

Abstract
NADH-cytochrome b5 oxidoreductase (Ncb5or) is an endoplasmic reticulum (ER)-associated redox enzyme involved in fatty acid metabolism, and phenotypic abnormalities of Ncb5or−/− mice include diabetes and lipoatrophy. These mice are lean and insulin-sensitive but become hyperglycemic at age 7 weeks as a result of β-cell dysfunction and loss. Here we examine early cellular and molecular events associated with manifestations of β-cell defects in Ncb5or−/− mice. We observe lower islet β-cell content in pancreata at age 4 weeks and prominent ER distention in β-cells by age 5 weeks. Ultrastructural changes progress rapidly in severity from age 5 to 6 weeks, and their frequency rises from 10% of β-cells at 5 weeks to 33% at 6 weeks. These changes correlate temporally with the onset of diabetes. ER stress responses and lipid load in Ncb5or−/− β-cells were assessed with isolated islets from mice at age 5 weeks. Expression levels of the stress marker protein Grp78/BiP and of phosphorylated eIF2α protein were found to be reduced, although their transcript levels did not decline. This pattern stands in contrast to the canonical unfolded protein response. Ncb5or−/− β-cells also accumulated higher intracellular levels of palmitate and other free fatty acids and exhibited greater reactive oxygen species production than wild-type cells. An alloxan-susceptible genetic background was found to confer accelerated onset of diabetes in Ncb5or−/− mice. These findings provide the first direct evidence that manifestations of diabetes in lean Ncb5or−/− mice involve saturated free fatty acid overload of β-cells and ER and oxidative stress responses.
Keywords: oxidative stress, ER stress, free fatty acids, diabetes, beta-cells
1. INTRODUCTION
Diabetes mellitus (DM) is characterized by increased blood glucose concentrations, and progressive dysfunction and loss of insulin-secreting pancreatic β-cells occurs during the pathogenesis of both Types 1 and Type 2 DM. Achieving a more complete understanding of mechanisms that govern β-cell survival, function, and responses to stress is thus an important objective in the quest to develop more effective means to prevent and treat DM.
The high rates of insulin biosynthesis and processing in pancreatic islet β-cells require an oxidizing environment in the endoplasmic reticulum (ER) for disulfide bond formation. Thus, β cells are sensitive to oxidative and ER stress induced by perturbations of protein synthesis, folding, or post-translational modification [1]. Accumulation of unfolded cargo proteins is sensed by Grp78/BiP (glucose-regulated protein 78/binding immunoglobulin protein), which triggers the unfolded protein response (UPR) via IRE1 (inositol-requiring enzyme 1), PERK (protein kinase RNA-like endoplasmic reticulum kinase) or ATF6 (activating transcription factor 6) pathways. UPR results in degradation of misfolded proteins, deceleration of protein synthesis, expansion of the ER network, and increased expression of chaperones to assist in protein folding [1,2]. Excessive, prolonged ER stress induces cell death via apoptosis that results from activation of JNK (c-jun N-terminal kinase), CHOP (C/EBP-homologous protein), and ATF3 (activating transcription factor 3) [3].
Lipid overload occurs in β-cells of humans and animals with obesity or Type 2 DM [4] and leads to both oxidative and ER stress [3,5]. Mechanistic studies with saturated fatty acid (SFA)-challenged β-cell lines in vitro suggest that some UPR markers, such as eIF2α (eukaryotic initiation factor 2α), ATF4 (activating transcription factor 4) and CHOP, are activated to respond to SFA-induced ER stress [6–8]. Dysregulated lipid metabolism precedes the onset of hyperglycemia in Type 1 DM patients [9] and may contribute to β-cell injury, but the mechanism by which this occurs is not currently understood.
We have generated a lean diabetic mouse model by disruption of the gene encoding NADH-cytochrome b5 oxidoreductase (Ncb5or) [10]. It is an ER-associated redox enzyme that is widely expressed in animal tissues [11,12] and might act as a redox partner of stearoyl-CoA desaturase (SCD) in cellular fatty acid metabolism [13,14]. Ncb5or−/− mice develop diabetes at about 7 weeks of age as a result of β-cell dysfunction and loss without apparent autoimmune manifestations, and they remain lean and insulin-sensitive before and after the onset of DM [10]. Ncb5or−/− mice exhibit glucose intolerance, reduced islet insulin content, and impaired glucose-stimulated insulin secretion as early as age 4 weeks but retain normal insulin sensitivity [10].
Prediabetic Ncb5or−/− mice exhibit profound changes in hepatic fatty acid metabolism. They include decreased specific activity of SCD, reduced content of monounsaturated fatty acids (MUFA) relative to SFA, diminished triacylglycerol (TAG) content, and elevated intracellular levels of free fatty acids (FFA) compared to wild-type (WT) mice. These abnormalities are accompanied by increased hepatic content of mitochondria, accelerated catabolism, and enhanced expression of markers of ER and oxidative stress [14]. Ncb5or−/− mice that receive WT islet transplants remain normoglycemic until age 12 weeks but exhibit the same abnormalities in lipid composition and metabolism that are observed in non-transplanted Ncb5or-null mice [13]. In mice lacking both Ncb5or and CHOP, which is a pro-apoptotic participant in ER stress responses, the onset of diabetes is delayed by about two weeks compared to Ncb5or mice with a functional CHOP gene. This suggests that ER stress responses are involved in β-cell loss in Ncb5or−/− mice [15].
Here we report studies of the early cellular and molecular events associated with β-cell dysfunction and loss in Ncb5or-null diabetes. We provide the first direct evidence of progressive ultrastructural changes that include ER distention, of elevated intracellular levels of SFA, and of increased expression of markers of oxidative stress in Ncb5or−/− β-cells. Our findings also suggest that the ER stress response in Ncb5or−/− β-cells is distinct from the canonical UPR.
2. MATERIALS AND METHODS
2.1. Materials
All reagents were obtained from Sigma (St. Louis, MO) with the following exceptions. Chemicals used for electron microscopy were purchased from Electron Microscopy Sciences (Hatfield, PA), culture media and serum from Invitrogen (Carlsbad, CA), and oligonucleotides from Integrated DNA Technologies (Coralville, IA).
2.2. Animals, diet, glucose tolerance test (GTT), and blood glucose measurement
The Ncb5or−/− line was generated in BALB/cAnN strain as described previously [10]. We produced C57BL/6 Ncb5or−/− mice using a strategy of outcross and then backcross to C57BL/6 for more than 10 generations. For most studies, C57BL/6 Ncb5or−/− mice were compared to C57BL/6 mice with intact Ncb5or genes. The animal experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center and the Children’s Hospital in Boston. Mice were handled in accordance with guidelines issued by the National Institutes of Health. Animals were housed in a pathogen-free facility on a 12-hr light/dark cycle with ad libitum access to water and standard chow, which was either TD8604 (Harlan Teklad, Madison, WI) or Prolab Isopro RMH 3000 (LabDiet, PMI Nutrition International, St. Paul, MN; for ALR and ALS colonies in Boston). Whole blood glucose levels from tail vein were measured with a OneTouch Ultra glucometer (LifeScan, Milpitas, CA). Mice were fasted for 4 hr. before intraperitoneal injection of glucose (1 g/kg body weight) for glucose tolerance testing (GTT), as previously described [10].
