

Abstract
Although the molecular changes that characterize gliomas have been studied, the pathogenesis of tumor development remains unclear. p21 contributes to gliomagenesis by stabilizing cyclin D1-cdk4 kinase complexes, suggesting that cyclin D1 and cdk4 may also be required for glial tumor development. In this study, we used a mouse model to attempt to confirm this hypothesis, finding that cyclin D1 and cdk4 played active roles in not only the tumor but also the tumor microenvironment. Loss of cdk4 blocked tumor development, but loss of cyclin D1 did not prevent gliomas from developing. Instead, loss of cyclin D1 impeded progression to higher stages of malignancy. Enforcing expression of cyclin D1 was insufficient to correct the progression defect observed in cyclin D1 deficient animals. In contrast, restoration of cdk4 in the cdk4 deficient animals restored cell proliferation and tumor formation, although at lower tumor grades. Notably, the failure of tumors in the cyclin D1 and cdk4 deficient animals to progress to higher grades was correlated with a failure to fully activate microglia in the tumor microenvironment. Moreover, when PDGF-transformed glial cells were engrafted orthotopically into the mice, the tumors that formed progressed to high grades in wild type mice but not cyclin D1 deficient animals. Together, our findings establish that the cyclinD1-cdk4 axis is not only critical in glial tumor cells, but also in stromal-derived cells in the surrounding tumor microenvironment that are vital to sustain tumor outgrowth.
Keywords: glioma, cyclin, cdk, tumor-associated microglia
INTRODUCTION
Despite impressive advances in imaging, diagnosis and treatment, neurologically destructive glial brain tumors remain amongst the most lethal and intractable forms of cancer, with a median prognosis of less than 18 months (1). Although a number of critical molecular alterations underpinning gliomas have been defined, the ways in which these changes orchestrate tumor formation are still unclear. One of the hallmarks of cancer is the capacity for autonomous cell growth through the elaboration of autocrine signaling loops. In malignant oligodendroglioma, over-expression of PDGF and its cognate receptor are commonly found (2, 3). This constitutive growth factor signaling loop leads to excessive cellular proliferation, which is a fundamental property of malignant glial tumors (4) representing an unshackling of the checkpoints that control the cell cycle.
In most cell types, the cyclin D1-cdk4 axis is the primary gateway through which mitogenic information is channeled. Extracellular growth signals result in the de novo synthesis of cyclin D1 and assembly with its catalytic partner, cdk4, prior to accumulation of the complex in the nucleus (5–7). Whilst the CIP/KIP family of cell-cycle inhibitors had been reported to associate with active cyclin D1-cdk4/6 complexes in cycling cells (8), stabilizing (9) and anchoring them in the nucleus (10, 11), whether this activity contributes to cell proliferation is controversial. The strongest evidence that cdk-inhibitor dependent assembly and stabilization of cyclin D1-cdk4 can contribute to cell proliferation came from a revolutionary approach in tumor modeling, in which the critical role of the cdk inhibitor p21 in PDGF-induced glioma could was mapped genetically to this activity (12). An important prediction stemming from this work is that cyclin D1 and cdk4 should also be required for tumor development.
Here we report the contribution of cyclin D1 and cdk4 to PDGF-induced gliomagenesis. We show that deletion of cdk4 abolishes glioma-associated morbidity and tumor formation, whereas ablation of cyclin D1 impedes tumor progression to higher grades. Surprisingly, tumor cell of origin-specific reconstitution of these gene products did not restore tumor-related morbidity or correct the difference in tumor grade. These results demonstrate the necessity of cyclin D1 and cdk4 for the development of aggressive gliomas, and further, suggest that cyclin D1-cdk4 can contribute to the progression of glial tumors by also influencing non-tumor cells. Histological examination pointed to a defect in the activation of tumor associated microglia (TAMs) in both cyclin D1 and cdk4 deficient mice as well as when tumor cells were transplanted into cyclin D1 knockout mice. We suggest that cyclin D1-cdk4 is a critical regulator of microglial activation, and that in its absence these cells are unable to support the development of glial tumor cells to more malignant states.
MATERIALS AND METHODS
Mice
Cyclin D1, cdk2 and cdk4 knockout mice were generously provided by Piotr Sicinski (Dana-Faber Cancer Institute, USA), Mariano Barbacid (CNIO, Spain) and Hiroaki Kiyokawa (Northwestern University, USA), respectively (13–15).
Cell culture, transfection with RCAS viruses and in vivo transduction of glial cells
The culture of chicken DF-1 fibroblasts used to propagate RCAS viruses, transfection of these cells with RCAS viral plasmids and intracranial injections used to infect nestin-positive glial stem and progenitor cells have all been described previously (12).
Histology and Immunohistochemistry
Morbid animals were euthanized and the brains removed and fixed overnight in 10% neutral buffered formalin (Fisher). The brains were dehydrated in graded alcohol, embedded in paraffin and 5 μm sections used for analysis. Sections were stained with haemotoxylin and eosin (H & E) or used for immunohistochemical analysis performed on an automated Ventana machine (Discovery, Ventana Medical Systems Inc.). The antibodies we used are indicated in the supplementary methods section.
