

Abstract
The prion protein displays a unique structural ambiguity in that it can adopt multiple stable conformations under physiological conditions. In our view, this puzzling feature resulted from a sudden environmental change in evolution when the prion, previously an integral membrane protein, got expelled into the extracellular space. Analysis of known vertebrate prions unveils a primordial transmembrane protein encrypted in their sequence, underlying this relocalization hypothesis. Apparently, the time elapsed since this event was insufficient to create a “minimally frustrated” sequence in the new milieu, probably due to the functional constraints set by the importance of the very flexibility that was created in the relocalization. This scenario may explain why, in a structural sense, the prion protein is still en route toward becoming a foldable globular protein.
Keywords: protein folding, energy landscape, structure prediction, protein evolution
Transmissible spongiform encephalopathies, or prion diseases, such as scrapie of sheep, BSE of cattle, and Creutzfeldt–Jakob syndrome of humans, belong to a distinct class of neurodegenerative disorders with a unique pathomechanism (1–3). According to the generally accepted “protein only” hypothesis, an aberrantly folded form of a host-encoded protein [prion protein (PrP)] is responsible for the genesis and transmission of disease. The existence of PrP in at least two extremely different structural states—the benign, cellular form of unknown function (PrPC) and the pathogenic scrapie state (PrPSc)—is incompatible with the current landscape theory of protein folding, which states that globular proteins possess a rather smooth, funnel-like conformational energy landscape that ensures their efficient folding into a unique, stable native state (4, 5). Multiple stable conformations of proteins in other cases are only seen in amyloidoses (6), a class of diseases in which the aberrant conformations of “ordinary” globular proteins are stabilized in fibrillar aggregates (amyloids). In fact, initially it appeared that prion diseases are accompanied by the formation of amyloid deposits. In preparations enriched for PrPSc, rod-shaped particles that fulfilled the morphological and tinctorial criteria for amyloid were seen by electron microscopy (7) and were subsequently seen in some animal and human prion diseases (8). Later, however, it was determined that other forms of prion disease develop without amyloid formation (cf. ref. 9) and infectivity of scrapie preparations increases significantly on treatment that disperses prion rods (10). Radiation inactivation studies showed infectious prion size to be about 55 kDa [i.e., dimer of PrP (11)] and prion rods were found to be an artifact of PrPSc purification (12). Thus, there are various lines of evidence to suggest that PrP is fundamentally different from amyloid and has the genuine structural capacity to attain distinct conformations without aggregation (cf. also refs. 3 and 9). Thus, the prion protein represents a structural conundrum, the existence of which raises serious questions.
In our opinion, the answers reside in the evolutionary history of the prion protein. The fossil of a transmembrane (TM) protein in the sequence of known vertebrate PrPs suggests that it used to be an integral membrane protein probably expelled to the extracellular space by a mutation. Apparently, this environmental change gave rise to other stable conformations of comparable free energy but, for reasons discussed in the paper, did not create an evolutionary pressure sufficient to select against all but one of these to result in a unique structure.
PrP Looks to Be of Membraneous Origin
To demonstrate that prion sequences harbor remnants of a transmembrane protein, their sequences have been analyzed by four principally different methods developed for predicting the localization of TM helices. The methods, each of which has a prediction accuracy above 90%, were: TOPPRED (13), DAS (14, 15), PHDhtm (16), and HMMTOP (17). Predictions with these algorithms were made via the Internet by using the corresponding servers.§ For analysis, prion sequences with sequence similarity to human PrP were collected by a blast search (18) using the nonredundant database at the National Institutes of Health. The resulting prion proteins represent a total of about 50, of which most show a high similarity to the human sequence; six of them, with identity less than 90% (cf. Fig. 1), were used in the prediction studies. The number of predicted transmembrane segments can be seen in Table 1. Evidently, prion sequences show a high propensity for membrane localization: the methods mostly predict three transmembrane segments for full-length sequences, with one or two such segments predicted only for those proteins for which processing (i.e., N- and C-terminal truncation) is considered in the database. For a comparison, the number of transmembrane segments have also been predicted for known globular and membrane proteins by using the same algorithms (Table 1). A representative set of 523 globular proteins with known atomic coordinates were selected from the PDB database (19, 20) and 158 transmembrane proteins with established membrane topology were collected as described (17). To make sure that the predictions are unbiased, each major structural class of globular proteins is duly represented as follows: 77 all α, 91 α + β, 141 αβ, 98 all β, and 116 other.