2.3. Generation of Ncb5or−/− mice on alloxan resistant (ALR) and alloxan susceptible (ALS) genetic backgrounds
The ALR and ALS inbred strains were initially developed from outbred CD-1 (ICR) mice with successive generations selected for resistance or for increased sensitivity to the diabetogenic effects of alloxan, which imposes oxidant stress on β-cells [16]. Tracking gene polymorphisms that distinguish B6 from the ALR or ALS genotypes allowed us to insert the Ncb5or−/− allele into ALR or ALS genetic backgrounds more rapidly. Progeny depleted in C57BL/6 polymorphisms were selected by screening each generation of mice with 72 microsatellite markers covering all 19 autosomes and the × chromosome. The first generation was bred into the ALR or ALS background to produce the first backcrossed generation (N1). The contaminating B6 genome was calculated to be 18.8–20.6% (ALR) and 15.6–17.9% (ALS) in N1 breeders. These mice were crossed with pure ALR or ALS mice to generate N2 pups. Contaminating genome in subsequent generations of breeders was: N2, 11.7–12.1% (ALR), 8.6–11.9% (ALS); N3, 5.8–6.9% (ALR), 4.4–7.1% (ALS); N4, 2.9% (ALR), 4.2–4.4% (ALS); N5, 0% (ALR and ALS). Two subsequent generations were created by backcrossing mice with 0% contaminating genome into pure ALR or ALS strains, with analysis initiated on pups generated from N7 breeding pairs.
2.4. Histology and confocal immunofluorescence analyses
Formalin-fixed, paraffin-embedded pancreata were used for histology with hematoxylin/eosin (H & E) staining and for confocal immunofluorescence analyses with Histostain-Plus kit and antibodies (Invitrogen) against insulin (α), glucagon (β), and somatostatin (δ) as described [17]. Confocal images were taken using a Nikon C1si confocal microscope. Individual α-, β-, and δ-cells were counted with the NIH Image J software.
2.5. Electron microscopy
Transmission electron microscopy (TEM) was performed in the EM core facility at the University of Kansas Medical Center. Mice underwent whole body perfusion through the heart for fixation of the pancreas as previously described [10]. Islets (10–15 per animal) were dissected for further processing. TEM micrographs from non-overlapping sections of islets were taken on a JEOL 100 CXII transmission electron microscope operating at 80 kV. Negatives were scanned and digital pictures were evaluated. About 50–300 nucleus-positive β-cells per sample were examined for the frequency and severity of distended ER (swollen ER sacs) and counted in a double-blind fashion (3–4 islets per sample, 3 mice per group). To assess the content of β-cells and non-β-cells, the same TEM grids were used to build digital islet maps (low magnification, × 600). Depending on islet sizes, 3–4 individual islet maps were used to count islet cell composition. Fifty to 500 non-redundant nucleus-positive cells per islet were counted and categorized as β- or non-β-cells based on distinct morphology of subcellular granules. These numbers were then used to calculate cell-type percentages. The NIH Image J software was used to measure relative areas of nuclei, mitochondria and cytosol in β-cells from TEM grids of the same magnification (x 1900).
2.6. Islet isolation
Mouse islets were isolated with a collagenase infiltration method [18] using Liberase (Roche Applied Science, Indianapolis, IN). Typical islet yields (all sizes) were ca. 350 per WT mouse or ca. 150–200 per Ncb5or−/− mouse. Islets were used directly or stored as pellets at −80°C.
2.7. qRT-PCR
Approximately 300–500 islets were used to prepare total RNA that was then reverse transcribed into cDNA. Quantitative real-time PCR was performed with SYBR Green PCR Master Mix on a 7900HT thermocycler (Applied Biosystems, Foster City, CA) as previously described [14]. The expression level of each target gene was normalized against 18S rRNA internal reference to obtain ΔCt, which was used to calculate the transcript level, 2ΔCt. Primer sequences are available upon request.
2.8. Immunoblot analyses
Isolated islets were lysed in Triton-X buffer (25 mM HEPES, 50 mM KCl, 6% glycerol, 5 mM EDTA, 5 mM EGTA, 0.5% Triton-X100, 50 mM NaF, 40 mM glycerol phosphate, and 25 mM sodium pyrophosphate with protease inhibitors) and total protein content was determined with BCA reagents (Pierce, Rockford, IL). Approximately 10–15 μg islet protein was loaded per lane onto 4–15% Ready Gel (Bio-Rad, Hercules, CA) for SDS-PAGE and transferred onto PVDF membranes (GE Healthcare Life Sciences, Piscataway, NJ) for immunoblot analysis with antibodies against phosphorylated and total eIF2α (Cell Signaling Technology, Danvers, MA), Grp78/BiP (BD Biosciences, Franklin Lakes, NJ), ATF6 (Alexis, Farmingdale, NY), and GAPDH (glyceraldehyde 3-phosphate dehydrogenase; Sigma, St. Louis, MO). After binding of horseradish peroxidase–conjugated secondary antibody, signals were detected with an enhanced chemiluminescence (ECL) kit, exposed to CL-XPosure film (Pierce, Rockford, IL), and quantified with the Adobe Photoshop software with normalization against GAPDH or β-actin.
2.9. Lipid profiling
A total of 150–350 islets from each mouse were used for lipid profiling as previously described [14]. Briefly, islets were suspended in ice-cold lysis buffer (18 mM Tris-HCl, 300 mM mannitol, 50 mM EGTA, pH 7.6, with protease inhibitors) and sonicated for 10 one-second pulses. Protein content was determined with BCA reagent (Pierce), and lipid content was normalized to that value. Standard C17:0-FFA and TAG (NuCheck Prep, Elysian, MN) were added to each islet sample before lipid extraction, and concentrated extracts were analyzed by thin layer chromatography (TLC). Fatty acid methyl esters (FAMEs) were prepared by transmethylation and then analyzed on a Varian GC-MS system. Supelco 37 FAME standards (Sigma) were used to confirm FAME identities. Individual FAME species were quantified relative to C17:0 internal standard, and the resultant values were used to calculate the sum of total FAs and desaturation index (DI).
2.10. Detection of cellular reactive oxygen species (ROS)
Cellular ROS were detected by staining islets with 2′,7′-dichlorofluorescein diacetate (DCFH-DA, Sigma), as modified from previous report [19]. Hand-picked islets were recovered at 37°C for 2 hours in RPMI 1640, 10% FBS, 11 mM glucose before pre-incubation with 5.5, 11 or 16.7 mM glucose for 60 minutes. DCFH-DA was added directly to islets (20 μM final concentration) and, after a 20-minute incubation (in the dark), the fluorescence (excitation 488 nm/emission 530 nm) was visualized with a FluoView 300 confocal microscope (Olympus, Center Valley, PA).
2.11. Statistical analysis
Levels of significance for differences among groups were determined using Analysis of Variance (ANOVA). P value <0.05 was considered statistically significant. Standard errors of the mean are specified by error bars in the figures.
3. RESULTS
3.1. Dysfunction and loss of β-cells in Ncb5or−/− mice
Our previous study demonstrated that β-cell dysfunction in prediabetic Ncb5or−/− mice manifests by reduced islet insulin content and impaired glucose-stimulated insulin secretion [10]. Those defects were also observed in the studies described here with Ncb5or−/− mice at 5 weeks of age (not shown).
