods of time allowed piecing together a sequence of events leading to aldi formation (Figure 4A and SI. 7). Depending on their apparent size, complexity, density of the internal structure and the time course of appearance, we divided aldis into early (Figure 4B) and late stage (Figure 4C). Based on object counts, aldis accumulated with time, to become the most frequent organelle observed in clofazimine-treated cells (Figure 4D).
Figure 4.
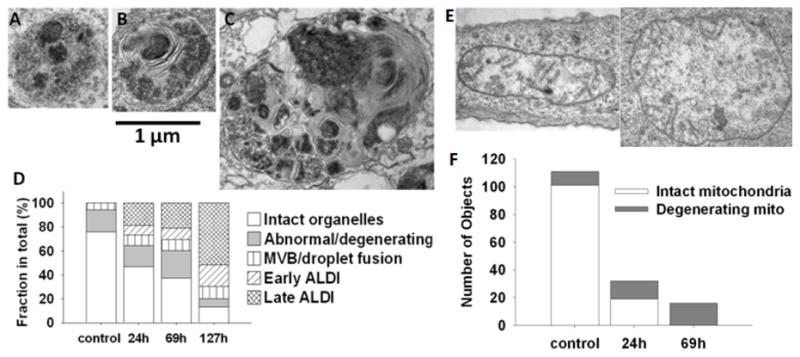
Temporal analysis of aldi biogenesis and mitochondria degeneration. A) Representative image of osmiophilic and granular multivesicular body that appears during the first 24 hr incubation. B) Early aldi, showing internal lamellae and heterogeneous appearance C) More complex aldi representative of those that appeared at latter time points. D) Measured frequency of different organelles, at various time points after beginning of clofazimine incubation. E) TEM images of abnormal/degenerating mitochondria in clofazimine treated cells. F) Frequency of normal vs. degenerating mitochondria changes during drug treatment. For TEM organelle counts, morphological features were manually scored from 6 or more cells.
5. Additional structural and functional evidence for a mitochondrial origin of aldis
In TEMs, mitochondria caught our attention, which were prominent in untreated cells but seldom observed in clofazimine-treated cells. We confirmed the number of intact mitochondria (Figure 4E) decreased significantly upon drug treatment, concomitant with an increase in the number of organelles with morphological features resembling degenerating mitochondria (Figure 4F). Staining cells with a mitochondria-specific, membrane potential-sensitive probe, MitoTracker® Red CMXRos (MTR, Figure 5), revealed that incubation with clofazimine led to mitochondrial membrane depolarization, with diffuse cytoplasmic MTR staining. In untreated cells, normal MTR signal was observed in association with cytoplasmic organelles possessing vermiform appearance, typical for mitochondrial labeling. After clofazimine incubation, MTR background signal in cytoplasm was increased, with fewer organelles showing specific mitochondria morphology. After 21 hours, small clofazimine-containing vesicles (observed using the epifluorescence eGFP channel) corresponded to MTR-labeled mitochondria (observed under rhodamine channel). However, the larger clofazimine inclusions which lacked clofazimine fluorescence did not stain with MTR. Accordingly, we inferred that some of the membranes in aldis could originate from degenerating mitochondria.
Figure 5.

