

Abstract
The integration of pharmaceutical nanocarriers with phage display techniques is emerging as a new paradigm for targeted cancer nanomedicines. We explored the direct use of landscape phage fusion proteins for the self-assembly of phage-derived binding peptides to liposomes for cancer cell targeting. The primary purpose of this study was to elucidate the targeting mechanism with a particular emphasis on the relative contributions of the two motifs that make up the landscape phage fusion protein (a binding peptide and the phage pVIII coat protein) to the targeting efficiency. Using transmission electron microscopy and dynamic light scattering, we confirmed the formation of phage-liposomes. Using FACS analysis, fluorescence microscopy, and fluorescence photospectrometry, we found that liposomes modified with MCF-7-specific phage fusion proteins (MCF-7 binding peptide, DMPGTVLP, fused to the phage PVIII coat protein) provided a strong and specific association with target MCF-7 cancer cells but not with co-cultured, non-target cells including C166-GFP and NIH3T3. The substitution for the binding peptide fused to phage pVIII coat protein abolished the targeting specificity. The addition of free binding peptide, DMPGTVLP, competitively inhibited the interaction of MCF-7-specific phage-liposomes with target MCF-7 cells but showed no reduction of MCF-7-associated plain liposomes. The proteolysis of the binding peptide reduced MCF-7 cell-associated phage-liposomes in a proteinase K (PK) concentration-dependent manner with no effect on the binding of plain liposomes to MCF-7 cells. Overall, only the binding peptide motif was involved in the targeting specificity of phage-liposomes. The presence of phage pVIII coat protein did not interfere with the targeting efficiency.
Keywords: Tumor targeting, Phage display, Landscape phage, Phage coat protein pVIII, Drug delivery, Liposome, Breast cancer
Introduction
Delivery of a sufficient quantity of anticancer therapeutics to tumors with a low impact on normal tissues is essential for the success of cancer therapy1. Within this conceptual frame, cancer-targeted nanomedicines have been developed. These efforts often involve the use of various pharmaceutical nanocarriers, such as liposomes, polymers, proteins, polymeric micelles and lipoplex/polyplex complexes2–4. These nanosized carriers are capable of passive targeting by preferential accumulation of nanocarrier-associated drugs in tumors via the “enhanced permeability and retention” effect, a unique physiological phenomenon occurring in tumor microenvironments as a result of active angiogenesis and impaired lymphatic drainage5. In addition, molecular signatures on the surface of tumor cells distinguish them from healthy tissues6, allowing for the design of ligand-mediated “active” tumor targeting2, 3, 7, 8. The targeting ligands guide pharmaceutical nanocarriers to specific cancer cells and mediate their intracellular delivery via an endocytic pathway. The integration of passive targeting of pharmaceutical nanocarriers with tumor cell recognition and/or cell-internalizing ligands is a sophisticated strategy with the synergistic advantages of pharmacokinetic-driven/biodistribution-driven passive targeting and active molecular targeting7. Ongoing challenges in the development of cancer-targeted nanomedicines include the identification of novel tumor-selective ligands and the development of approaches for their integration with pharmaceutical nanocarriers.
The merger of nanotechnology with the combinatorial phage display technique represents a new paradigm for advanced cancer-targeted nanomedicines9–11. Phage display technology provided a promising way for the discovery of novel targeting ligands specific for cancer cells in which prior knowledge about receptors targeted is not necessary. Instead, the identification of targeting ligands is accomplished by an affinity screening of phage display peptide libraries based on their abilities to interact specifically with target cancer cells of interest10–12. This high throughput methodology has led to the discovery of a plethora of cancer-specific ligands9, 13, 14. The usual scheme for the combination of a phage-derived targeting ligand and a liposomal drug carrier requires multiple steps: a chemical synthesis of the identified peptide sequence followed by a modification of the synthesized peptide with a hydrophobic moiety for its anchorage in the liposome membrane15, 16. Although, a wide range of chemical conjugations have been developed, the modification of ligands still faces many challenges. In some cases, the introduction of a reactive group for cross-linking a ligand with a hydrophobic anchor results in a reduced cell binding. Harsh reactive conditions may inactivate proteins as well.7 Chemical conjugation of a targeting moiety to a pharmaceutical nanocarrier usually requires a complex reaction scheme, and the attachment of each requires specific efforts.
As an alternative, we have proposed a landscape phage protein-based approach, in which chemical modifications are bypassed by a direct use of the landscape phage fusion protein, an intermediate in the phage display process with binding peptide fused to the N-terminus of the phage pVIII coat protein17–19. The rational for this approach is based on an intrinsic property of the phage pVIII coat protein as an amiphiphilic membrane protein, which can navigate into bacterial membranes during the phage infection and assembly20,21. This approach exploits this capacity for spontaneous insertion of the phage pVIII coat proteins into the liposomal bilayers that resembles their residences within bacterial membranes22, 23, and accordingly, tethers the fused binding peptides on the liposomal surface17–19. Our early study was a proof-of-concept showing, in particular, that the landscape phage protein-based approach could impart doxorubicin-loaded liposomes with a selective cancer cell targeting and enhanced tumor cell killing17, 18.