Figure 1.

The localization of TM helices in globular, transmembrane, and prion proteins. For comparison, 6–6 homologs of three TM and three globular proteins that have a similar relative similarity dendogram to that of the selected six prion proteins were selected (proteins are given by SWISS-PROT accession no. or ID). For each protein, TM helices are predicted by four prediction methods and are shown by color coding as follows: TOPPRED (green), DAS (red), PHDhtm (blue), and HMMTOP (yellow). A TM region (defined as in the legend of Table 2) is boxed if predicted by three (gray) or four (black) methods. Please note that there are only 11 globular proteins of 523 for which at least three methods predict a TM region (cf. Table 2); only three could be found with a relative similarity dendogram as shown.
Table 1.
The number of experimentally observed versus predicted transmembrane helices in globular, transmembrane, and prion proteins
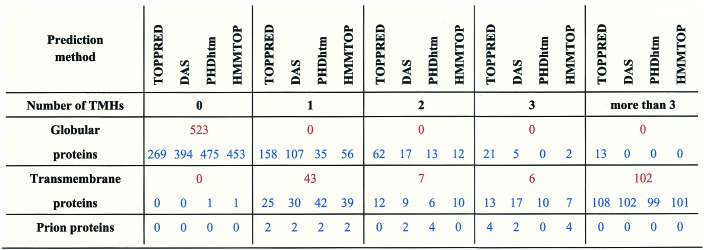
Predictions by four different algorithms were made via the Internet by using the corresponding servers for 523 globular proteins, 158 membrane proteins with known transmembrane topology, and six prion proteins. The methods used and proteins selected are given in the text. For each structural class, the number of proteins with a given number of transmembrane helices (TMHs) observed (red) or predicted (blue) is shown.
The comparison of data in Table 1 indicates that the prion protein differs from globular proteins. For the globular proteins the methods overwhelmingly predict zero or at the most one transmembrane segment; prediction of a TM helix for these latter is rarely common for the different methods. For the membrane proteins, on the other hand, there is only a single case for which no transmembrane region is predicted at all; for 156 of 158 proteins the number and localization of such regions are rather accurately assigned. In light of these, the finding that none of the six (and consequently, none of the 50) prion proteins is predicted to lack transmembrane helices entirely, seems to be of high predictive value. Three of the four methods predict three such helices for all full-length sequences, invariably in the regions 2–22, 114–134, and 232–252 (human PrP numbering). Although the prion sequences are related, this analysis places the prion protein among the transmembrane proteins and not the globular ones. There are three lines of considerations that support the significance of this finding.
First, the four methods are based on different principles that makes their agreement highly significant: TOPPRED uses a hydropathy plot analysis improved by the “positive-inside” rule; DAS relies on that the amino acid composition of transmembrane segments is more conservative than the sequence itself; the PHDhtm method is based on an artificial neural network that is taught on a TM-protein database, whereas HMMTOP utilizes a hidden Markov model to find the topology of maximum likelihood for a given protein. Of these four, TOPPRED and HMMTOP rely on a priori principles and involve no statistical parameterization; thus, their prediction is not biased by the choice of an initial set of proteins. On the other hand, DAS and PHDhtm incorporate direct or indirect comparison of the query sequence with known TM proteins; therefore, their results are of a statistical nature and may be influenced by the curious behavior of PrP that may make it defy rules that apply to “normal” TM (and globular) proteins. Given their differences, the exceptional match of predictions by all four methods over TM regions in PrP gives our conclusion a very strong credit.