Clofazimine associates with mitochondria, followed by dissipation of the mitochondrial membrane potential. A) MitoTracker Red (MTR) staining of untreated vs. clofazimine treated cells. Arrows at 21 hr treatment indicate clofazimine fluorescence (green channel) colocalized with functional mitochondria (labeled with MTR signal in red channel). However, the larger (older) clofazimine inclusions do not stain with MTR. (B) Clofazimine binds to isolated mitochondria. As a control, the same experiment was repeated with storage buffer alone and no mitochondria (w/o mito). Under control conditions, clofazimine signal was not detected in the pellet after centrifugation. Error bar represents standard deviation from representative experiment. Student’s T-test show statistically significant differences in both supernatant and pellet (p<0.05; N=2). C) Oxygen consumption rate (OCR) differences of clofazimine-treated cells (◆) vs. control cells (
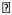 ). Various inhibitors of mitochondrial respiration were sequentially applied in the presence or absence of clofazimine at pre-determined time points, as indicated in x-axis. The OCR difference was calculated as the measured OCR level minus the baseline OCR level (at the 27th minute). Error bars indicate s.e.m; N=3 (the error bar may be smaller than marks). Statistical significance for each treatment was tested comparing the last data point before vs. the new treatment’s last data point: 4th point vs. 11th point for clofazimine effect, 11th vs. 14th for oligomycin, 14th vs. 17th for FCCP, and 17th vs. 20th for Antimycin A. For every treatment, p < 0.05.
). Various inhibitors of mitochondrial respiration were sequentially applied in the presence or absence of clofazimine at pre-determined time points, as indicated in x-axis. The OCR difference was calculated as the measured OCR level minus the baseline OCR level (at the 27th minute). Error bars indicate s.e.m; N=3 (the error bar may be smaller than marks). Statistical significance for each treatment was tested comparing the last data point before vs. the new treatment’s last data point: 4th point vs. 11th point for clofazimine effect, 11th vs. 14th for oligomycin, 14th vs. 17th for FCCP, and 17th vs. 20th for Antimycin A. For every treatment, p < 0.05.
Consistent with a mitochondrial origin of these induced, drug-membrane complexes, clofazimine avidly partitioned into isolated mitochondria in vitro (Figure 5B). In clofazimine- treated cells, mitochondrial respiratory function was perturbed by clofazimine treatment (Figure 5C). Although basal oxygen level consistently ranged between 132 – 141 mmHg, oxygen consumption rate (OCR) was affected by clofazimine treatment, which reflects a change in mitochondrial respiration responsible for the production of ATP through the reduction of O2. In positive control experiments, OCR varied in response to the standard pharmacological inhibitors of mitochondrial respiration (Figure 5C,
): 1) oligomycin inhibited the ATP synthase (complex V), resulting in a decrease in OCR; 2) carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) disrupted the electrochemical gradient across the mitochondrial membrane, increasing the OCR; and, 3) Antimycin A blocked complex I and III in electron transfer chain, decreasing the OCR. In the experimental conditions, addition of clofazimine (◆) increased the OCR, suggesting a disruption of the proton gradient across the mitochondrial inner membrane, similar to FCCP addition. Furthermore, after clofazimine treatment, the OCR became unresponsive to oligomycin or FCCP, whereas Antimycin remained fully active. This was consistent with clofazimine interfering with the mitochondrial membrane potential, similar to FCCP.
Discussion
Based on our observations, the intracellular accumulation of clofazimine is accompanied by the formation of massive, intracellular drug-membrane aggregates. Morphologically, these aggregates do not resemble the crystal-like drug inclusions that are observed in patients after months of clofazimine treatment 8–11, but they may be a precursor for such inclusions since similar structures can be also be observed in electron micrographs of clofazimine-treated tissues. Because clofazimine has weakly basic amine groups with reported pKa ranging from 7.87 to 9.1123, the drug is expected to be protonated and more water soluble in acidic environments. However, the drug is also expected to become deprotonated and less soluble in neutral or alkaline environments, which can lead to the formation of supersaturated solution once the drug is absorbed and distributes throughout the body. It is possible that a lipoprotein carrier could be involved in cellular uptake24 and that the low pH environment of lysosomes could lead to ion-trapping4, 13. However, based on electron microscopy, the mechanism ultimately responsible for sequestering clofazimine appears to be the formation of aldis: drugmembrane aggregates that resembled autophagosomes in morphology. Biochemical studies with isolated mitochondria revealed that clofazimine accumulates in mitochondria, and functional studies confirmed that clofazimine interacts with mitochondria inside cells, suggesting at least some of the membranes in these aggregates may originate from degenerating mitochondria.