Generation of whole-brain lysates
As soon as animals showed signs of morbidity they were euthanized, and the brains rinsed in PBS, snap-frozen in liquid nitrogen, and ground into a fine powder using a mortar and pestle. Lysates prepared by sonication in 50mM Tris-Cl, pH 7.4; 250mM NaCl; 5mM EDTA; 0.5% NP-40; 10 μg/ml leupeptin; 10 μg/ml aprotinin; 10 μg/ml soybean trypsin inhibitor; 2mM PMSF. Clarified lysates (13,000 × g, 5 minutes, 4°C) were aliquoted, snap-frozen in liquid nitrogen, and stored at −80°C.
Western blotting
20–80 μg of whole-brain lysate was resolved by SDS-PAGE and transferred to PVDF membranes (Millipore) for immunoblotting as described (8) except that bound antibodies were detected with western SuperSignal Pico ECL chemiluminescence (Pierce).
Statistical analysis
Log-rank analysis was used to assess differences in survival based on Kaplan-Meier curves. As considerable regional heterogeneity exists within glial tumors, differences in tumor cell proliferation (as determined by Ki-67 staining) were examined using a logit model with generalized estimating equations to correct for correlations within mice.
RESULTS
Cyclin D1 contributes to PDGF-induced gliomagenesis
Given the exciting discovery that the cdk inhibitor p21 facilitated the nuclear accumulation of cyclin D1-cdk4 complexes to drive the development of PDGF-induced glioma (12), we wanted to probe this further by asking if cyclin D1 and cdk4 were required for tumor formation. Consistent with this possibility, we found cyclin D1 was over-expressed in both human glioma as well as PDGF-driven tumors developing in nestin-tvA mice (Supplemental Fig. 1). Thus, we first set out to determine whether cyclin D1 was required for gliomagenesis. To accomplish this, we crossed the nestin-tvA transgene into a cyclin D1 knockout background (14). These mice were subsequently challenged with RCAS-PDGF and followed for 11 weeks for symptoms of glioma (macrocephaly, lethargy, seizures). Previous studies had shown that more than 90% of wild type nestin-tvA mice developed a glial tumor by this time when challenged with RCAS-PDGF (12). The absence of cyclin D1 reduced the onset of glioma-associated morbidity in a dose-dependent manner (Fig. 1A). Somewhat surprisingly, these animals developed histologically identifiable glial tumors at the same frequency as wild type animals (Fig. 1B). It is known from studies of the human disease that tumor grade is a strong prognostic marker of survival in glioma patients (CBTRUS registry; www.cbtrus.org); thus, we looked at the grade of those tumors that developed in wild type and cyclin D1 knockout mice.
Figure 1. Cyclin D1 contributes to PDGF-induced gliomagenesis.

(A) Kaplan-Meier curve. The indicated mice were followed for 11 weeks following infection with RCAS-PDGF at post-natal day 1–3. wild type; n=27, cyclin D1+/−; n=43, cyclin D1−/−, n=18. cyclinD1+/+ versus cyclinD1−/−; log rank, p=0.02. (B) Gross tumor formation was determined by H & E stain. The percentage of wild type and cyclin D1−/− mice with histologically identifiable glioma is shown. (C) Oligodendrogliomas from wild-type (n=24) and cyclin D1−/− knockout (n=14) mice were graded as low (L), moderate (M) or high (H) according to Kleihues and Cavanee (2000). (D) Tumor cellularity. Due to the diffuse and invasive nature of glioma, cellularity is analyzed as a distribution of the number of nuclei within each 400X field of tumor. The total number of nuclei within each of 120 fields from 4 wild type and 4 cyclin D1−/− tumors was counted and plotted. (E) Ki-67 staining. Sections from three wild type and cyclin D1−/− knockout mice were stained and examined microscopically (representative images are shown on the right). Cell counts were obtained in 90 fields. The number of Ki-67 positive and total nuclei per field was quantified using Metamorph, with the ratio of these two numbers designated the Ki-67 labeling index. cyclinD1+/+ versus cyclinD1−/−, p=0.01. A representative image is shown to the right.
Glial tumors from wild type and cyclin D1 knockout mice were graded according to criteria from the World Health Organization (Kleihues and Cavanee, 2000, Supplemental Fig. 2). In wild type mice, greater than 80% of the tumors were moderate- or high-grade. In contrast, all of the tumors in cyclin D1 knockout mice were low-grade (Fig. 1C). This was confirmed by examining two other parameters correlated with histological grade: tumor cellularity (Fig. 1D) and proliferation (Fig 1E). The difference in proliferation remained apparent even when comparing the low-grade gliomas arising in cyclin D1 knockout animals to the few low-grade gliomas observed in wild-type animals (data not shown). Taken together, these results highlight a critical role for cyclin D1 in the progression of PDGF-induced glial tumors from low-grade to more malignant states.
cdk4 is essential for gliomagenesis
The importance of cdk4 activity for the growth of glioma has been suggested by pharmacological inhibition of cdk4 in cell lines and in mice carrying human GBM xenografts (16–18). To assess if cdk4 contributes to glioma formation, we challenged nestin-tvA wild type and cdk4 knockout animals (13) with RCAS-PDGF and monitored them for symptoms of glial tumors. Strikingly, removal of cdk4 abolished glioma-associated morbidity (Fig. 2A) and tumor formation (Fig. 2B). Although we did not detect tumors by H&E staining, we observed scattered cyclin D1 positive cells and occasionally an isolated focus of such cells (data not shown). These foci did not stain with Ki-67 (data not shown). Whilst such foci were generally not seen in sections from uninfected animals, we could not say whether they might progress into tumors if they coalesced in some fashion. To rule out the possibility that the number of target cells that could be infected by RCAS-PDGF was reduced in cdk4−/− mice, we confirmed that nestin-positive progenitors were expressed in the brains of these animals at levels equivalent to that seen in wild type animals (Figs. 2C and 2D). Thus, cdk4 was critical for the rapid and timely development of glial tumors.