No overt morphologic abnormalities were observed in Ncb5or−/− islets compared to WT at age 4 weeks upon microscopic analyses of H&E-stained pancreatic sections (Fig. 1A–B). By age 6 weeks, however, Ncb5or−/− islets appeared irregular and contained some vacuolated cells (Fig. 1D). Confocal immunofluorescence analyses of islet β-, α- and δ-cells (Fig. 1E–H) revealed that WT islets contained 75% β-cells at ages 4 and 6 weeks (Fig. 1I), consistent with published values for rodent islets [20,21]. In contrast, β-cells comprised only 58% and 43% of the total islet cell population in Ncb5or−/− mice at ages 4 and 6 weeks, respectively (Fig. 1I). At age 6 weeks, only 26% of Ncb5or−/− β-cells (non-α, non-δ) exhibited immunoreactive insulin signals with intensities similar to WT, although nearly all Ncb5or−/− β-cells did so at age 4 weeks (not shown). By age 6 weeks, Ncb5or−/− islets exhibited marked morphological abnormalities that included the appearance of α-cells and δ-cells in the central region of islets (Fig. 1G–H) rather than in their usual peripheral locations, a pattern previously observed in islets of overtly diabetic Ncb5or−/− mice [10].
FIG. 1.
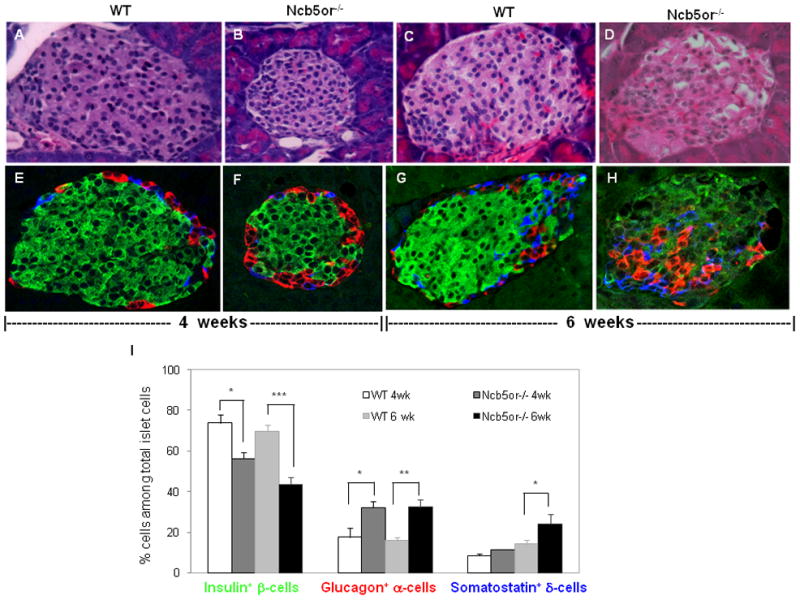
(A–D) H & E staining and (E–H) confocal immunofluorescence analyses against insulin (β-cells, green), glucagon (α-cells, red) and somatostatin (δ-cells, blue) of representative islet sections from Ncb5or−/− and WT mice at age 4 and 6 weeks. (I) Average percentages of each cell type (n=3 mice per group; 5 islets per mouse section). *, p<0.05. **, p<0.01. ***, p<0.005.
3.2. Distended ER and increased mitochondrial content in Ncb5or−/− β-cells
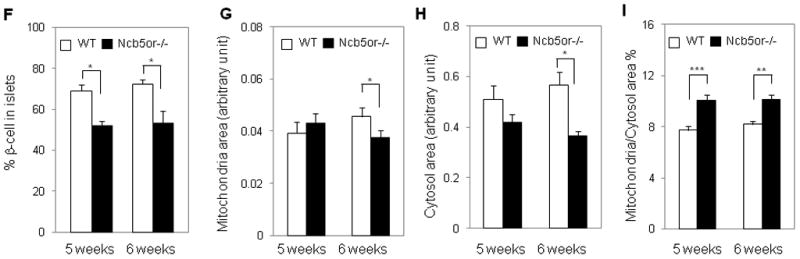
Transmission electron microscopic (TEM) studies of pancreatic sections revealed no ultrastructural differences between WT and Ncb5or−/− β-cells at age 3 weeks (not shown). By age 5 weeks (Supplemental Fig. 1A–D) or 6 weeks (Fig. 2A–D), Ncb5or−/− islets did exhibit ultra-structural abnormalities, and distention of ER (dER) was the most prominent of them and increased in severity at age 6 weeks. Ribosome-like structures attached to the surface of some dER sacs were observed at higher magnification (not shown). Compared to WT, Ncb5or−/− β-cells with dER contained substantially fewer insulin granules (or were “degranulated”), and their ER lumens were less electron-dense. The percentage of dER+ β-cells divided by total Ncb5or−/− islet β-cells increased from 10% at age 5 weeks to 33% at age 6 weeks (Fig. 2E), and this was associated with increasing dER severity and declining insulin granule content (Fig. 2C–D). Such dER structures were not observed in WT mice at age 5 or 6 weeks and hence reflected ER stress in Ncb5or−/− β-cells. This was further examined at the molecular level as described below. TEM analyses also revealed that, compared to WT, the percentage of β-cells among total islet cells was lower in Ncb5or−/− mice (Fig. 2F), consistent with the confocal immunofluorescence analyses.
FIG. 2.
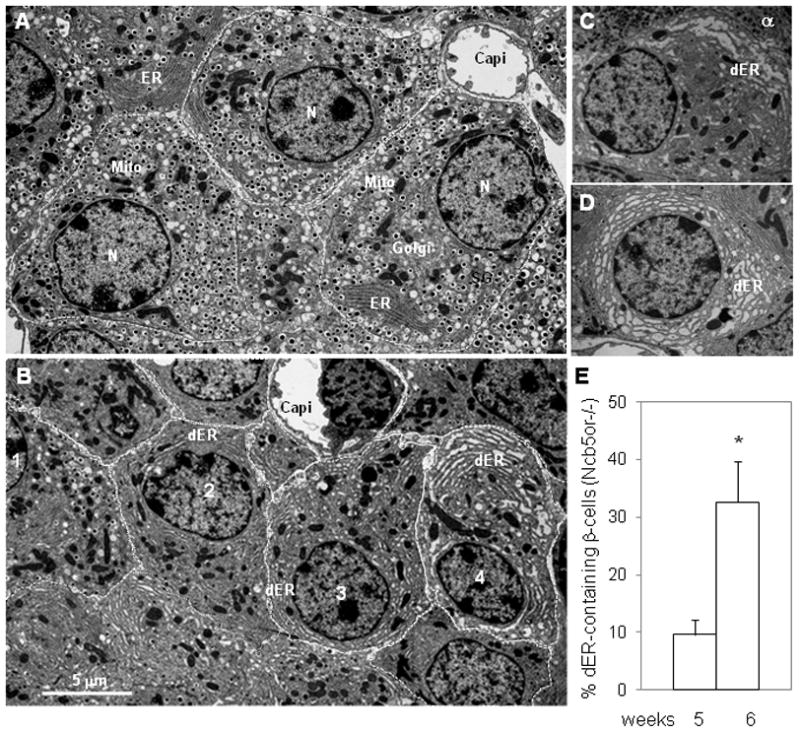
(A–D) TEM analyses of ER stress in Ncb5or−/− β-cells. ER distention (dER, swollen ER sacs) was observed in β-cells of Ncb5or−/− mice at age 6 weeks (B–D) but not in WT (A). Fewer insulin granules were found in dER-containing β-cells. Subcellular structures are labeled as: capi, capillary; dER, distended ER; ER, endoplasmic reticulum; mito, mitochondria; N, nucleus; SG, secretary granules. Different extent of ER distention was observed among β-cells in (B, #1–4), (C, where one α-cell was visible) and (D). (E) Frequency of dER-containing β-cells among endocrine cells in Ncb5or−/− islets at age 5 and 6 weeks.