In the continuous presence of clofazimine, the formation of drug-membrane aggregates can destabilize organelle structure and function, inducing membranes to coalesce, fuse and condense, and give rise to aldis. The formation of drug-membrane aggregates can serve as a sink to facilitate the continuous accumulation of clofazimine inside cells, without clofazimine crystals forming intracellularly. With supersaturated clofazimine solutions forming in the gastrointestinal tract, clofazimine can be absorbed into the body and partition into cellular lipids and the phospholipid membranes that form the boundaries of intracellular organelles. Upon prolonged dosing, aldis can accumulate and may serve as an intracellular clofazimine depot. Presumably, aldis may continue to form and grow as long as the drug is not toxic to cells, and the rate of drug-membrane aggregate formation does not exceed the cells’ capacity to synthesize new membranes and regenerate its degenerating organelles.
Morphologically, aldis resembled autophagosomes or mitophagosomes 22. However, aldis lacked the double membrane that normally surrounds autophagosomes. Unlike autophagosomes and mitophagosomes, aldis appeared completely filled with condensed lipid membrane aggregates, instead of the typical, degenerating vesicular membrane cargo. In the natural degradation of mitochondria (and other organelles) 22, 25, 26, a membrane structure termed phagophore is observed to engulf the degenerating organelle. Later, the mitophagosomes fuse with lysosomes to form autophagolysosomes 22, 25, 26 which degrade the membrane contents for intracellular recycling. Unlike autophagolysosomes or mitophagosomes, aldis appeared to grow continuously by coalescence and condensation. Considering the possibility that aldis were related to multilamellar bodies resulting from phospholipidosis 14, 27, a toxicological phenotype associated with certain classes of lipophilic drugs 28, 29, clofazimine did not lead to a measurable increase in phospholipid levels in MDCK cells.
To conclude, the ability of clofazimine to interact with mitochondria and induce the formation of intracellular drug membrane aggregates may be worth considering not only as a drug side effect, but also in terms of potential therapeutic applications. Indeed, while such phenotypic changes are generally reported as drug side effects, they could also have cytoprotective functions30, 31. For example, interaction of clofazimine with mitochondria could be useful to attenuate mitochondrial damage associated with oxidative stress that occurs during ischemia-reperfusion injury 32, 33. Today, there is an increasing interest in probing drug-membrane interactions as a determinant of the cellular disposition of lipophilic small molecule drugs6, 34. Experimental evidence indicates that chloroquine35 and amiodarone28, 36 accumulate in association with multilamellar bodies. However, characterizing the formation of drug-membrane complexes within cells has been a challenge. In this context, the ability to use a combination of chemical imaging technologies including Raman, fluorescence, and absorbance to study clofazimine’s intracellular disposition, together with the ability to perform electron microscopic observations on the resulting drug inclusions, constitutes a major advance in terms revealing the appearance of prominent drug-membrane aggregates that result from drug-membrane interactions as they occur within cells.
Supplementary Material
Acknowledgments
The project was supported by grant number GM007767 from NIGMS for J.B., and by NIH grant RO1GM078200 to G.R.R. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. We thank Dorothy Sorenson (TEM, University of Michigan Microscopy and Imaging Analysis Lab), Peng Zou (LC/MS), Dr. Arun K. Das (Michigan Nutrition and Obesity Research Center, NIH grant, DK089503), Katherine Overmyer (Seahorse, Burant Lab), and the engineers and scientists at WITec (Raman confocal microscopy) for technical assistance. We thank Dr. Charles Burant, Dr. Nair Rodriguez-Hornedo, and Dr. David E. Smith for insightful comments.