Figure 2. cdk4 is critical for glioma development.
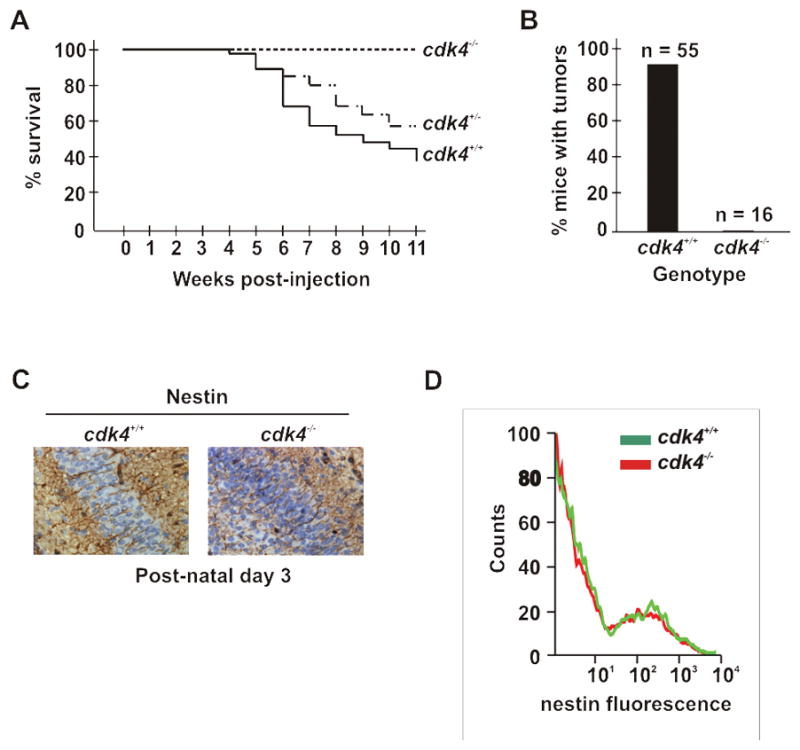
(A and B) Cdk4 deficiency prevents RCAS-HA-PDGF induced tumor development in nestin-tvA mice. These panels are arranged as described in the legend to figure 1. (A) Survival. wild type; n=55, cdk4+/−; n=91, cdk4−/−; n=16. cdk4+/+ versus cdk4−/−; log rank, p=0.0002. (B) Gross tumor formation was determined by H&E staining, and confirmed by the complete lack of cyclin D1 immunoreactivity in the brains of these animals (data not shown). (C and D) Nestin expression is unaffected by deletion of cdk4. (C) Immunohistochemical analysis of neonatal brains showing nestin expression (brown deposits) as long filaments extruded between apposed cells. (D) Flow cytometry. Neonatal brains were removed and single cell suspensions generated prior to labeling with anti-nestin antibodies and analysis by flow cytometry. Overlaid fluorescence intensity curves for representative mice are shown.
Glial-specific expression of cdk4 restores progression of PDGF-induced glioma in cdk4 knockout mice
We next asked if we could reconstitute tumor development by restoring cdk4 expression in nestin-positive progenitors. To accomplish this, we challenged cdk4 knockout mice with RCAS-vectors expressing PDGF and cdk4. Glial cell-specific expression of cdk4 did not change glioma-associated morbidity from the level observed in the cdk4 knockout animals (Fig. 3A); however, these mice did develop glial tumors at the same frequency as wild type animals (Fig. 3B). The bulk of the tumor cells were positive for an oligodendrocyte marker, OLIG2, although tumors also contained trapped neurons (NeuN-positive) and astrocytes (GFAP-positive) (Fig. 3C). These staining patterns and the characteristic nuclear haloing of the tumor cells indicated that these were oligodendrogliomas. Though the Ki-67 index in the reconstituted tumor cells were similar to that seen in wild-type tumor cells (Fig. 3D), these tumors were still low-grade based on their histological appearance and cellularity these tumors (Fig. 3E). Together, these results indicate that cdk4 promotes gliomagenesis both by increasing the proliferation of nestin-positive progenitors and through another mechanism unrelated to its expression in glial tumor cells.
Figure 3. Expression of cdk4 in cdk4 knockout glial cells restores development of glial tumors, but not progression.