While phage display techniques have enabled the identification of target ligands for a wide spectrum of diseases in a high-throughput fashion, their timely translation into biomedical application has been challenged by the lack of rapid and efficient methods for their attachment to pharmaceuticals and pharmaceutical carriers. The landscape phage protein-based approach presented here, provides a new paradigm for the self-assembly of phage fusion protein with pharmaceutical nanocarriers, such as liposomes17–19 and polymeric micelles24. With this novel approach, the phage pVIII coat protein, the same as used in the conventional phage display, acts as an universal genetic support for various binding peptides, which are displayed on the surface of bacteriophage11 and are subsequently screened against different targets of interest11, 25. The phage pVIII coat protein also serves as a universal anchorage to tether binding peptides to the surface of liposomes and allowing for the site-specific targeting of the liposome-loaded drugs to the pathological area17–19, 26. Thus this approach represents a potential “one-fit-all” scheme for the development of targeted nanomedicines against a wide variety of diseases.
On the other hand, the phage pVIII coat protein on its own may potentially interact with cells and trigger a non-specific binding since its membranephilic, highly hydrophobic transmembrane motif can interact with biological membranes as well as with liposomal membranes23, 27, 28, Therefore, the key task to allow the applicability of this novel approach in various disease conditions is to preclude the possibility that the phage pVIII coat protein motif itself may interfere with the selective targeting. Ideally, only the binding peptide motif should be involved in specific cell binding.
We have used the phage display platform to screen different phage-derived fusion proteins with specificity towards different targets including MCF-7 breast cancer cells, PC3 prostate cancer cells, and streptavidin,17, 18, 24, 26 and have applied the same preparation protocol for the incorporation of different landscape phage fusion proteins into the liposomal membranes in such a fashion that the targeting peptides are exposed on the outer surface of the liposomes. The resultant phage-liposome formulations showed a high degree of binding affinity and specificity towards the targets 17–19. Still, the exact mechanisms underlying targeting remain incompletely understood. In particular, it is unclear whether only the binding peptide contributes to the targeting specificity or if the phage pVIII coat protein is involved in some way as well.
Hence, the primary purpose of this study was to elucidate the targeting mechanism of the landscape phage fusion protein-based approach with a particular emphasis on the relative contributions to the targeting efficiency of the two motifs that make up the landscape phage fusion protein. In examination of the roles of binding peptide and phage pVIII coat protein in the targeting phenomenon, we have found the involvement of only the binding peptide in the target specificity of phage-liposomes with the phage pVIII coat protein being not involved in the specific binding phenomenon.
Experimental Section
Materials and Reagents
L-α-phosphatidylcholine (ePC); 1, 2-dipalmitoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt; DPPG); 1, 2-dioleoyl-3-trimethylammonium-propane (chloride salt; DOTAP); 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (polyethylene glycol) 2000] (ammonium salt; PEG2k-PE); cholesterol (98%) (CHOL), and 1, 2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt, Rho-PE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). Sodium cholate and proteinase K were purchased from Sigma (St. Louis, MO); Free binding peptide DMPGTVLP was custom–synthesized by Biomatik Corporation (Cambridge, Canada). Fluor Mounting Medium was from Trevigen Inc (Gaithersburg, MD). Streptavidin-coated plates were purchased from Thermo Fisher Scientific Inc. MCF-7 (HTB 22™) human breast adenocarcinoma cells, and control NIH3T3 (CRL-1658™) mouse fibroblasts cells and C166-GFP (cells expressing the green fluorescence protein, CRL-2583™) mouse yolk sac endothelial cells were obtained from the ATCC (Manassas, VA). All cells were grown as recommended by ATCC at 37°C, 5% CO2.
Preparation of liposomes
Phage-liposomes were prepared using a post-insert protocol as described before17, 29. Briefly, a lipid mix composed of ePC: CHOL: DPPG: DOTAP: PEG2k-PE: Rho-PE at % molar ratio of 44: 30: 20: 2: 3: 1 in chloroform was evaporated to remove the solvent, followed by the hydration of the lipid film obtained using PBS buffer (pH 7.4). After bath-sonication for 30 min and following extrusion through 200 nm polycarbonate membrane 10 times, the preformed plain liposomes were incubated with phage fusion proteins at lipid-to-phage protein weight ratios of 200:1 or 100:1 and at a 15mM final concentration of sodium cholate. After the overnight incubation at 37°C, the remaining sodium cholate was removed using dialysis at 4°C against PBS buffer (pH 7.4).
Size and size distribution of phage-liposomes
The size distribution of liposomes was determined by dynamic light scattering (DLS). Briefly, samples were diluted in PBS buffer (pH 7.4) followed by a measurement of size using a Beckman Coulter N4 Plus Particle analyzer (Beckman Coulter, Fullerton, CA) with a scattering angle of 90° and particle size range measurements of 1–1000nm.
Zeta potential of phage-liposomes
Aliquots of the liposomal preparations were diluted as needed with distilled water. Zeta potential was measured in a Zeta potential analyzer (Brookhaven, Holtstsville, NY).