Footnotes
Supporting Information Available
Supporting information figure SI. 1 – SI. 7. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Rajendran L, Knolker HJ, Simons K. Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov. 2010;9:29–42. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 2.Zhang X, Shedden K, Rosania GR. A cell-based molecular transport simulator for pharmacokinetic prediction and cheminformatic exploration. Mol Pharmaceutics. 2006;3:704–16. doi: 10.1021/mp060046k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng N, Tsai HN, Zhang X, Rosania GR. The subcellular distribution of small molecules: from pharmacokinetics to synthetic biology. Mol Pharmaceutics. 2011 doi: 10.1021/mp200092v. (Submitted for publication) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, Vanhoof F. Lysosomotropic Agents. Biochemical Pharmacology. 1974;23:2495. doi: 10.1016/0006-2952(74)90174-9. [DOI] [PubMed] [Google Scholar]
- 5.Zheng N, Zhang X, Rosania GR. Effect of phospholipidosis on the cellular pharmacokinetics of chloroquine. J Pharmacol Exp Ther. 2010;336:661–71. doi: 10.1124/jpet.110.175679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balaz S. Modeling kinetics of subcellular disposition of chemicals. Chem Rev. 2009;109:1793– 899. doi: 10.1021/cr030440j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horobin RW, Trapp S, Weissig V. Mitochondriotropics: A review of their mode of action, and their applications for drug and DNA delivery to mammalian mitochondria. Journal of Controlled Release. 2007;121:125–136. doi: 10.1016/j.jconrel.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee DK, Ellard GA, Gammon PT, Waters MF. Some observations on the pharmacology of clofazimine (B663) Am J Trop Med Hyg. 1974;23:1110–5. doi: 10.4269/ajtmh.1974.23.1110. [DOI] [PubMed] [Google Scholar]
- 9.Atkinson AJ, Jr, Sheagren JN, Rubio JB, Knight V. Evaluation of B.663 in human leprosy. Int J Lepr Other Mycobact Dis. 1967;35:119–27. [PubMed] [Google Scholar]
- 10.Conalty ML, Barry VC, Jina A. The antileprosy agent B. 663 (Clofazimine) and the reticuloendothelial system. Int J Lepr Other Mycobact Dis. 1971;39:479–92. [PubMed] [Google Scholar]
- 11.Sukpanichnant S, Hargrove NS, Kachintorn U, Manatsathit S, Chanchairujira T, Siritanaratkul N, Akaraviputh T, Thakerngpol K. Clofazimine-induced crystal-storing histiocytosis producing chronic abdominal pain in a leprosy patient. Am J Surg Pathol. 2000;24:129–35. doi: 10.1097/00000478-200001000-00016. [DOI] [PubMed] [Google Scholar]
- 12.McDougall AC. Electron microscope studies of the antileprosy drug B663 (clofazimine; Lamprenel) Int J Lepr Other Mycobact Dis. 1974;42:1–12. [PubMed] [Google Scholar]
- 13.Goldman SD, Funk RS, Rajewski RA, Krise JP. Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis. 2009;1:1445–59. doi: 10.4155/bio.09.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reasor MJ, Hastings KL, Ulrich RG. Drug-induced phospholipidosis: issues and future directions. Expert Opin Drug Saf. 2006;5:567–83. doi: 10.1517/14740338.5.4.567. [DOI] [PubMed] [Google Scholar]
- 15.Chen VY, Posada MM, Blazer LL, Zhao T, Rosania GR. The role of the VPS4a-exosome pathway in the intrinsic egress route of a DNA-binding anticancer drug. Pharmaceutical Research. 2006;23:1687–1695. doi: 10.1007/s11095-006-9043-0. [DOI] [PubMed] [Google Scholar]
- 16.Ferrick D, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today. 2008;13:268–74. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, Ferrick DA, Nicholls DG, Brand MD. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–78. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–8. [PubMed] [Google Scholar]