(A) Reconstitution of nestin-positive progenitors in cdk4 knockout mice with cdk4 does not change survival from knockout levels following challenge with PDGF. Six neonatal cdk4 knockout nestin-tvA mice were injected intracranially with DF-1 chicken cells expressing RCAS-HA-PDGF and RCAS-cdk4, either singly or in combination. Mice were subsequently followed and assessed as described in the legend to Figure 1A. (B) Gross tumor formation was determined by H & E staining as described in the legend to Figure 1B. (C) Tumors in reconstituted cdk4 knockout mice were classified oligodendroglioma by immunostaing for OLIG2, GFAP and NeuN. Brown deposits represent positive staining. (D) Ki-67 staining. Positive proliferating cells marked by Ki67 stain are brown. (E) Tumor cell reconstitution of cdk4 does not correct the progression defect in glioma. Tumors from the wild type mice and cdk4 knockout mice in which cdk4 expression was reconstituted in oligodendrocytes were graded and plotted as described in the legend to figure 1C.
Sequestration of p27 by cdk4 is not sufficient to explain its tumor-promoting activity
Our results strongly indicate that both cyclin D1 and cdk4 contribute to glial tumor formation. One of the mechanisms by which cyclin D1-cdk4 facilitates proliferation is through sequestration of the cell-cycle inhibitor, p27 (13, 19–21). Thus, we asked whether removal of p27 in cdk4 knockout mice would similarly re-establish PDGF-driven gliomagenesis. Whereas p27 deficiency enhanced glioma formation and associated morbidity in this model (22), ablation of one or both alleles of p27 in a cdk4 knockout background does not restore gliomagenesis, and no readily identifiable tumors were found in any mouse in this cohort (data not shown). Given that sequestration of p27 by the cyclin D1-cdk4 complex is thought to relieve inhibition of the cyclin E-cdk2 kinase (23), we also examined whether cdk2 was required for gliomagenesis. Studies by others have indicated a role for cdk2 in the proliferation and expansion of the glial cell lineage (24–26). However, deletion of cdk2 did not profoundly affect morbidity, glial tumor formation or grade (Supplemental Fig. 3). In sum, these results suggest that neither sequestration of p27 by the cyclin D1-cdk4 complex nor the presence of cdk2 play an essential role in glial tumor formation.
Glial-specific reconstitution of cyclinD1 does not restore progression of PDGF-induced glioma to more malignant stages
Given that we could restore tumor development in cdk4 knockout animals by reconstituting the tumor cell-of-origin with cdk4, but were unable to restore the malignancy of such cells, we asked whether glial cell-specific re-expression of cyclin D1 would affect malignant progression of tumors in cyclin D1 knockout animals following challenge with RCAS-PDGF. For these experiments, we made use of a nuclear-stabilized and activated mutant of cyclin D1, D1T286A, in which mutation of threonine 286 to alanine prevents its phosphorylation and nuclear export by GSK3β (12, 27). The progression of disease in wild type mice expressing this allele of cyclin D1 and PDGF is accelerated compared to mice expressing PDGF alone (data not shown). However, co-infection with RCAS-PDGF and RCAS-D1T286A did not accelerate morbidity of animals any more than infection with RCAS-PDGF alone (Fig. 4A). All of the tumors arising in the co-infected knockout animals expressed cyclin D1 protein (Fig. 4A, inset). Furthermore, tumor cell specific reconstitution of cyclin D1 did not affect the grade of the tumors. These mice developed low-grade tumors based on histology, cellularity and cell proliferation (Figs. 4B, C, D and E). Therefore, it appears that whilst cyclin D1-cdk4 can promote the initial formation of glial tumors in a tumor cell-specific manner, progression to higher grades depended on another role of these proteins, one not carried out within the tumor cell.
Figure 4. Expression of cyclin D1 in cyclin D1 knockout glial cells does not restore progression of PDGF-mediated gliomagenesis.

(A) Neonatal cyclin D1 knockout (n=9) nestin-tvA mice were injected intracranially with DF-1 chicken cells expressing RCAS-HA-PDGF or RCAS-cycD1-T286A, either singly or in combination. Mice were subsequently followed over the course of 11 weeks for the development of symptoms of gliomagenesis, at which time they were sacrificed. The original cyclinD1 knockout survival curve is depicted for comparison: cyclinD1−/− versus cyclinD1−/− + D1-T286A; log rank, p=0.41. Inset; Immunohistochemistry shows re-expression of the exogenous cyclinD1 allele in cyclinD1 knockout glial tumor cells following gene transfer of activated cyclinD1-T286A in cyclinD1 knockout animals. Tumors from wild type animals challenged with PDGF alone do not show reactivity. (B) Gross tumor formation was determined by H & E stain. (C) Exogenous cyclin D1 does not correct the histological grade of oligodendroglioma. (D) Re-expression of cyclin D1 does not affect tumor cellularity. The original plot for cyclin D1 knockout mice is shown for comparison. (E) Re-expression of cyclin D1 does not affect Ki-67 staining. The original cyclinD1 knockout data is plotted for comparison. cyclinD1−/− versus cyclinD1−/− + D1-T286A; log rank p = 0.93.