(F) The β-cell content in islets of Ncb5or−/− and WT mice at age 5 and 6 weeks.
(G) Mitochondrial area and (H) cytosol area in β-cells from Ncb5or−/− and WT mice at age 5 and 6 weeks were counted to calculate “% mitochondria: cytosol” (I). WT (n=4) and Ncb5or−/− mice (n=3), 10 intact β-cells per animal, were analyzed for each age. Results are expressed as mean ± S.E. *, p<0.05. **, p<0.01. ***, p<0.005. White-bar: wild-type (WT); Black-bar: Ncb5or−/−.
We have previously observed hypertrophic and hyperplastic mitochondria in β-cells from diabetic Ncb5or−/− mice at age 7 weeks [10]. To determine whether such changes began before the onset of diabetes, we measured areas occupied by mitochondria (Fig. 2G), cytosol (Fig. 2H), nuclei, and entire Ncb5or−/− and WT β-cells (Supplemental Fig. 1E–F). All of those areas (Fig. 2G–H and Supplemental Fig. 1E–F) remained constant in WT β-cells between ages 5 and 6 weeks, but mitochondrial and cytosolic areas declined with age in Ncb5or−/− β-cells and, by age 6 weeks, were lower than WT by 20% and 33%, respectively (Fig. 2G-H). The ratio of the mitochondrial and cytosolic areas was significantly higher in Ncb5or−/− β-cells (10%) than in WT (8%) both at ages 5 and 6 weeks (Fig. 2I). These values are about one-third of those in hepatocytes for Ncb5or−/− (33%) and WT (22%) mice at the same ages [14]. Neither β-cells nor hepatocytes from prediabetic Ncb5or−/− mice exhibited discernible abnormalities in mitochondrial morphology.
3.3. ER stress response in Ncb5or−/− β-cells
To evaluate ER stress in Ncb5or−/− β-cells, we measured transcript levels of ER stress response genes in Ncb5or−/− islets at age 5 weeks. These values were compared to those at age 3 weeks, which is an age that precedes manifestations of β-cell dysfunction and at which abnormal ER stress is not expected. Transcripts measured include those encoding ATF6α [22], Grp78/BiP [23], Grp94 (glucose-regulated protein of 94 kDa) [24], ATF3 [25], CHOP [26], eIF2α [27], and IRE1-activated splicing of Xbp1 (X-box binding protein 1) [28].
At age 3 weeks, Ncb5or−/− and WT islets exhibited similar levels of these ER stress response gene transcripts (Table 1) and of marker proteins (Fig. 3). By age 5 weeks, Ncb5or−/− islets contained higher levels of BiP, Grp94, ATF6α, and Xbp1s (spliced) transcripts than did WT islets, although levels of transcripts for ATF3, Xbp1t (total), eIF2α, and CHOP were similar between the two genotypes (Table 1). Immunoblot analyses revealed that Ncb5or−/− islets contained lower levels of BiP and phosphorylated eIF2α proteins compared to WT at age 5 weeks, but islets of the two genotypes contained similar levels of activated ATF6α at that age (Fig. 3). It is worth noting that exposure to the reducing agent dithiothreitol (DTT) suppressed eIF2α phosphorylation (Fig. 3A) in WT islets, in addition to activating ATF6α as previously reported [29].
Table 1.
Transcript levels of genes involved in ER and oxidative stress response in islets of Ncb5or−/− and wild-type (WT) mice at age 3 and 5 weeks (n=4 and 6 each, respectively).
| 3 weeks | 5 weeks | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pathway | Gene | WT | Ncb5or−/− | Fold increase in Ncb5or−/− | p | WT | Ncb5or−/− | Fold increase in Ncb5or−/− | p |
| Endocrine Synthesis | Ins2 | 433000±114000 | 238000±57000 | 0.6 | NS | 32900±7900 | 67100±15000 | 2.0 | 0.09 (NS) |
| Glgn | 4.7±1.3 | 3.7±0.8 | 0.8 | NS | 5460±1430 | 16500±3400 | 3.0 | 0.02 | |
| ER Stress | BiP | 140±26 | 74.8±24.6 | 0.5 | NS | 9.3±2.7 | 46.0±5.2 | 5.0 | 0.001 |
| Grp94 | 541±80 | 319±78 | 0.6 | NS | 63.5±11.4 | 133±19 | 2.1 | 0.02 | |
| eIF2α | 1150±190 | 1380±190 | 1.2 | NS | 86.7±18.8 | 62.7±16.6 | 0.7 | NS | |
| ATF6α | 59.2±15.3 | 60.3±13.9 | 1.0 | NS | 8.9±1.7 | 21.1±4.2 | 2.4 | 0.02 | |
| Xbp1s | 79.9±16.9 | 72.4±11.4 | 0.9 | NS | 11.1±2.1 | 20.1±1.7 | 1.8 | 0.03 | |
| Xbp1t | 221±38 | 397±95 | 1.8 | NS | 65.9±13.7 | 91.4±11.4 | 1.4 | NS | |
| ATF3 | 0.58±0.14 | 2.6±1.5 | 4.5 | 0.09 (NS) | 1.7±0.6 | 4.7±1.7 | 2.7 | NS | |
| CHOP | 264±47 | 399±94 | 1.5 | NS | 191±24 | 160±33 | 0.8 | NS | |
| Oxidative Stress | Ero1α | 24.3±6.5 | 13.8±3.4 | 0.6 | NS | 1.3±0.5 | 4.3±0.6 | 3.3 | 0.03 |
| Hmox1 | 17.7±3.2 | 18.4±1.7 | 1.0 | NS | 0.40±0.10 | 1.7±0.4 | 4.2 | 0.004 | |
All values are mean ± S.E. and relative to 18S (×106). NS: non-significant.
FIG. 3.
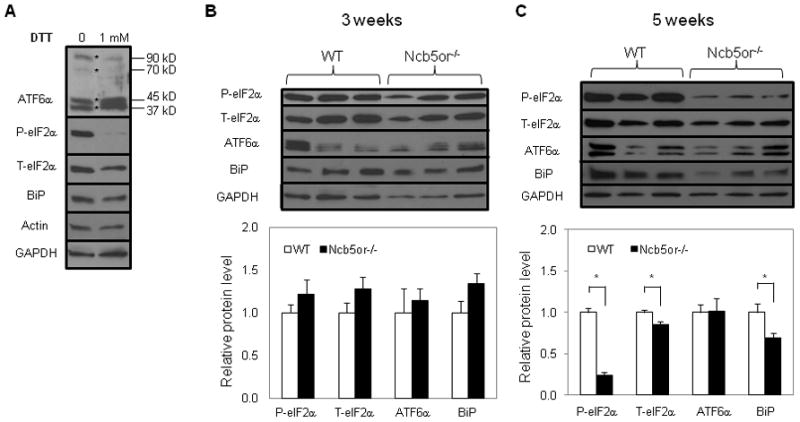
Immunoblot analyses of ATF6, BiP and eIF2α proteins in total extract of (A) WT islets at age 5 weeks that were treated with dithiothreitol (DTT) for 2 hours and of untreated Ncb5or−/− and WT islets at age 3 weeks (B) and 5 weeks (C). Intensities of immunoblot signals were quantified against loading control glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which was shown to be similar to β-actin (A). WT (n=5) and Ncb5or−/− islets (n=4) were analyzed and results are expressed as mean ± S.E. *, p<0.05. T, total; P, phosphorylated. White-bar: wild-type (WT); Black-bar: Ncb5or−/−.