Transmission electron microscopy
A JEOL JEM-1010 transmission electron microscope (JEOL USA, Inc., Peabody, MA) was used to observe the morphology of phage-liposomes. Briefly, samples of phage-liposomes (10 μl) were dropped on a copper grid with formvar and carbon coating, followed by negative staining with 1% uranyl acetate. After air-drying at room temperature, samples were imaged using a transmission electron microscopy operating at an accelerating voltage of 80 kV.
FACS analysis of the binding affinity of phage-liposomes
MCF-7 cells were cultured in MEM supplemented with 10% FBS and 1% antibiotics until 70–80% confluence, unless otherwise stated. For the analysis of the binding affinity of phage-liposomes to target cells, MCF-7 were treated with 140 μM of Rhodamine-labeled phage-liposomes with a varying weight lipid-to-phage protein ratio of 200:1 or 100:1 or with control plain liposomes for 1 h at 37°C. For the competitive inhibition by the free binding peptide, DMPGTVLP, MCF-7 were pretreated with free peptides at a concentration of 2.1mM for 1 h followed by a co-incubation with either MCF-7-specific phage-liposomes or plain liposomes at the lipid concentration of 56.5 μM or 113 μM for an additional 20 min at 37°C. As control groups, MCF-7 cells were also treated with MCF-7-specific phage-liposomes alone or with plain liposomes alone at the lipid concentration of 56.5 μM or 113 μM for 20 min at 37°C. The competitive inhibition ratio was defined as the difference in the geomean of cell binding between phage-liposomes and phage-liposomes in the presence of free peptide, divided by the difference in the geomean of cell binding between phage-liposomes and untreated control cells.
To follow the effect of proteinase K (PK) treatment on the interaction of liposomes with target cells, MCF-7 cells were incubated with 140 μM of phage liposomes or 140 μM of plain liposomes with or without a 30 min pretreatment by PK at a final concentration of 0.25 mg/ml for 1 h. After a 2× PBS washing, cells were detached and collected by centrifugation. The cell pellets were re-suspended in 100 μl of PBS containing 4% paraformaldehyde. The collected cells were analyzed with a BD FACSCalibur flow cytometer (Becton Dickinson Company). Cell-associated Rhodamine-labeled liposomes were observed as the red fluorescence indicated by a right shift of the population on the x axis (FL2-H, Red).
Fluorescence microscopy
MCF-7 cells or NIH3T3 cells were co-cultured with C166 endothelial cells expressing GFP at a 1:1 ratio and seeded in 6 well microplates in MEM with 10% FBS and 1% antibiotics until 70–80% confluence. Cells were incubated with 170 μM of Rhodamine-labeled phage-liposomes or plain liposomes for 30 min at 37°C. After 3× PBS washing, cells were imaged using a fluorescence microscope (Nikon, Japan) at 40× magnification with FITC or TRITC filter. All images were taken with a CCD camera. The data was collected using SPOT software and exported as tagged image files (TIF).
Fluorescence photospectrometric analysis
For the determination of streptavidin–associated liposomes, Rhodamine-labeled liposomes modified with either streptavidin-specific or MCF-7-specific phage fusion protein were incubated with streptavidin-coated 96-well microplates at varying concentrations for 15 min, followed by 2× PBS washing to remove the liposomes not associated with streptavidin. The florescence of streptavidin-associated Rhodamine-labeled liposomes was measured at wavelengths with EX/EM of 525/590 nm in a fluorescence spectrometer (Bio-Tek, Winooski, VT).
For the assessment of the effect of the PK treatment on the association of liposomes with MCF-7 cells, MCF-7 cells were grown in 96-well microplates in MEM supplemented with 10% FBS and 1% antibiotics until 70–80% confluence. Then, MCF-7 cells were incubated with varying concentrations of MCF-7-specific phage-liposomes or plain liposomes with or without a 30 min pretreatment with varying concentrations of PK (0.063–0.375 mg/ml) for 1 h, followed by 2× PBS washing to remove liposomes not associated with MCF-7 cells. The florescence of MCF-7 cell-associated Rhodamine-labeled liposomes was measured at wavelengths with EX/EM of 525/590 nm in a fluorescence spectrometer (Bio-Tek, Winooski, VT).
Statistical analysis
The statistical significance of the results was analyzed using SPSS software (version 17). Differences between experimental groups were compared using an ANOVA followed by the Bonferroni post hoc test. The results were considered statistically significant if the p value was less than 0.05.
RESULTS
Characterization of MCF-7-specific phage-liposomes
The morphology of phage-liposomes revealed using a TEM with a magnification of 163000×, showed monodisperse particles with uniform, spherical shapes (Figure 1). Results from the dynamic light scattering analysis were consistent with those from TEM analysis with the liposome size distribution within 130 to 230 nm (Table 1).
Figure 1. Transmission electron micrograph of MCF-7-specific phage-liposomes.

(Magnification = 163000×).
Table 1.