Expression of PDGFR-α and downstream PDGF signaling is unaffected by the deletion of cyclinD1
Glioma formation and progression is dose-dependent and the amount of PDGF signaling dictates tumor development and grade (28). We therefore interrogated the levels of PDGFR-α expression in glial tumors arising in wild-type and cyclin D1 knockout mice to determine whether the difference in tumor grade might reflect altered expression of PDGFR. PDGFR-α was expressed in both the tumors arising in wild type and cyclin D1 knockout mice (Supplemental Fig. 4A). Consistent with this, phosphorylation of ERK1/2 (Ras pathway effector) and p70S6K (Akt pathway effector) were not affected by cyclin D1 deficiency (Supplemental Fig. 4B). Thus, the ability of glial tumor cells to respond to PDGF at the level of the receptor appears to be unaffected by the absence of cyclin D1. Collectively, this further enforces the notion that tumor cell specific expression of either cyclin D1 or cdk4 was insufficient to correct the defects in morbidity and malignancy in the respective knockout animals, leading us to consider the possibility that the absence of these genes might perturb some aspect of the microenvironment in which glioma arise.
The activation of tumor-associated microglia is perturbed in cyclin D1 and cdk4 deficient mice
The development of cancer relies on the dynamic interplay between the mutant tumor cell and other normal cells in the microenvironment (29). Since glial cell-specific expression of either cyclin D1 or cdk4 was insufficient to correct the defects in morbidity and malignancy in the respective knockout animals, it was possible that the absence of these genes affected recruitment or activation of another cell type in the tumor microenvironment. Given the complete absence of tumors arising in nestin-tvA;cdk4 knockout animals challenged with RCAS-PDGF, we focused on asking whether there were cell types missing in the tumors that arose in nestin-tvA;cyclin D1 knockout animals.
Angio- and vasculogenesis are critical determinants for the transition of tumors, including glioma, to higher grade (30, 31), and the loss of cell-cycle regulators in endothelial cells and stem cell progenitors will impair tumor neoangiogenesis and haematopoietic cell proliferation (32–34). However, there was no obvious difference in CD34 expression when we stained tumors arising in wild type and cyclin D1 knockout mice (Supplemental Fig. 4C), indicating that the absence of cyclin D1 did not prevent the establishment of a coherent tumor vasculature.
We also detected GFAP-positive astrocytes (Fig. 5A; i and ii), NeuN-positive neurons (Fig. 5A; iii and iv), and Iba1-positive microglia (Fig. 5B) in the tumors regardless of genotype. However, whereas the number and intensity of the GFAP- and NeuN- positive cells in tumors was comparable between wild type and mutant mice, the morphology and intensity of the Iba1 stained cells was very different.
Figure 5. The absence of cyclin D1 does not affect the infiltration of stromal-derived cells into glial tumors, but alters the activation state of tumor-associated microglia.
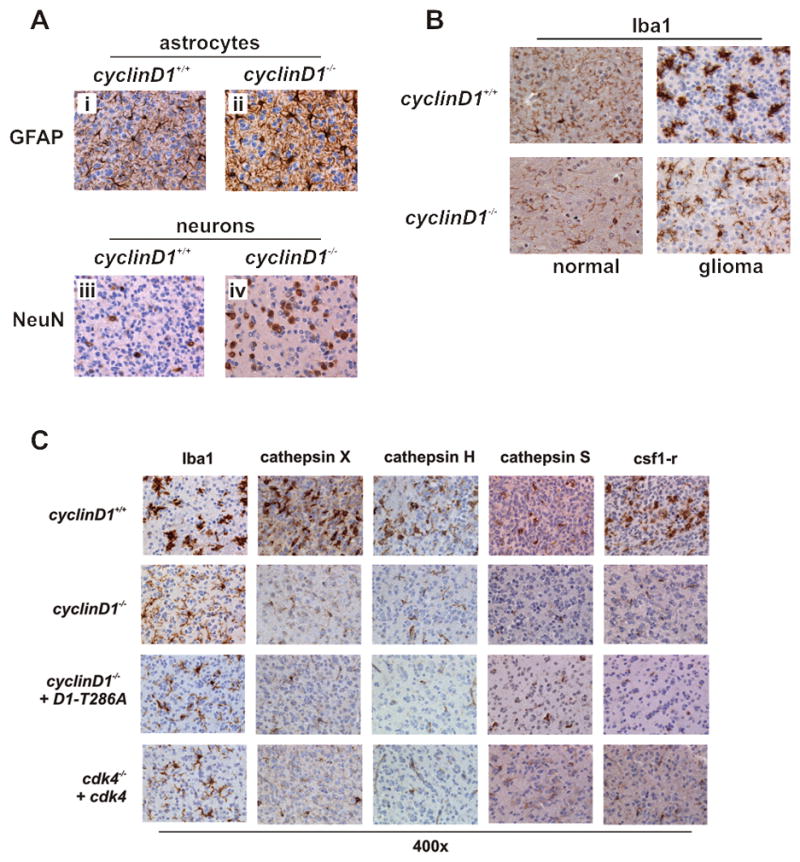
(A) Tumors from wild type and cyclin D1 knockout animals symptomatic for glioma were fixed in formalin and stained with antibodies to GFAP (reactive astrocytes; i and ii) and NeuN (neurons; iii and iv). (B) TAMs, marked by Iba1 staining. (C) Gliomas arising in the cyclin D1 knockout mice and cyclinD1 and cdk4 knockout mice in which the cyclinD1-cdk4 axis has been reconstituted in the tumor cell-of-origin were assessed for microglia activation by Iba1, cathepsin X, H and S and CSF1R immunohistochemistry. Activation status of TAMs TAMs in cyclinD1 knockout gliomas express and secrete reduced levels of cathepsin X.