3.4. Oxidative stress in Ncb5or−/− β-cells
Compared to WT, Ncb5or−/− β-cells from mice at 5 weeks of age contained higher intracellular levels of ROS, as reflected by oxidation of the fluorescent indicator DCFA, upon incubation with varied concentrations of glucose, and the magnitude of the ROS signal increased with the glucose concentration over the range 5.5 mM to 16.7 mM (Fig. 4). Insulin secretion rises from basal to nearly maximally stimulated values over this range of glucose concentrations. Consistent with this finding, transcript levels for the oxidative stress response genes Ero1α (endoplasmic oxidoreductin-1α) [30] and Hmox1 (heme oxygenase 1) [31] were found to be higher in Ncb5or−/− than in WT islets at age 5 weeks (Table 1).
FIG. 4.
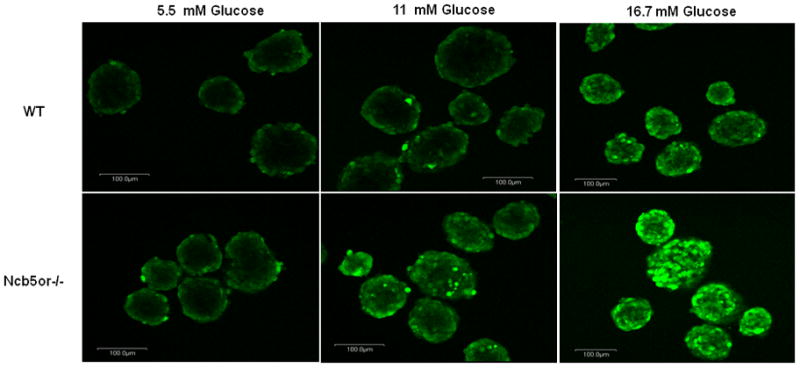
DCF staining of higher H2O2 levels in Ncb5or−/− islet β-cells. Islets from Ncb5or−/− and WT mice at age 5 weeks were incubated with 5.5, 11 and 16.7 mM glucose for one hour prior to DCF staining. A total of three pairs (n=3) were analyzed and representative images were shown. Signals in peripheral are likely resulted from physical barrier for penetration of dye.
3.5. Elevated intracellular levels of free saturated fatty acids (SFA) in Ncb5or−/− β-cells
Analyses of lipid content revealed that Ncb5or−/− islets exhibited significantly higher levels of palmitic acid and total FFA compared to WT at age 5 weeks (Table 2). There was also a trend toward higher TAG content in Ncb5or−/− islets that did not achieve statistical significance, but no differences were observed between genotypes in FFA or TAG SCD indices that reflect the ratio of monounsaturated to saturated fatty acid content (Table 2). This indicates that elevation of intracellular FFA occurs in Ncb5or−/− β-cells by the time that ultrastructural abnormalities and manifestations of ER and oxidative stress have appeared. The values for islet content of lipid species observed here fall within the range of values for rodent islets reported by others [32]. No difference was observed between the two genotypes for islet phospholipid content or SCD indices (not shown).
Table 2.
Lipid content and desaturation indices in islets of Ncb5or−/− and WT mice at age 5 weeks. Levels of total (Total), saturated (SFA) and monounsaturated (MUFA) fatty acids in intracellular free fatty acid (FFA) and triacylglycerol (TAG) pools were quantified against protein content.
| FFA | TAG | |||||
|---|---|---|---|---|---|---|
| WT | Ncb5or−/− | p | WT | Ncb5or−/− | p | |
| Total (nmole/mg) | 176±21 | 365±79 | 0.05 | 33.7±3.5 | 66.5±18.0 | 0.11 (NS) |
| SFA (nmole/mg) | 128±18 | 264±53 | 0.04 | 24.5±3.2 | 35±11 | 0.11 (NS) |
| MUFA (nmole/mg) | 31.9±3.3 | 69.1±21.5 | 0.13 (NS) | 4.8±0.5 | 10.6±3.1 | 0.10 (NS) |
| C16:0 (nmole/mg) | 60±8 | 120±23 | 0.04 | 10.5±1.7 | 19.1±5.6 | 0.18 (NS) |
| C18:0 (nmole/mg) | 41±5 | 87±20 | 0.06 (NS) | 6.3±0.5 | 13.3±4.1 | 0.12 (NS) |
| C16:1 (nmole/mg) | 3.5±0.3 | 6.4±1.6 | 0.13 (NS) | 0.5±0.1 | 1.2±0.6 | NS |
| C18:1 (nmole/mg) | 25.8±2.2 | 55.7±18.1 | 0.14 (NS) | 3.3±0.2 | 7.6±2.3 | 0.10 (NS) |
| DI (C16:1/C16:0) | 0.062±0.009 | 0.053±0.008 | NS | 0.055±0.019 | 0.059±0.017 | NS |
| DI (C18:1/C18:0) | 0.622±0.023 | 0.640±0.134 | NS | 0.526±0.044 | 0.629±0.098 | NS |
All values are mean ± S.E. (n=5). DI: desaturation index. NS: non-significant.
3.6. Accelerated onset of diabetes in Ncb5or−/− mice with an alloxan-susceptible genetic background
We generated strains of Ncb5or−/− mice that were relatively resistant to the diabetogenic oxidant alloxan (ALR) or that exhibited enhanced sensitivity (ALS) to the agent in order to examine further the role of oxidative stress in Ncb5or−/− β-cell dysfunction. We found that ALS-Ncb5or−/− mice exhibited an accelerated onset of impaired glucose tolerance (age 4.5 weeks) and overt diabetes (age 5.5 weeks) (Fig. 5A–D) compared to Ncb5or−/− mice with the ALR or neutral B6 genetic background (Fig. 5E). These findings suggest that the ALS genetic background, which confers increased sensitivity of β-cells to oxidant stress, exacerbates the diabetes-prone phenotype of Ncb5or deficiency.
FIG. 5.