Characterization of liposomes.
| Liposomes | Phage-liposomes | Plain liposomes |
|---|---|---|
| Characteristics | ||
| Mean size (nm) | 187 ± 51.7 | 209.8 ± 60.2 |
| Polydispersity | 0.063 | 0.02 |
| ζ-potential (mV) | −28.47 ± 7.82 | −39.48 ± 3.71 |
The binding affinity of MCF-7-specific phage-liposomes to target MCF-7 cells
The association of MCF-7-specific phage-liposomes with target tumor MCF-7 cells was determined using FACS analysis in comparison with control plain liposomes (Figure 2A, B). Phage-liposomes showed a significantly stronger binding with MCF-7 cells than did plain liposomes. The binding affinity increased with an elevated density of phage proteins present in liposome formulations (Figure 2B).
Figure 2. Binding of MCF-7-specific phage-liposomes by MCF-7 cells.

(A) Representative FACS histogram, and (B) Geomean analysis on MCF-7 cell-associated liposomes, showing uptake of MCF-7-specific phage-liposomes by MCF-7 cells; (mean ± SD, n=3). Colors of lines in (A): Red: untreated; Blue: plain liposomes; Pink: MCF-7-specific phage-liposomes with 0.5% (w/w) phage fusion protein; Green: MCF-7-specific phage-liposomes with 1% (w/w) phage fusion protein.
The binding selectivity of MCF-7-specific phage-liposomes to target MCF-7 cells
To investigate whether the MCF-7-specific phage-liposomes bind selectively to targeted cancer cells rather than to normal cells, we designed a co-culture model, in which target cancer MCF-7 cells were co-grown with non-target, non-cancer endothelial cells C166 expressing the GFP (C166-GFP). As a negative control, another co-culture model with non-target, non-cancer NIH3T3 and C166-GFP cells was used. C166-GFP cells could be visualized due to their green fluorescence with fluorescence microscopy and therefore could be distinguished from the co-cultured MCF-7 or NIH3T3 cells. The treatment with Rhodamine-labeled MCF-7-specific phage-liposomes of the co-cultures of MCF-7 and C166-GFP cells has clearly showed specific binding of phage-liposomes to MCF-7 cells but not to C166-GFP cells as showed by the lack of co-localization of red and green fluorescence (Figure 3A). Control plain liposomes showed little, if any, association with both MCF-7 and C166-GFP cells (Figure 3B), indicating that the phage protein mediated the selective targeting of phage-liposomes to MCF-7 cells. To further confirm these results, a negative control co-culture model composed of NIH3T3 and C166-GFP cells was also treated with Rhodamine-labeled MCF-7-specific phage-liposomes or plain liposomes, and neither liposome formulation showed an association with non-targeted NIH3T3 or C166-GFP cells (Figure 3C & 3D).
Figure 3. Binding selectivity of MCF-7-specific phage-liposomes in co-cultures.

(A) MCF-7-specific phage-liposomes and (B) plain liposomes in a co-culture of target MCF-7 and non-target C166-GFP cells. (C) MCF-7-specific phage-liposomes and (D) plain liposomes in a co-culture of non-target of NIH3T3 and C166-GFP cells.
Dependence of the targeting specificity on the binding peptide
Since the MCF-7-specific phage protein is a hybrid fusion protein, in which an 8-mer binding peptide (DMPGTVLP) was fused genetically to the phage pVIII coat protein, we sought to elucidate exactly which segment – the binding peptide or the phage pVIII coat protein – contributes to the specific targeting. A streptavidin-specific phage fusion protein with an 8-mer binding peptide (VPEGAFSS) fused to the same phage pVIII coat protein was selected as a negative control for these experiments. While the MCF-7-specific phage-liposomes bound selectively to target MCF-7 cells co-cultured with C166-GFP cells (Figure 4A), negative control streptavidin-specific phage-liposomes associated with both MCF-7 cells and C166-GFP cells without preference (Figure 4B), indicating that the binding peptide, DMPGTVLP, rather than the phage pVIII coat protein is responsible for the targeting specificity. Meanwhile, streptavidin-specific phage-liposomes showed a significantly higher binding to target of their own – streptavidin coated on the microplates, than did MCF-7-specific phage-liposomes (Figure 4C), further indicating that the binding peptide sequence plays the key role in targeting selectivity of phage-liposomes.
Figure 4. The binding peptide and the selectivity of targeting.

(A) MCF-7-specific phage-liposomes containing binding peptide (DMPGTVLP) fused to the N-terminus of phage PVIII coat protein, showing specific binding to MCF-7 cells co-cultured with C166-GFP cells; (B) Control streptavidin-specific phage-liposomes containing binding peptide (VPEGAFSS) fused to N-terminus of phage pVIII coat protein, showing a non-preferential binding to co-cultured MCF-7 cells and C166-GFP cells. (C) Streptavidin-specific phage-liposomes showing significantly higher binding to streptavidin coated on a microplate than MCF-7-specific phage-liposomes (mean ± SD, n=8). Brocade bar: Streptavidin-specific phage-liposomes; Grey Bar: MCF-7-specific phage-liposomes.