Second, a simple statistical analysis can be performed to calculate the probability that a given protein is correctly classified with respect to its membrane localization. As seen (Table 2), the chance of correct classification is already rather high if prediction of a protein to contain at least one TM region (for definition, see the legend to Table 2) is made by only one method. This probability, of course, improves tremendously on requiring agreement of more methods: by combining all four, the chance that any given protein is correctly classified to belong to either the globular or the TM class is very high (99.6%); this number applies to our conclusion that PrP is a transmembrane protein with three probable membrane-spanning regions.
Table 2.
The percentage of proteins correctly predicted to fall into the globular or transmembrane class
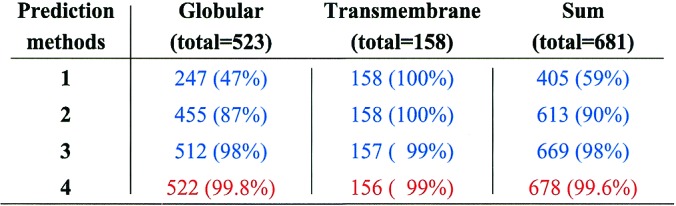
523 globular and 158 TM proteins have been selected as given in the text. A TM region is defined as that part of the sequence for which a 15-residue segment is invariably predicted as a TM helix by the given number (1 to 4) of prediction methods. For each case the number and percentage of proteins predicted to have no (for globular proteins) or to have (for transmembrane proteins) TM region(s) is calculated. The 11 globular proteins that have TM region(s) by at least three methods are: 1bia, 1ciy, 1din, 1frp, 1lci, 1lcp, 1oxa, 1phg, 1ytb, 2adm, and 2pia; the only such protein by four methods is 1din. The sum of total numbers yields the percent probability of classifying any randomly selected protein correctly in terms of the class it belongs to.
Third, the four methods have all been developed to predict TM regions in integral membrane proteins and have never been optimized to distinguish between globular and TM proteins. Our studies, however, demonstrate that this distinction is nevertheless feasible with the methods. Fig. 1 shows that for each of the homologs of selected TM proteins they almost invariably predict TM helices where, and only where, they actually are. In contrast, in the rare cases of known globular proteins for which TM regions are predicted at all, a quite varied picture emerges: most often a given segment is predicted by one method, but not by the others; in other cases it is predicted for one or two homologs, but not for the others. Thus, the four prediction methods together have a significant potential in classifying proteins in terms of their membrane localization. A single look at the perfect agreement of the localization of TM helices in PrP homologs makes it clear that this protein fits in with TM proteins and not with globular ones. As PrP today is clearly an extracellular protein under normal conditions (21), its relocalization from the membrane at some point of evolution seems evident.
Other Evidence for the Membraneous Origin of PrP
As seen, the case for PrP as a primordial integral membrane protein is rather convincing and probably explains its structural ambiguity. Other evidence also argues that this view is correct.
The strongest point is that the biogenesis of prions seems to recall memories of its membraneous past: even today, PrP sometimes confounds its cellular destination and behaves like a membrane protein. Studies of its translocation at the endoplasmic reticulum (ER) membrane have revealed that both in a cell-free translation system and in vivo it is synthesized in more than one topologic form (22). The secretory form is fully translocated into the ER lumen and gets transported to the cell surface where it is located under normal physiological conditions. The remainder, however, gets stuck with a region between residues 113 and 135 spanning the membrane [termed TM1 (23)], which is exactly the position of the second transmembrane helix we predict for the ancestral prion protein. The relative amounts of the different forms is very sensitive to mutations (22), or to the translation system applied (24), which attests to the propensity of PrP toward membrane localization. Thus, there is ample evidence that PrP has an intimate and intricate relationship with the membrane even today. Conservation of TM topology as predicted (Fig. 1) shows that this might be an ancient trait of PrP that probably existed before the separation of mammals, birds, and reptiles. Furthermore, alignment of all PrP sequences reveals that the middle TM region (TM1) is absolutely conservative (9): a continuous stretch of 16 amino acids (113–128 in human PrP) shows no variation at all. The last common ancestor of the species studied had to have a PrP with exactly the same sequence here, which is of very strong TM character: the TM origin of PrP looks certain.