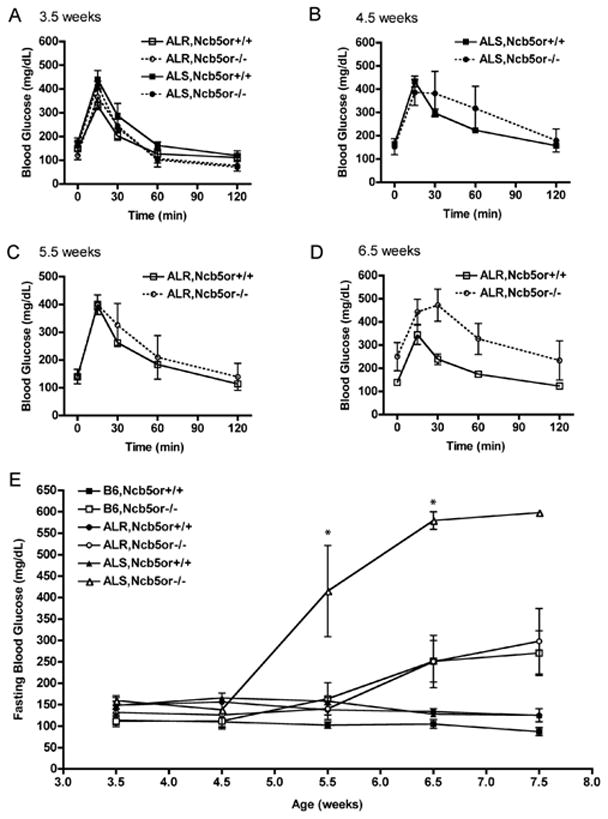
(A–D) GTT and (E) fasting blood glucose levels of ALR and ALS mice. Glucose was administered intraperitoneally following fasting of ALR and ALS mice. Blood glucose was measured at the indicated times. Clear square (ALR,Ncb5or+/+; n=8), clear circle (ALR,Ncb5or−/−; n=5), black square (ALS,Ncb5or+/+; n=8), black circle (ALS,Ncb5or−/−; n=4). GTT curves were generated for ALS mice at age 3.5 and 4.5 weeks (A,B) and ALR at age 3.5, 5.5 and 6.5 weeks (A,C,D). (E) Fasting blood glucose was measured starting at 3.5 weeks (post-weaning) through 8 weeks of age. Black square (B6,Ncb5or+/+; n=8), white square (B6,Ncb5or−/−; n=8), black circle (ALR,Ncb5or+/+; n=8), white circle (ALR,Ncb5or−/−; n=5), black triangle (ALS,Ncb5or +/+; n=8), white triangle (ALS,Ncb5or−/−; n=4).
4. DISCUSSION
Here we provide the first direct evidence that β-cell dysfunction in Ncb5or−/− mice, which remain lean but develop insulinopenic diabetes in the setting of normal insulin sensitivity, is temporally associated with increased intracellular levels of palmitic acid and total fatty acids and with manifestations of both ER and oxidative stress responses in β-cells.
Ncb5or−/− mice display mild growth retardation after birth but no abnormalities in islets up to the age of 3 weeks. By age 4 weeks, islets from Ncb5or−/− mice have lower β-cell content than WT islets, but Ncb5or−/− mice exhibit no insulin insufficiency and normal glucose tolerance under basal conditions, presumably as a result of enhanced insulin sensitivity conferred by their lower body weights and fat contents. By age 5 weeks, Ncb5or−/− mice display overt β-cell dysfunction that is associated with increased intracellular levels of free saturated fatty acids, manifestations of oxidative and ER stress responses, reduced β-cell size, and ER distention, and the latter two abnormalities worsen rapidly over the course of the next week. By age 7 weeks, Ncb5or−/− mice develop overt hyperglycemia in association with a reduction in measures of islet function and in β-cell content. Compromised fatty acid metabolism that results in accumulation of free saturated fatty acids in β-cells is thus among the earliest abnormalities in Ncb5or−/− islets, precedes severe manifestations of oxidative and ER stress responses and overt islet dysfunction, and may be among the causative factors in the later deterioration of β-cell function and β-cell loss.
The accumulation of saturated fatty acids in Ncb5or−/− islets appears to reflect an intrinsic β-cell defect because WT and Ncb5or−/− mice at 5 weeks of age exhibit similar levels of circulating FFA and other lipids [14]. The SCD index of FFA and TAG, which reflects the relative abundance of monounsaturated and saturated fatty acids, does not differ in islets of WT and Ncb5or−/− mice at age 5 weeks, even though levels of SCD1 and SCD2 transcripts are higher in Ncb5or−/− islets. We have reported similar findings in hepatocytes of Ncb5or−/− mice compared to WT at this age [14]. No abnormalities in islet phospholipid content or fatty acid composition are observed in Ncb5or−/− mice. This is also the case for Zucker diabetic fatty rat, which is a widely studied rodent model of lipotoxic diabetes [33]. Although we have not observed any perturbation in the net TAG content of islets from Ncb5or−/− mice, it is possible that altered rates of TAG synthesis and disposal might represent a mechanism to protect β-cells from lipid overload in vivo, and the contribution of Ncb5or to fatty acid metabolism may differ between islets and liver. The latter possibility is currently being investigated in mice with tissue-restricted Ncb5or deficiency.
By age 5 weeks, islet β-cells from Ncb5or−/− exhibit prominent ER distention. The frequency and severity ER distention increase rapidly between ages 5 and 6 weeks, and at the latter age, the average β-cell size decreases significantly just before the onset of overt diabetes at age 7 weeks. The abnormal ER structures in Ncb5or−/− β-cells resemble those in CHO cells and in β-cells incubated with high concentrations of palmitic acid in vitro and that develop fatty acid overload and oxidative injury [8,34,35]. In each of these cases, the cells have enlarged ER lumens with increased electron transparency, which suggests a lower protein content and stands in contrast to the electron-opaque ER lumens engorged with aggregates of unfolded proteins that are observed in PERK-null [36] and Akita mice [37]. In the latter, a missense mutation of a critical cysteine residue in insulin blocks disulfide bond formation during maturation.
The ER stress response in Ncb5or−/− β-cells appears to be different from the canonical UPR. Levels of ATF6α and BiP transcripts are higher in Ncb5or−/− than in WT islets, but islet levels of ATF6α and BiP proteins are similar in the two genotypes. Increased levels of these proteins are expected as a result of the canonical UPR [22,38,39]. Moreover, phosphorylation of eIF2α is reduced in Ncb5or−/− β-cells, whereas increased eIF2α phosphorylation occurs with protective ER stress responses, such as that observed in Ncb5or−/− hepatocytes [14] and in SFA-treated hepatocytes [40] and β-cells in culture [6,35,41]. DTT-induced redox stress in islet β cells also results in reduced eIF2α phosphorylation, and this pattern resembles that produced by Ncb5or deficiency.
The ratio of the intracellular content of FFA divided by that for TAG is higher for β-cells than for hepatocytes, and β-cells have a lower mitochondrial content than hepatocytes. This suggests that β-cells have reduced capacity to channel excess FFA into synthesis of TAG and might therefore incur increased susceptibility to ER and oxidative stress induced by saturated fatty acid overload. Although mitochondria in Ncb5or−/− β-cells and hepatocytes exhibit no obvious morphologic and or functional abnormalities, both cell types do exhibit increased manifestations of oxidative stress responses than do the corresponding WT cells. Many types of ROS cause oxidation of the fluorescent indicator DCFA, thus it cannot identify specific molecular species of ROS or the subcellular locus of their generation. The observed pattern of ROS production in Ncb5or−/− β-cells nonetheless resembles that of glucose-stimulated mitochondrial superoxide production in isolated islets [19]. Among others, candidate ROS sources also include: 1) Pathways involved in GSIS that are associated with increased glycolytic flux, ratio of [ATP] to [ADP], and intracellular [Ca2+] [42]; 2) Peroxisomal β-oxidation [43]; and 3) Oxidative protein folding in ER, such as that mediated by Ero1α [44], although the latter is unlikely to be the major source of oxidative stress in Ncb5or−/− β-cells in view of their reduced insulin synthesis.