Competitive inhibition of the binding of MCF-7-specific phage-liposomes to MCF-7 cells by free binding peptide, DMPGTVLP
To further confirm the role of the binding peptide, DMPGTVLP, in the target specificity of MCF-7-specific phage-liposomes, we carried out a competitive inhibition experiment using the synthetic free binding peptide, DMPGTVLP. The FACS analysis clearly showed that the pretreatment by the excess of free binding peptide for 1h at the molar ratio of free peptide to liposome-bound phage fusion protein of ca. 56,620: 1, significantly reduced the binding of MCF-7-specific phage-liposomes to target MCF-7 cells (Figure 5A & B), but did not affect the binding of plain liposomes to MCF-7 cells (Figure 5C & D). As the molar ratio of free peptide to liposome-bound phage fusion protein decreased from 56,620: 1 to 28,310: 1, the competitive inhibition ratio decreased from 24.3% to 17.7 %. As the constant molar ratio of 56,620: 1, the increase in MCF-7 cell confluence from ca. 75% to ca. 90% resulted in a reduction in cellular binding from 24.3% to 21%.
Figure 5. Competitive inhibition of binding of MCF-7-specific phage-liposomes to MCF-7 cells by free peptide (DMPGTVLP).
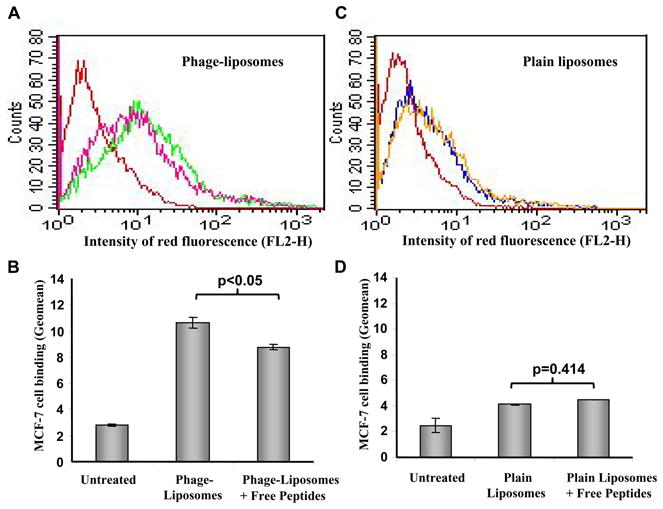
(A) Representative histogram of FACS analysis and (B) Geomean analysis of the binding of MCF-7-specific phage-liposomes to MCF-7 cells in the presence and absence of free peptide. (C) Representative histogram of FACS analysis and (D) Geomean analysis of the binding of plain liposomes to MCF-7 cells in the presence and absence of free peptide. Colors of lines in A, C: Red: untreated; Pink: MCF-7-specific phage-liposomes in the presence of free peptide; Green: MCF-7-specific phage-liposomes. Blue: plain liposomes; Yellow: plain liposomes in the presence of free peptide (mean ± SD, n=3).
The effect of proteinase K (PK) treatment on the binding of MCF-7-specific phage-liposomes to target MCF-7 cells
In previous studies, we found that PK treatment of phage-liposomes digested the N-terminus of the phage fusion protein but had no effect on its C-terminus, indicating that phage fusion protein resides within liposome membranes in such a way that N-terminus is exposed on the outer-surface of phage-liposomes while the C-terminus is immersed within the liposomal membrane17, 19. We further examined the effect of PK digestion on the binding of MCF-7-specific phage-liposomes to MCF-7 cells. FACS analysis showed that PK degradation of DMPGTVLP peptide fused to the N-terminus of MCF-7-specific phage protein, significantly reduced the quantity of MCF-7 cell-associated phage-liposomes to a level comparable statistically to that of plain liposomes (Figure 6A & B), but the PK treatment had no significant effect on the binding of plain liposomes to MCF-7 cells (Figure 6C & 6D). In addition, a spectrophotometric assay confirmed these results (Figure 6E), and target cell binding of MCF-7 phage-liposomes decreased as the concentration of PK increased (Figure 6F).
Figure 6. The effect of the degradation of the N-terminus of the phage fusion protein by proteinase K (PK) on the binding of rhodamine-labeled, MCF-7-specific phage-liposomes to target MCF-7 cells.

(A) Representative FACS histograms, and (B) geomean analysis showing that PK treatment results in a significant decrease in MCF-7 cell-associated MCF-7-specific phage-liposomes to a level comparable statistically to that of plain liposomes. (C) Representative FACS histogram and (D) geomean analysis showing that PK treatment has no effect on MCF-7 cell-associated plain liposomes. Red: Untreated cells; Blue: Plain liposomes; Pink: Phage- liposomes; Green: Phage-liposomes with PK treatment. Yellow: Plain liposomes with PK treatment. (E) Fluorescence photometric data confirming that PK treatment results in a decrease in MCF-7 cell-associated MCF-7-specific phage–liposomes to the level comparable to plain liposomes, but has no effect on MCF-7-associated plain liposomes. Grey bar: MCF-7-phage-liposomes; Brocade bar: Phage-liposomes with PK treatment. White bar: plain liposomes; Black bar: Plain liposomes with PK treatment. (F) The quantity of MCF-7 cell-associated MCF-7- specific phage–liposomes decreases with an increase in the PK concentration. (Mean ± SD, n=3).