We noted that the TAMs (tumor associated microglia) in tumors arising in cyclin D1 knockout animals had a predominantly ramified appearance, with processes extending away from the cell body and into the adjacent stroma (Fig. 5B). This is typical of cells residing in a surveillance state (35). On the other hand, TAMs in tumors arising in wild type mice were far more compact and ameboid, indicative of fully engaged and reactive cells (35). We then looked at the expression of the cathepsins X, H, and S, as well as the CSF1 receptor, additional markers associated with the activation of microglia (36–38). The intensity of each of these markers was reduced in the tumors arising in cyclin D1 knockout mice compared to that seen in wild type mice (Fig. 5C). Additionally, TAMs in gliomas arising in tumor-cell reconstituted cyclin D1 and cdk4 knockout animals also exhibited the staining pattern and morphology associated with a lower activation state (Fig. 5C). This indicates that TAMs recruited into the tumor bulk of cyclin D1 and cdk4 knockout animals exist in a lower activation state compared to those recruited into the tumor bulk in wild type mice.
While there is a strong correlation between the activation of TAMs and tumor progression in this model as well as others (39–41), whether this is a consequence of tumor progression or contributes to tumor progression is not clear. To address this, we attempted to carry out transplantation experiments. We isolated tumor cells from moderate-grade tumors arising in nestin-tvA;Ink4a/Arf knockout mice challenged by infection with RCAS-PDGF and transplanted these orthotopically into the brains of wild type and cyclin D1 knockout recipients. Nine wild type mice and five cyclin D1 knockout recipients were followed for six weeks and tumors isolated from morbid animals or at the end of the experiment. Although we found no difference in morbidity between wild type and cyclin D1-deficient recipient animals, seven of the nine gliomas that arose in the wild type animals were high-grade, and the resident TAMs were fully activated based on their morphology and staining with Iba1 and cathepsin X (Supplemental Figure 5). On the other hand, three of the four tumors that arose in cyclin D1 knockout animals were moderate grade and the activation of the TAMs appeared between the resting state observed in cyclin D1 knockout mice and the activated state seen in the wild type mice (Supplemental Figure 5). Because microglia are derived from resident cells of the CNS and not hematopoietic cells infiltrating from the circulation (42, 43), we were unable to ask if reconstituting cyclin D1 deficient animals with wild type microglia was able to support tumor progression to higher states of malignancy in cyclin D1 knockout mice. However, on the basis of the outcomes above, we conclude that cyclin D1 and cdk4 are required for maturation and activation of microglia, an event that contributes to the progression of this disease.
DISCUSSION
Malignant gliomas remain an almost invariably destructive and rapidly lethal form of cancer despite substantial progress in the development of new diagnostic and treatment modalities. It is clear that a more detailed understanding of the underlying molecular and cellular basis of this disease is required to identify critical new targets that might be amenable to therapeutic intervention.
The impact of the cyclin D1-cdk4 axis on tumor development and progression resides in its ability to phosphorylate the Rb tumor suppressor, which pushes cells through the G1-S phase boundary (13, 20, 23, 44). Deregulation of the cyclin D1-cdk4 cell-cycle axis is a common feature in glioma, with levels of proliferation increasing as tumors progress towards higher states of malignancy (45, 46). However, direct evidence that cyclin D1 and cdk4 can contribute to gliomagenesis has been lacking.
Using the RCAS-PDGF/nestin-tvA model of glioma, we found that both cyclin D1 and cdk4 are necessary for glioma development and progression. Whereas cdk4 knockout mice were completely refractory to glioma, tumors formed in cyclin D1 knockout animals. Reconstituting cyclin D1 or cdk4 expression in the nestin-positive progenitors that give rise to the tumors could increase the proliferation of the tumor cells in low-grade lesions; however, this was insufficient to support progression to higher-grade. Likewise, the stroma of cyclin D1 deficient mice was unable to support progression of transplanted tumor cells. This tumor cell independent stromal effect was correlated with a failure of the tumor associated microglia to enter a strongly activated state, both by morphologic and immunohistochemical criteria. On the basis of these observations, we concluded that cyclin D1 and cdk4 have roles in both the tumor cell and in the development of the surrounding stroma.
The traditional view of cancer as an autonomously growing aggregation of mutant cells has been superseded by one in which the tumor acts more insidiously, actively subverting the surrounding tissue to support its growth and proliferation. This is especially true in glial tumors, where development, progression and ultimately, invasion, relies on extracellular cues derived from stromal cells, including astrocytes, neurons and brain macrophages, recruited into the growing tumor mass, (29, 40). This dynamic interplay between tumor and stroma, coordinated by the tumor cells themselves, creates a permissive environment in which progression to malignancy is favored. For example, brain microglia stimulated by exposure to glioma cell conditioned media or infiltrating into tumor sites have been shown to actively secrete numerous factors, including immunosuppressive cytokines such as TGF-β, mitogens, cathepsins and metalloproteinases (41, 47, 48). Such microglia are polarized toward a phenotype that is pro-growth and immunosuppressive, leading to a state in which tumor cell growth and diffusion throughout the brain parenchyma becomes more likely (49). All of these processes can contribute to the ability of glial tumor cells to grow and invade into surrounding tissue, while simultaneously avoiding clearance by immunological sentinels in the brain (38, 50, 51). With TAMs comprising an active component of the tumor microenvironment, understanding the mechanisms that govern the responsiveness of these cells to external cues is important.