Pancreatic islet β-cells are susceptible to alloxan, which is a glucose analog capable of generating ROS via auto-oxidation of its thiol-reduced product [45]. The hypersensitivity of β cells to alloxan is attributable, at least in part, to unusually low expression of anti-oxidant enzymes, including superoxide dismutase, catalase and glutathione peroxidase in β-cells compared to other tissues, such as liver [46–48]. The ALS genetic background imposes additional deficiencies in β-cell oxidative defense mechanisms, and this exacerbates β-cell injury in Ncb5or−/− mice and results in accelerated onset of diabetes. The ALR background has been shown to confer resistance both to chemically induced [49–51] and spontaneous autoimmune [52] diabetes through amplification of antioxidant defense mechanisms in β-cells and other tissues that include overexpression of antioxidant enzymes and elevated levels of glutathione [49,50]. In contrast to the effect of the ALS genetic background to exacerbate the diabetes-prone phenotype of Ncb5or−/− mice, the ALR background does not affect the age of onset of diabetes in Ncb5or−/− mice, which indicates that the increased antioxidant capacity of the ALR background is insufficient to rescue Ncb5or−/−β-cell function.
Ncb5or deficiency appears to affect β-cell maturation and maintenance, as reflected by reduced islet β-cell content of Ncb5or−/− mice observed as early as 4 weeks of age and by their increased islet α-cell content with unchanged circulating glucagon levels (W.F. Wang, unpublished results). Assessment of β-cell proliferation rates by Ki-67 staining yields similar values for islets from Ncb5or−/− and WT mice [10]. Although both apoptosis and necrosis have been demonstrated β-cells incubated with saturated fatty acids such as palmitate [53], necrosis may be the dominant mode of β-cell loss in Ncb5or−/− mice. Ncb5or−/− β-cells that contained distended ER structures do not exhibit markers of apoptosis, such as TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining [10], caspase 8 activation, or pyknosis (W.F. Wang, unpublished results). Thus, lean Ncb5or-null mice represent a novel model for characterizing pathways involved in maintenance of β-cell function and viability and in protection against lipid-induced stress.
Supplementary Material
TEM analysis of ultrastructure changes in β-cells of Ncb5or−/− mice at age 5 weeks (A–D). Distinctive ER distention (dER, vacuole-like structures) was visible in β cells from Ncb5or−/− mice (B–D) whereas none in WT (A). Subcellular structures are labeled as: dER, distended ER; ER, endoplasmic reticulum; mito, mitochondria; N, nucleus; SG, secretion granules. Total cellular (E) and nuclear areas (F) in WT (n=4) and Ncb5or−/− mice (n=3), 10 intact β-cells per animal, were measured by the NIH Image J software. Results are expressed as mean ± S.E. *, p<0.05. White-bar: wild-type (WT); Black-bar: Ncb5or−/−.
Highlights.
We characterize early events in beta-cell dysfunction in a lean diabetes mouse.
We examine beta-cell lipids, endoplasmic reticulum and oxidative stress.
Severity and abundance of distended endoplasmic reticulum worsens over time.
Elevated levels of saturated fatty acids are associated with cellular stress.
Manifestation of diabetes correlates to endoplasmic reticulum and oxidative stress.
Acknowledgments
Authors thank S. Janette Williams for training with islet isolation, Barbara Fegley for help with electron microscopy, and Ronald MacGregor for critical reading of the manuscript. This work is supported by grants from NIH RO1-DK067355 (H.F.B. and H.Z.) and RO1-AI056374 (C.E.M), the Juvenile Diabetes Research Foundation 1-2005-121 (H.F.B.) and 3-2005-232 (K.L.), the American Diabetes Association 7-04-RA-15 (H.F.B.), the Emilie Rosebud Diabetes Research Foundation (L.S.B.), and a fellowship from the China Scholarship Council (Y.G.). J.T. is supported by NIH R37-DK34388, P41-RR00954, P60-DK20579, and P30-DK56341. Core facilities at KUMC are supported by NIH HD02528.
The abbreviations used are
- DI
desaturation index
- ER
endoplasmic reticulum
- FA
fatty acids
- FAME
fatty acid methyl esters
- FBS
fetal bovine serum
- FFA
free fatty acids
- GC
gas chromatography
- GTT
glucose tolerance test
- MS
mass spectrometry
- MUFA
monounsaturated fatty acid
- NADH
nicotinamide adenine dinucleotide (reduced)
- PBS
phosphate buffered saline
- ROS
reactive oxygen species
- SFA
saturated fatty acid
- TEM
transmission electron microscopy
- TAG
triacylglycerol
- UPR
unfolded protein response
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 2.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 3.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci U S A. 1994;91:10878–10882. doi: 10.1073/pnas.91.23.10878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl 1):S119–124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 6.Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, Flamez D, Boyce M, Yuan J, Eizirik DL. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J Biol Chem. 2007;282:3989–3997. doi: 10.1074/jbc.M607627200. [DOI] [PubMed] [Google Scholar]
- 7.Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, Moore F, Rasschaert J, Cardozo AK, Bellomo E, Overbergh L, Mathieu C, Lupi R, Hai T, Herchuelz A, Marchetti P, Rutter GA, Eizirik DL, Cnop M. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. 2008;121:2308–2318. doi: 10.1242/jcs.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diakogiannaki E, Welters HJ, Morgan NG. Differential regulation of the endoplasmic reticulum stress response in pancreatic beta-cells exposed to long-chain saturated and monounsaturated fatty acids. J Endocrinol. 2008;197:553–563. doi: 10.1677/JOE-08-0041. [DOI] [PubMed] [Google Scholar]
- 9.Oresic M, Simell S, Sysi-Aho M, Nanto-Salonen K, Seppanen-Laakso T, Parikka V, Katajamaa M, Hekkala A, Mattila I, Keskinen P, Yetukuri L, Reinikainen A, Lahde J, Suortti T, Hakalax J, Simell T, Hyoty H, Veijola R, Ilonen J, Lahesmaa R, Knip M, Simell O. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med. 2008;205:2975–2984. doi: 10.1084/jem.20081800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie J, Zhu H, Larade K, Ladoux A, Seguritan A, Chu M, Ito S, Bronson RT, Leiter EH, Zhang CY, Rosen ED, Bunn HF. Absence of a reductase, NCB5OR, causes insulin-deficient diabetes. Proc Natl Acad Sci U S A. 2004;101:10750–10755. doi: 10.1073/pnas.0404044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu H, Qiu H, Yoon HW, Huang S, Bunn HF. Identification of a cytochrome b-type NAD(P)H oxidoreductase ubiquitously expressed in human cells. Proc Natl Acad Sci U S A. 1999;96:14742–14747. doi: 10.1073/pnas.96.26.14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu H, Larade K, Jackson TA, Xie J, Ladoux A, Acker H, Berchner-Pfannschmidt U, Fandrey J, Cross AR, Lukat-Rodgers GS, Rodgers KR, Bunn HF. NCB5OR is a novel soluble NAD(P)H reductase localized in the endoplasmic reticulum. J Biol Chem. 2004;279:30316–30325. doi: 10.1074/jbc.M402664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larade K, Jiang Z, Zhang Y, Wang W, Bonner-Weir S, Zhu H, Bunn HF. Loss of NCB5OR results in impaired fatty acid desaturation, lipoatrophy and diabetes. J Biol Chem. 2008;283:29285–29291. doi: 10.1074/jbc.M804645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu M, Wang W, Frontera JR, Neely MC, Lu JJ, Aires D, Hsu FF, Turk J, Swerdlow RH, Carlson SE, Zhu H. Ncb5or deficiency increases fatty acid catabolism and oxidative stress. J Biol Chem. 2011;286:11141–11154. doi: 10.1074/jbc.M110.196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Larade K, Jiang ZG, Ito S, Wang W, Zhu H, Bunn HF. The flavoheme reductase Ncb5or protects cells against endoplasmic reticulum stress-induced lipotoxicity. J Lipid Res. 2010;51:53–62. doi: 10.1194/jlr.M900146-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ino T, Kawamoto Y, Sato K, Nishikawa K, Yamada A, Ishibashi K, Sekiguchi F. Selection of mouse strains showing high and low incidences of alloxan-induced diabetes. Jikken Dobutsu. 1991;40:61–67. doi: 10.1538/expanim1978.40.1_61. [DOI] [PubMed] [Google Scholar]
- 17.Huang HH, Novikova L, Williams SJ, Smirnova IV, Stehno-Bittel L. Low insulin content of large islet population is present in situ and in isolated islets. Islets. 2011;3:1–8. doi: 10.4161/isl.3.1.14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacGregor RR, Williams SJ, Tong PY, Kover K, Moore WV, Stehno-Bittel L. Small rat islets are superior to large islets in in vitro function and in transplantation outcomes. Am J Physiol Endocrinol Metab. 2006;290:E771–779. doi: 10.1152/ajpendo.00097.2005. [DOI] [PubMed] [Google Scholar]
- 19.Bindokas VP, Kuznetsov A, Sreenan S, Polonsky KS, Roe MW, Philipson LH. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J Biol Chem. 2003;278:9796–9801. doi: 10.1074/jbc.M206913200. [DOI] [PubMed] [Google Scholar]
- 20.Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A. 2006;103:2334–2339. doi: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M. Islet architecture: A comparative study. Islets. 2009;1:129–136. doi: 10.4161/isl.1.2.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 24.Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol. 2002;22:8506–8513. doi: 10.1128/MCB.22.24.8506-8513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 28.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 29.Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. 2007;27:1027–1043. doi: 10.1128/MCB.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabibbo A, Pagani M, Fabbri M, Rocchi M, Farmery MR, Bulleid NJ, Sitia R. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J Biol Chem. 2000;275:4827–4833. doi: 10.1074/jbc.275.7.4827. [DOI] [PubMed] [Google Scholar]
- 31.Strandell E, Buschard K, Saldeen J, Welsh N. Interleukin-1 beta induces the expression of hsp70, heme oxygenase and Mn-SOD in FACS-purified rat islet beta-cells, but not in alpha-cells. Immunol Lett. 1995;48:145–148. doi: 10.1016/0165-2478(95)02459-x. [DOI] [PubMed] [Google Scholar]
- 32.Peyot ML, Guay C, Latour MG, Lamontagne J, Lussier R, Pineda M, Ruderman NB, Haemmerle G, Zechner R, Joly E, Madiraju SR, Poitout V, Prentki M. Adipose triglyceride lipase is implicated in fuel- and non-fuel-stimulated insulin secretion. J Biol Chem. 2009;284:16848–16859. doi: 10.1074/jbc.M109.006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu FF, Bohrer A, Wohltmann M, Ramanadham S, Ma Z, Yarasheski K, Turk J. Electrospray ionization mass spectrometric analyses of changes in tissue phospholipid molecular species during the evolution of hyperlipidemia and hyperglycemia in Zucker diabetic fatty rats. Lipids. 2000;35:839–854. doi: 10.1007/s11745-000-0593-z. [DOI] [PubMed] [Google Scholar]
- 34.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology. 2006;147:3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- 36.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 40.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 41.Chambers KT, Weber SM, Corbett JA. PGJ2-stimulated beta-cell apoptosis is associated with prolonged UPR activation. Am J Physiol Endocrinol Metab. 2007;292:E1052–1061. doi: 10.1152/ajpendo.00274.2006. [DOI] [PubMed] [Google Scholar]
- 42.Fridlyand LE, Philipson LH. Does the glucose-dependent insulin secretion mechanism itself cause oxidative stress in pancreatic beta-cells? Diabetes. 2004;53:1942–1948. doi: 10.2337/diabetes.53.8.1942. [DOI] [PubMed] [Google Scholar]
- 43.Elsner M, Gehrmann W, Lenzen S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes. 2011;60:200–208. doi: 10.2337/db09-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tu BP, Weissman JS. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell. 2002;10:983–994. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 45.Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. 2008;51:216–226. doi: 10.1007/s00125-007-0886-7. [DOI] [PubMed] [Google Scholar]
- 46.Grankvist K, Marklund SL, Taljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J. 1981;199:393–398. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- 48.Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 49.Mathews CE, Leiter EH. Resistance of ALR/Lt islets to free radical-mediated diabetogenic stress is inherited as a dominant trait. Diabetes. 1999;48:2189–2196. doi: 10.2337/diabetes.48.11.2189. [DOI] [PubMed] [Google Scholar]
- 50.Mathews CE, Leiter EH. Constitutive differences in antioxidant defense status distinguish alloxan-resistant and alloxan-susceptible mice. Free Radic Biol Med. 1999;27:449–455. doi: 10.1016/s0891-5849(99)00084-2. [DOI] [PubMed] [Google Scholar]
- 51.Mathews CE, Leiter EH, Spirina O, Bykhovskaya Y, Gusdon AM, Ringquist S, Fischel-Ghodsian N. mt-Nd2 Allele of the ALR/Lt mouse confers resistance against both chemically induced and autoimmune diabetes. Diabetologia. 2005;48:261–267. doi: 10.1007/s00125-004-1644-8. [DOI] [PubMed] [Google Scholar]
- 52.Mathews CE, Graser RT, Savinov A, Serreze DV, Leiter EH. Unusual resistance of ALR/Lt mouse beta cells to autoimmune destruction: role for beta cell-expressed resistance determinants. Proc Natl Acad Sci U S A. 2001;98:235–240. doi: 10.1073/pnas.98.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes. 2001;50:1771–1777. doi: 10.2337/diabetes.50.8.1771. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TEM analysis of ultrastructure changes in β-cells of Ncb5or−/− mice at age 5 weeks (A–D). Distinctive ER distention (dER, vacuole-like structures) was visible in β cells from Ncb5or−/− mice (B–D) whereas none in WT (A). Subcellular structures are labeled as: dER, distended ER; ER, endoplasmic reticulum; mito, mitochondria; N, nucleus; SG, secretion granules. Total cellular (E) and nuclear areas (F) in WT (n=4) and Ncb5or−/− mice (n=3), 10 intact β-cells per animal, were measured by the NIH Image J software. Results are expressed as mean ± S.E. *, p<0.05. White-bar: wild-type (WT); Black-bar: Ncb5or−/−.