Discussion
The molecular signatures of tumor cells provide a rational basis for the identification of their-specific ligands and for targeted cancer therapies25, 30. Phage display technology is emerging as a powerful tool for discovery of cancer cell-specific peptide ligands10, 13, 31. The merger of phage display technology with pharmaceutical nanocarriers has resulted in novel targeted cancer nanomedicines11, 13. The landscape phage protein-based approach reported provides a simple and straight-forward way to the fusion of both technologies without a chemical conjugation.
In the landscape phage protein-based approach, the phage fusion proteins self-assemble with the liposomal drug carriers to create targeted nanoparticles with components coexisting in a compatible and synergistic form. The presence of the phage protein does not disturb the liposome morphology. Indeed, we observed that their mix resulted in uniform, spherical particles with a liposomal morphology revealed in the TEM micrographs. The modification of liposomes with phage protein provided liposomes with the ability to bind target cells stronger compared to the non-modified liposomes. The binding affinity of phage-liposomes depends on the density of phage protein present in the liposomes, further supporting the role of phage protein in the mediation of liposomal association with target cells.
Our early studies showed that the landscape phage fusion proteins interacted with liposomes in such a way that their C-termini were buried in the liposomal bilayers exposing only their N-termini fused with binding peptide on the outer surface of liposomes17, 19. The resultant topology is derived from the membraneotropic nature of phage pVIII coat protein. While a Nout/Cin terminal orientation of phage fusion protein within the liposomal bilayers provides a reasonable interpretation as to why phage fusion protein-modified liposomes retained the target specificity of the binding peptide and why the presence of the phage pVIII coat protein motif does not interfere with the targetability, this study provides more direct evidence that the binding peptide motif provides the target specificity rather than the phage pVIII coat protein motif. We found that liposomes modified with MCF-7-specific phage fusion protein [the MCF-7-binding peptide sequence (DMPGTVLP) fused to phage PVIII coat protein], selectively recognized target MCF-7 cells rather than non-target, normal cells including C166-GFP and NIH3T3, while liposomes modified with an irrelevant phage fusion protein [the streptavidin-binding peptide sequence (VPEGAFSS) fused to the same phage PVIII coat protein], did not show any target specificity towards MCF-7 cells. Conversely, it was the streptavidin-specific liposomes but not MCF-7-specific liposomes that showed the stronger binding to streptavidin-coated microplates. As shown in an early study of ours26, the streptavidin-specific liposomes consistently recognized streptavidin-conjugated colloidal gold nanoparticles. These results indicated that targeting specificity of the landscape phage fusion protein-based approach depends on the binding peptide motif.
This conclusion was further strengthened by the observation that an excess of the free binding peptide (DMPGTVLP) competitively inhibited the binding of MCF-7-specific phage-liposomes to target MCF-7 cells. Furthermore, the proteinase K treatment of phage-liposomes to cleave the targeted peptide sequence displayed on the N-terminus of phage fusion protein led to a significant decrease in association of phage-liposomes with target cells, confirming that the remaining phage coat protein segment within liposomal bilayer after the PK treatment is not essential to the targetability of phage-liposomes. As a matter of fact, we previously observed that liposomes modified with the Wide-type phage pVIII coat protein (no fused binding peptide) behaved like non-modified plain liposomes which lack the ability to discriminate between target and non-target cells18. Collectively, these results suggest that the phage pVIII coat protein motif does not interfere with the binding specificity of the target peptide motif.
The key aspect of the landscape phage protein-based approach is the assurance of the correct orientation of the phage fusion protein within the liposomal bilayer that maintains the target selectivity of the binding peptide and avoids the possibility of non-specificity induced by the phage pVIII coat protein itself. The approach explores the unique propensity of phage coat proteins to incorporate spontaneously into the bacterial, as well as liposomal, membranes22. The hydrophobic interaction supplemented by the electrophoretic force are believed to drive the spontaneous incorporation process 27. It is also well-established that both phage coat protein and their interaction with the lipid environment of the bacterial (E.coli) membrane contribute to the efficiency of the membrane insertion and topology of the phage coat membrane proteins27. It is notable that topology of phage coat protein within the bacterial membrane always follows the “positive inside rule” with their N-termini on the periplasma side and C-termini on the cytoplasm side32. Basic amino acid residues within the C-termini of phage coat protein dictate the Nout/Cin terminal orientation33. In addition to the phage coat protein, anionic lipids are also involved in the membrane translocation and determination of the Nout/Cin orientation. Factors, such as the physicochemical properties of lipid used in the liposome construct, density of phage fusion protein and preparation protocol, play important roles ensuring that only the binding peptide sequence is involved in the targeting specificity of the phage fusion protein-modified liposomes19.