- 19.O’Connor R, O’Sullivan JF, O’Kennedy R. The pharmacology, metabolism, and chemistry of clofazimine. Drug Metab Rev. 1995;27:591–614. doi: 10.3109/03602539508994208. [DOI] [PubMed] [Google Scholar]
- 20.Chernenko T, Matthäus C, Milane L, Quintero L, Amiji M, Diem M. Label-Free Raman Spectral Imaging of Intracellular Delivery and Degradation of Polymeric Nanoparticle Systems. ACS Nano. 2009;3:3552–3559. doi: 10.1021/nn9010973. [DOI] [PubMed] [Google Scholar]
- 21.Keren S, Zavaleta C, Cheng Z, de la Zerda A, Gheysens O, Gambhir SS. Noninvasive molecular imaging of small living subjects using Raman spectroscopy. Proc Natl Acad Sci U S A. 2008;105:5844–9. doi: 10.1073/pnas.0710575105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 23.Wan H, Holmen AG, Wang Y, Lindberg W, Englund M, Nagard MB, Thompson RA. High-throughput screening of pKa values of pharmaceuticals by pressure-assisted capillary electrophoresis and mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2639–48. doi: 10.1002/rcm.1229. [DOI] [PubMed] [Google Scholar]
- 24.Morrison NE, Marley GM. Clofazimine binding studies with deoxyribonucleic acid. Int J Lepr Other Mycobact Dis. 1976;44:475–81. [PubMed] [Google Scholar]
- 25.Fader CM, Colombo MI. Autophagy and multivesicular bodies: two closely related partners. Cell Death Differ. 2009;16:70–8. doi: 10.1038/cdd.2008.168. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alakoskela JM, Vitovic P, Kinnunen PK. Screening for the drug-phospholipid interaction: correlation to phospholipidosis. ChemMedChem. 2009;4:1224–51. doi: 10.1002/cmdc.200900052. [DOI] [PubMed] [Google Scholar]
- 28.Reasor MJ. Influence of a pre-existing phospholipidosis on the accumulation of amiodarone and desethylamiodarone in rat alveolar macrophages. Res Commun Chem Pathol Pharmacol. 1991;72:169–81. [PubMed] [Google Scholar]
- 29.Reasor MJ, McCloud CM, Beard TL, Ebert DC, Kacew S, Gardner MF, Aldern KA, Hostetler KY. Comparative evaluation of amiodarone-induced phospholipidosis and drug accumulation in Fischer-344 and Sprague-Dawley rats. Toxicology. 1996;106:139–47. doi: 10.1016/0300-483x(95)03175-f. [DOI] [PubMed] [Google Scholar]
- 30.Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, Thervet E, Anglicheau D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4:783–91. doi: 10.4161/auto.6477. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–8. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korde AS, Pettigrew LC, Craddock SD, Maragos WF. The mitochondrial uncoupler 2,4- dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem. 2005;94:1676–84. doi: 10.1111/j.1471-4159.2005.03328.x. [DOI] [PubMed] [Google Scholar]
- 33.Shao H, Li J, Zhou Y, Ge Z, Fan J, Shao Z, Zeng Y. Dose-dependent protective effect of propofol against mitochondrial dysfunction in ischaemic/reperfused rat heart: role of cardiolipin. Br J Pharmacol. 2008;153:1641–9. doi: 10.1038/bjp.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balaz S, Lukacova V. Subcellular pharmacokinetics and its potential for library focusing. J Mol Graph Model. 2002;20:479–90. doi: 10.1016/s1093-3263(01)00149-8. [DOI] [PubMed] [Google Scholar]
- 35.Zheng N, Zhang X, Rosania GR. The Effect of Phospholipidosis on the Cellular Pharmacokinetics of Chloroquine. J Pharmacol Exp Ther. 2010 doi: 10.1124/jpet.110.175679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee P, Kirk RG, Reasor MJ. X-ray microanalysis of cultured alveolar macrophages with phospholipidosis. Exp Mol Pathol. 1993;58:96–104. doi: 10.1006/exmp.1993.1008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.