A further point in support of an evolutionary relocalization of PrP from the membrane comes from studies aimed at understanding the TM topology of its membraneous form. Originally it has been noted that one form of PrP is a TM protein that spans the lipid bilayer twice with both its N and C terminus localized in the ER lumen (24, 25). Such a topology requires PrP to span the membrane not only at TM1 (113) but also at TM2, the amphipathic helix downstream (157) (23). This topology is consistent with the luminal attachment of the C-terminal glycosyl-phosphatidylinositol (GPI) anchor, but is very difficult to reconcile with the “positive-inside” rule, which states that the primary determinant of the orientation of a TM helix is the charge difference between its two immediate flanking regions, with the more positive side having a significant cytoplasmic preference (13, 26). The high value provides a very strong preference to the NcytoCexo (N terminus in the cytoplasm) orientation of TM1 (cf. also Fig. 2 for the most probable topology), in which the highly positive N-terminal flanking region of TM1 [stop transfer effector (STE)] is located in the cytoplasm; furthermore, the membrane insertion of TM2 is not too likely as all of the prediction methods have overlooked it. More recently, in fact, it was found that PrP actually exists in two different TM forms, each spanning the membrane at TM1 but having an opposite orientation (22). All these observations—along with the secretory form (27) and the noted sensitivity of transmembrane forms to the translation system (23, 24) and point mutations within STE and TM1 (22)—are clear signs of the “topological frustration” of PrP (28) (i.e., that its different parts dictate conflicting topologies). This topological “conundrum” has also been noted in the literature (21, 29). Such a situation, not surprisingly, is very rare for natural proteins (30), but can be brought about by planned mutations in a multispanning TM protein (28). It is logical to assume that a mutation of similar consequence occurred somewhere along PrP evolution and frustrated the TM topology of PrP. It is to be added that frustration of topology is physically closely related to frustration of three-dimensional structure; this universal ambiguity of PrP structure definitely points to a profound and sudden evolutionary change.
Figure 2.
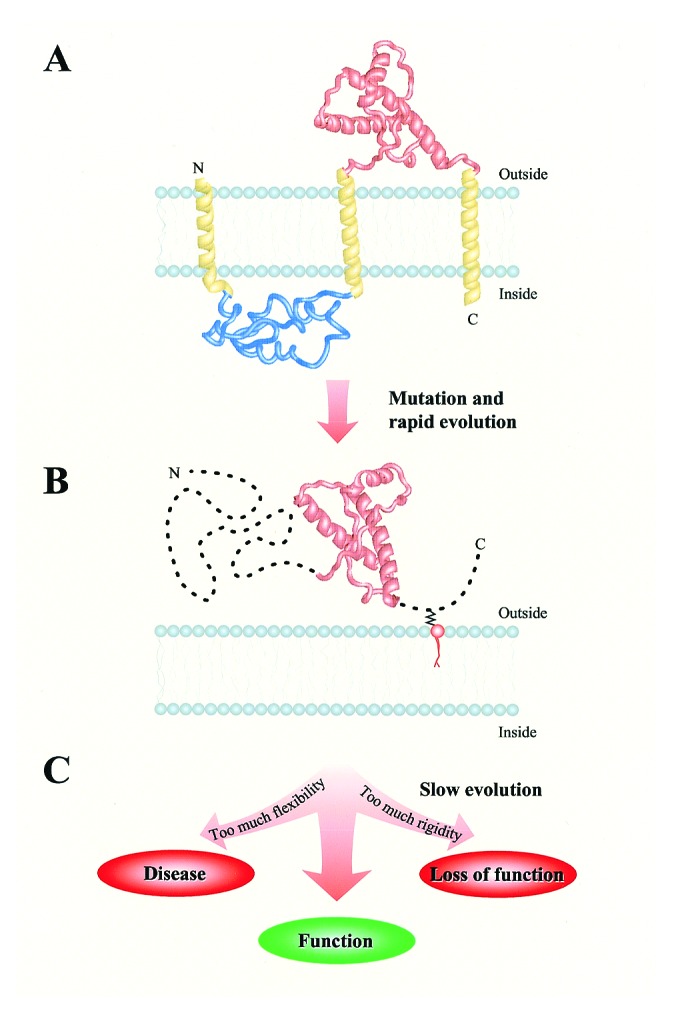
An outline of the possible evolutionary history of the prion protein. It is suggested that three major stages be considered. (A) The prion protein used to be an integral membrane protein with three transmembrane helices. Today's globular domain (NMR structure 1B10 from the PDB database, shown in red) resided entirely in the extracellular compartment. (B) A mutation caused PrP to relocalize from the membrane to the extracellular space. A frustrated sequence with a floppy structure was created. A short, rapid evolutionary drift toward a minimally frustrated sequence and globular structure commenced. Soon after, however, the progress was halted by establishing a function that relied on the very structural flexibility that resulted from the relocalization. (C) A slow sequential progress followed, as required by the need to balance between too much flexibility (disease) and too much rigidity (loss of function) of the structure.