Using the phage display technique for the discovery of disease recognition molecules avoids the necessity for the knowledge regarding cell surface receptors to which the targeting molecules bind. This merit enables the fast screening of target ligands and promotes the development of targeted nanomedicines. Still, the further elucidation of how MCF-7 specific phage fusion protein mediates cellular targeting could provide insight in understanding of the landscape-based phage-liposome formulation. This is a part of our ongoing efforts towards the identification of the exact targeting site.
Overall, the landscape phage fusion protein approach bypasses the need for chemical modification and makes liposomes cancer cell-targeted. Landscape phage fusion proteins joined with cancer cell-specific peptides obtained using the phage display technology self-assemble with liposomes and retain their targetability. The binding peptide sequence provides targeting of phage-liposomes to specific cancer cells, while the phage pVIII coat protein contributes the anchoring of the target peptide on the liposomal surface without loss of target specificity. The landscape phage fusion protein approach is expected to expand for the development of combinatorial targeted nanomedicines against a wide range of cancers and other pathologies using a one-fit-all scheme.
Acknowledgments
This work was supported by the NIH grant# R01 CA125063-01 and the Animal Health and Disease Research grant 2006–9, College of Veterinary Medicine Auburn University to Valery A. Petrenko and by the NIH grant #1U54CA151881 to Vladimir P. Torchilin
References
- 1.Allen TM. Ligand-Targeted Therapeutics in Anticancer Therapy. Nat Rev Cancer. 2002;2(10):750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 2.Lammers T, Hennink WE, Storm G. Tumour-Targeted Nanomedicines: Principles and Practice. Br J Cancer. 2008;99(3):392–397. doi: 10.1038/sj.bjc.6604483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torchilin VP. Drug Targeting. Eur J Pharm Sci. 2000;11 (Suppl 2):S81–91. doi: 10.1016/s0928-0987(00)00166-4. [DOI] [PubMed] [Google Scholar]
- 4.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 5.Torchilin V. Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv Drug Deliv Rev. 2010 doi: 10.1016/j.addr.2010.03.011. In press. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 7.Noble CO, Kirpotin DB, Hayes ME, Mamot C, Hong K, Park JW, Benz CC, Marks JD, Drummond DC. Development of Ligand-Targeted Liposomes for Cancer Therapy. Expert Opin Ther Targets. 2004;8(4):335–353. doi: 10.1517/14728222.8.4.335. [DOI] [PubMed] [Google Scholar]
- 8.Torchilin VP. Passive and Active Drug Targeting: Drug Delivery to Tumors as an Example. Handb Exp Pharmacol. 2010;197:3–53. doi: 10.1007/978-3-642-00477-3_1. [DOI] [PubMed] [Google Scholar]
- 9.Khandare JJ, Minko T. Antibodies and Peptides in Cancer Therapy. Crit Rev Ther Drug Carrier Syst. 2006;23(5):401–435. doi: 10.1615/critrevtherdrugcarriersyst.v23.i5.20. [DOI] [PubMed] [Google Scholar]
- 10.Aina OH, Liu R, Sutcliffe JL, Marik J, Pan CX, Lam KS. From Combinatorial Chemistry to Cancer-Targeting Peptides. Mol Pharm. 2007;4(5):631–651. doi: 10.1021/mp700073y. [DOI] [PubMed] [Google Scholar]
- 11.Petrenko V. Evolution of Phage Display: From Bioactive Peptides to Bioselective Nanomaterials. Expert Opin Drug Deliv. 2008;5(8):825–836. doi: 10.1517/17425247.5.8.825. [DOI] [PubMed] [Google Scholar]
- 12.Mori T. Cancer-Specific Ligands Identified from Screening of Peptide-Display Libraries. Curr Pharm Des. 2004;10(19):2335–2343. doi: 10.2174/1381612043383944. [DOI] [PubMed] [Google Scholar]
- 13.Krumpe LR, Mori T. The Use of Phage-Displayed Peptide Libraries to Develop Tumor-Targeting Drugs. Int J Pept Res Ther. 2006;12(1):79–91. doi: 10.1007/s10989-005-9002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown KC. Peptidic Tumor Targeting Agents: The Road from Phage Display Peptide Selections to Clinical Applications. Curr Pharm Des. 2010;16(9):1040–1054. doi: 10.2174/138161210790963788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holig P, Bach M, Volkel T, Nahde T, Hoffmann S, Muller R, Kontermann RE. Novel RGD Lipopeptides for the Targeting of Liposomes to Integrin-Expressing Endothelial and Melanoma Cells. Protein Eng Des Sel. 2004;17(5):433–441. doi: 10.1093/protein/gzh055. [DOI] [PubMed] [Google Scholar]