Our results suggest that cyclinD1-cdk4 can play two roles in the development of glioma: one, a tumor cell-autonomous role driving proliferation and the other, regulating the activity of non-tumor cells in the microenvironment, which can also affect malignant progression. Microglia are known to show a burst of cyclin D1 and cdk4 expression that precedes activation and correlates with proliferation and migration into regions exhibiting damage to neural integrity (52, 53). Consistent with this, we found that cyclin D1 and cdk4 knockout TAMs exhibited a ramified morphology (characterized by multiple processes extending from the central cell body into the adjacent tissue) associated with a reduced state of activation. Furthermore, other markers of TAM activation, including cathepsin X, H and S and CSF1R expression were reduced in knockout tumors relative to wild-type gliomas. Hence, in the absence of cyclin D1 or cdk4, TAMs may be unable to evolve into fully activated cells, which impedes progression to increased malignancy. How this occurs remains unclear. It is possible that cyclin D1 and cdk4 are required in TAMs or alternatively, are required in other stromal cells which facilitate activation of TAMs. In vitro experiments on isolated cells and the use of specific genetically engineered mouse models may clarify this in the future.
Toogood, Chin, and Waldman independently demosntrated that the cdk4 inhibitor drug, PD0332991, currently in phase II clinical trials, may be effective in halting the progression of glioma cell lines and xenografts. We are in the process of addressing whether PD0332991 can inhibit disease progression or reduce tumor burden in mice that have developed oligodendroglioma in situ and whether this is due to effects in the tumor cell, the microglia, or both. Current therapeutic strategies typically focus on direct inhibition of glial tumor proliferation and growth, even though compromising macrophage activity can also enhance chemotherapeutic efficiencies in other systems (reviewed in Nature 272: 303–4, 2011). Thus, cdk4 inhibitor therapies may be useful in targeting both the tumor cell and modulating the activity of a stromal cell-type that is critical to supporting malignant progression.
Supplementary Material
Acknowledgments
We thank Piotr Sicinski (Dana-Faber Cancer Institute, USA), Hiroaki Kiyokawa (Northwestern University, USA) and Mariano Barbacid (CNIO, Spain) who provided us the cyclin D1, cdk4, and cdk2 knockout mice, respectively. Katia Manova, Afsar Barlas, Voloidia Guoergovia and Alexander Baldys of the MSKCC molecular cytology core facility for critical technical assistance, Jason Huse (MSKCC) for grading the tumors, and Elyn Reidel (MSKCC) for performing the statistical analyses. We are grateful to Leny Gocheva, Gayle Carbajal, Dolores Hambardzumyan, Jim Finney, Massimo Squatrito and Nancy Yeh for their technical assistance and stimulating discussions. This work was supported by the NCI (NCI-CA89563) and the Golfers Against Cancer Fund (to A.K.), and an NCI Core Grant to Memorial Sloan-Kettering Cancer Center. D.C. is supported by a fellowship from the Brain Tumor Center (MSKCC, USA) and the Joel A. Gingras Jr. American Brain Tumor Association basic research fellowship.
Footnotes
Conflicts of interest: None
References
- 1.Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120–9. doi: 10.1038/35052535. [DOI] [PubMed] [Google Scholar]
- 2.Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer research. 2002;62:3729–35. [PubMed] [Google Scholar]
- 3.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & development. 2001;15:1913–25. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes & development. 2001;15:1311–33. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 5.Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1091–6. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsushime H, Ewen ME, Strom DK, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–34. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 7.Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–13. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- 8.Soos TJ, Kiyokawa H, Yan JS, et al. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 1996;7:135–46. [PubMed] [Google Scholar]
- 9.Cheng M, Olivier P, Diehl JA, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. The EMBO journal. 1999;18:1571–83. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes & development. 2000;14:3102–14. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alt JR, Gladden AB, Diehl JA. p21(Cip1) Promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. The Journal of biological chemistry. 2002;277:8517–23. doi: 10.1074/jbc.M108867200. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Yeh N, Zhu XH, et al. Somatic cell type specific gene transfer reveals a tumor-promoting function for p21(Waf1/Cip1) The EMBO journal. 2007;26:4683–93. doi: 10.1038/sj.emboj.7601886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–9. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sicinski P, Donaher JL, Parker SB, et al. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–30. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- 15.Ortega S, Prieto I, Odajima J, et al. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nature genetics. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- 16.Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Molecular cancer therapeutics. 2004;3:1427–38. [PubMed] [Google Scholar]