The Possible Evolutionary History of PrP
The foregoing sections have dealt with the evidence for a sudden environmental change during the evolution of PrP, although the more speculative issue of how it all occurred is also worth considering. Two questions in relation to the scenario put forward will be considered in depth: (i) Which part of PrP had undergone the mutation that caused the change and (ii) why had this mutation been accepted and fixed if it had such an apparently “frustrating” consequence on PrP structure and function?
For the exact site of the mutation there are two obvious candidates, but others cannot be excluded either. There is a good deal of evidence that cellular localization of PrP is very sensitive to changes in the STE-TM1 region. In addition, this region seems to be responsible for insertion of PrP in both putative TM orientations and also for the appearance of the secretory form (31). As TM1 is absolutely conservative, the STE region (or the translocation machinery that interacts with it) is the primary candidate for the mutation that caused relocalization. A further place to look is around or within the two terminal TM helices (2–22 and 232–252 in huPrP). These sequences seem to signal initiation of translocation and glycolipidation, respectively, and are usually cleaved off during maturation (3); this may not have always been so. A mutation, for example, that created a cleavage site for signal peptidase at the N-terminal TM helix could have a consequence we deal with today: the a loss of a permanent membrane anchor may have upset the entire process of PrP biogenesis. Underlying this—and in fact the entire relocalization scheme—is that these TM regions fall exactly where amino acid variability of PrP peaks (32). If these parts of the protein had been instrumental in stabilizing the structure in the membrane, they were inevitably subject to most of the sequential changes on relief of this constraint and the need to adapt to the new environment.
Irrespective of where exactly the mutation occurred, it is probably even more intriguing why it had been fixed despite the seemingly serious consequences. The simplest answer is to refer to observations that the consequences of the mutation have not been so serious as they appear at first glance. The outcome of relocalization and the loss of PrP function may in fact have been very marginal as the protein could have no vital function. Even today, knockout mice show no (33, 34) or very mild (35–37) functional deficit on losing PrP; this may apply equally well to the evolutionary relocalization. No evolutionary pressure arose, thus, to purge PrP so affected. Furthermore, the frustration of three-dimensional structure and the chance conversion to a lethal pathogenic state, PrPsc could also have very little evolutionary consequence. Because prion diseases usually have a late onset, their sporadic appearance is of low frequency, and their transmission by infection is rather unlikely (32), the mutation that caused relocalization could be passed on without being subject to much negative selection. Thus, mutation of PrP could be fixed by chance.