- 16.Lee TY, Lin CT, Kuo SY, Chang DK, Wu HC. Peptide-Mediated Targeting to Tumor Blood Vessels of Lung Cancer for Drug Delivery. Cancer Res. 2007;67(22):10958–10965. doi: 10.1158/0008-5472.CAN-07-2233. [DOI] [PubMed] [Google Scholar]
- 17.Wang T, D’Souza GG, Bedi D, Fagbohun OA, Potturi LP, Papahadjopoulos-Sternberg B, Petrenko VA, Torchilin VP. Enhanced Binding and Killing of Target Tumor Cells by Drug-Loaded Liposomes Modified with Tumor-Specific Phage Fusion Coat Protein. Nanomedicine (Lond) 2010;5(4):563–574. doi: 10.2217/nnm.10.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jayanna PK, Bedi D, Gillespie JW, DeInnocentes P, Wang T, Torchilin VP, Bird RC, Petrenko VA. Landscape Phage Fusion Protein-Mediated Targeting of Nanomedicines Enhances Their Prostate Tumor Cell Association and Cytotoxic Efficiency. Nanomedicine. 2010;6(4):538–546. doi: 10.1016/j.nano.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang T, Kulkarni N, Bedi D, D’Souza GG, Papahadjopoulos-Sternberg B, Petrenko VA, Torchilin VP. In Vitro Optimization of Liposomal Nanocarriers Prepared from Breast Tumor Cell Specific Phage Fusion Protein. Journal of Drug Targeting. 2011 doi: 10.3109/1061186X.2010.550920. In press. [DOI] [PubMed] [Google Scholar]
- 20.Hemminga MA, Vos WL, Nazarov PV, Koehorst RB, Wolfs CJ, Spruijt RB, Stopar D. Viruses: Incredible Nanomachines. New Advances with Filamentous Phages. Eur Biophys J. 2010;39(4):541–550. doi: 10.1007/s00249-009-0523-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn A. Major Coat Proteins of Bacteriophage Pf3 and M13 as Model Systems for Sec-Independent Protein Transport. FEMS Microbiol Rev. 1995;17(1–2):185–190. doi: 10.1111/j.1574-6976.1995.tb00201.x. [DOI] [PubMed] [Google Scholar]
- 22.Soekarjo M, Eisenhawer M, Kuhn A, Vogel H. Thermodynamics of the Membrane Insertion Process of the M13 Procoat Protein, a Lipid Bilayer Traversing Protein Containing a Leader Sequence. Biochemistry. 1996;35(4):1232–1241. doi: 10.1021/bi951087h. [DOI] [PubMed] [Google Scholar]
- 23.Geller BL, Wickner W. M13 Procoat Inserts into Liposomes in the Absence of Other Membrane Proteins. J Biol Chem. 1985;260(24):13281–13285. [PubMed] [Google Scholar]
- 24.Wang T, Petrenko VA, Torchilin VP. Paclitaxel-Loaded Polymeric Micelles Modified with MCF-7 Cell-Specific Phage Protein: Enhanced Binding to Target Cancer Cells and Increased Cytotoxicity. Mol Pharm. 2010;7(4):1007–1014. doi: 10.1021/mp1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sergeeva A, Kolonin MG, Molldrem JJ, Pasqualini R, Arap W. Display Technologies: Application for the Discovery of Drug and Gene Delivery Agents. Adv Drug Deliv Rev. 2006;58(15):1622–1654. doi: 10.1016/j.addr.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jayanna PK, Torchilin VP, Petrenko VA. Liposomes Targeted by Fusion Phage Proteins. Nanomedicine. 2009;5(1):83–89. doi: 10.1016/j.nano.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dalen A, de Kruijff B. The Role of Lipids in Membrane Insertion and Translocation of Bacterial Proteins. Biochim Biophys Acta. 2004;1694(1–3):97–109. doi: 10.1016/j.bbamcr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 28.Stopar D, Spruijt RB, Wolfs CJ, Hemminga MA. Protein-Lipid Interactions of Bacteriophage M13 Major Coat Protein. Biochim Biophys Acta. 2003;1611(1–2):5–15. doi: 10.1016/s0005-2736(03)00047-6. [DOI] [PubMed] [Google Scholar]
- 29.Wang T, Yang S, Petrenko VA, Torchilin VP. Cytoplasmic Delivery of Liposomes into MCF-7 Breast Cancer Cells Mediated by Cell-Specific Phage Fusion Coat Protein. Mol Pharm. 2010;7(4):1149–1158. doi: 10.1021/mp1000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sapra P, Tyagi P, Allen TM. Ligand-Targeted Liposomes for Cancer Treatment. Curr Drug Deliv. 2005;2(4):369–381. doi: 10.2174/156720105774370159. [DOI] [PubMed] [Google Scholar]
- 31.Li ZJ, Cho CH. Development of Peptides as Potential Drugs for Cancer Therapy. Curr Pharm Des. 2010;16(10):1180–1189. doi: 10.2174/138161210790945913. [DOI] [PubMed] [Google Scholar]
- 32.von Heijne G, Gavel Y. Topogenic Signals in Integral Membrane Proteins. Eur J Biochem. 1988;174(4):671–678. doi: 10.1111/j.1432-1033.1988.tb14150.x. [DOI] [PubMed] [Google Scholar]
- 33.Andersson H, Bakker E, von Heijne G. Different Positively Charged Amino Acids Have Similar Effects on the Topology of a Polytopic Transmembrane Protein in Escherichia Coli. J Biol Chem. 1992;267(3):1491–1495. [PubMed] [Google Scholar]