- 17.Michaud K, Solomon DA, Oermann E, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer research. 70:3228–38. doi: 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiedemeyer WR, Dunn IF, Quayle SN, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proceedings of the National Academy of Sciences of the United States of America. 107:11501–6. doi: 10.1073/pnas.1001613107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geng Y, Yu Q, Sicinska E, Das M, Bronson RT, Sicinski P. Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:194–9. doi: 10.1073/pnas.011522998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong W, Pollard JW. Genetic evidence for the interactions of cyclin D1 and p27(Kip1) in mice. Mol Cell Biol. 2001;21:1319–28. doi: 10.1128/MCB.21.4.1319-1328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 22.See WL, Miller JP, Squatrito M, Holland E, Resh MD, Koff A. Defective DNA double-strand break repair underlies enhanced tumorigenesis and chromosomal instability in p27-deficient mice with growth factor-induced oligodendrogliomas. Oncogene. 29:1720–31. doi: 10.1038/onc.2009.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 24.Ghiani C, Gallo V. Inhibition of cyclin E-cyclin-dependent kinase 2 complex formation and activity is associated with cell cycle arrest and withdrawal in oligodendrocyte progenitor cells. J Neurosci. 2001;21:1274–82. doi: 10.1523/JNEUROSCI.21-04-01274.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science (New York, NY) 1993;262:2050–4. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 26.Belachew S, Aguirre AA, Wang H, et al. Cyclin-dependent kinase-2 controls oligodendrocyte progenitor cell cycle progression and is downregulated in adult oligodendrocyte progenitors. J Neurosci. 2002;22:8553–62. doi: 10.1523/JNEUROSCI.22-19-08553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes & development. 1997;11:957–72. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 28.Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer research. 2004;64:4783–9. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 29.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nature reviews. 2009;9:239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 31.Quon H, Hasbini A, Cougnard J, Djafari L, Lacroix C, Abdulkarim B. Assessment of tumor angiogenesis as a prognostic factor of survival in patients with oligodendroglioma. J Neurooncol. 2010;96:277–85. doi: 10.1007/s11060-009-9961-x. [DOI] [PubMed] [Google Scholar]
- 32.Vidal A, Zacharoulis S, Guo W, et al. p130Rb2 and p27kip1 cooperate to control mobilization of angiogenic progenitors from the bone marrow. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6890–5. doi: 10.1073/pnas.0405823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chien WM, Garrison K, Caufield E, Orthel J, Dill J, Fero ML. Differential gene expression of p27Kip1 and Rb knockout pituitary tumors associated with altered growth and angiogenesis. Cell Cycle. 2007;6:750–7. doi: 10.4161/cc.6.6.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soeiro I, Mohamedali A, Romanska HM, et al. p27Kip1 and p130 cooperate to regulate hematopoietic cell proliferation in vivo. Mol Cell Biol. 2006;26:6170–84. doi: 10.1128/MCB.02182-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature neuroscience. 2007;10:1387–94. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 36.Gocheva V, Zeng W, Ke D, et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes & development. 2006;20:543–56. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma J, Tanaka KF, Yamada G, Ikenaka K. Induced expression of cathepsins and cystatin C in a murine model of demyelination. Neurochem Res. 2007;32:311–20. doi: 10.1007/s11064-006-9183-y. [DOI] [PubMed] [Google Scholar]
- 38.Markovic DS, Glass R, Synowitz M, Rooijen N, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. Journal of neuropathology and experimental neurology. 2005;64:754–62. doi: 10.1097/01.jnen.0000178445.33972.a9. [DOI] [PubMed] [Google Scholar]
- 39.Ghosh A, Chaudhuri S. Microglial action in glioma: a boon turns bane. Immunol Lett. 131:3–9. doi: 10.1016/j.imlet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583–93. doi: 10.1093/jnci/djm187. [DOI] [PubMed] [Google Scholar]
- 41.Watters JJ, Schartner JM, Badie B. Microglia function in brain tumors. J Neurosci Res. 2005;81:447–55. doi: 10.1002/jnr.20485. [DOI] [PubMed] [Google Scholar]
- 42.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature neuroscience. 2007;10:1538–43. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 43.Mildner A, Schmidt H, Nitsche M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nature neuroscience. 2007;10:1544–53. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 44.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature reviews. 2009;9:153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 45.Fiano V, Ghimenti C, Schiffer D. Expression of cyclins, cyclin-dependent kinases and cyclin-dependent kinase inhibitors in oligodendrogliomas in humans. Neuroscience letters. 2003;347:111–5. doi: 10.1016/s0304-3940(03)00615-3. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Zhao M, Huang AY, Fei Z, Zhang W, Wang XL. The effect of cyclin D expression on cell proliferation in human gliomas. J Clin Neurosci. 2005;12:166–8. doi: 10.1016/j.jocn.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 47.Markovic DS, Vinnakota K, Chirasani S, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12530–5. doi: 10.1073/pnas.0804273106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40:252–9. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- 49.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in immunology. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 50.Gocheva V, Wang HW, Gadea BB, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes & development. 24:241–55. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu A, Wei J, Kong LY, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-oncology. 12:1113–25. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kato H, Takahashi A, Itoyama Y. Cell cycle protein expression in proliferating microglia and astrocytes following transient global cerebral ischemia in the rat. Brain research bulletin. 2003;60:215–21. doi: 10.1016/s0361-9230(03)00036-4. [DOI] [PubMed] [Google Scholar]
- 53.Wiessner C, Brink I, Lorenz P, Neumann-Haefelin T, Vogel P, Yamashita K. Cyclin D1 messenger RNA is induced in microglia rather than neurons following transient forebrain ischaemia. Neuroscience. 1996;72:947–58. doi: 10.1016/0306-4522(95)00601-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.