Several points can be raised, however, that indicate positive selection also contributed to fixation of the mutation. One interesting consideration comes from the current view of different TM forms of PrP in pathogenesis. It is thought that the prion protein under normal conditions is fully translocated into the ER lumen and gets transported to the cell surface from where it can be released by cleaving the glycosyl-phosphatidylinositol (GPI) anchor (21). Membrane-integrated forms are only seen in cell-free translation systems (23, 24), in transgenic mice expressing mutant or chimeric constructs (22), and at least one form of an inherited human prion disease, Gerstmann–Sträussler–Sheinker (GSS) disease (22). As noted, PrP can be synthesized in two principal TM forms in which TM1 is inserted into the membrane in opposite orientations. Interestingly, the expression of one form (CtmPrP, for which TM1 is inserted in NcytoCexo orientation) in transgenic mice correlates well with neurodegenerative changes resembling genetic prion diseases (22); the TM form of opposite topology appears to have no pathogenic effect. The mutation A117V in GSS also causes the accumulation of this pathogenic TM form, thus it may be the cause of the neuropathological changes. Intriguingly, as seen in Fig. 2, this potentially pathogenic orientation is predicted for the primordial PrP. If correlation of prion disease with the prevalence of this form is correct, ancient species with this form may actually have been more prone to prion disease than today's species following the evolutionary relocalization event—the chance of PrPC to convert to PrPSc is probably much less than it used to be. No doubt, the mutation that made PrP leave the membrane could have an immediate evolutionary advantage and was fixed quickly because of positive selection.
This contention might be further supported by structural studies implying that flexibility of PrP structure may have evolutionary advantages per se: as noted, TM1 is absolutely conservative (9); still it appears to be the most flexible part of the molecule on many accounts. The flexibility of this region is evident from the different accessibility of epitopes here to specific antibodies (38): this is the likely site of α-helix → β-sheet transition implicated in PrPSc formation (39, 40); NMR studies show considerable disorder around this region (41–43) and different prion strains exhibit different cleavage sites in this region (4). TM1's exceptional flexibility is also supported by the existence of miniprions, PrP deletion mutants corresponding to this central segment with a capacity to PrPSc formation and infection (44, 45). This remarkable structural plasticity and the strong sequence conservation (9) are hard to reconcile, especially because this region abounds with alanines and glycines (i.e., with amino acids of no chemical functionality). The only way around this dilemma is that flexibility and/or the ability to undergo conformational change is intimate to the function of PrP (9); such a unique behavior may explain selection against mutations affecting this region (i.e., amino acid substitutions toward a more definite globular function).
In fact, in a recent paper (46) we have argued that the plastic nature of its structure and the self-sustaining autocatalytic propagation of its alternative conformations may form the basis of the normal cellular function of PrP: this protein may constitute a molecular switch that determines phenotype and operates in diverse functions possibly including even memory processes. The feasibility of this gain-in-function scenario is underlined by the operation of prion switches in lower eukaryotes (47, 48). In yeast it has been found that two non-Mendelian heritable genetic elements can be traced back to the genuine prion-like propagation of altered conformations of chromosomally encoded cytoplasmic proteins. These proteins are regulatory effectors of function, one of which (Sup35) allows cells to exploit preexisting genetic variation to adapt to a changing environment (49). The structural and functional analogy of these proteins and the mammalian prions is underlined by physical studies that show that a particular region within them can switch between two alternative states and acquire stable prion conformations (50, 51); this region is noted for its sequential conservation. The vertebrate prion protein may have stopped halfway en route to becoming a globular protein for the same reason.
There is one more point that may help conceive how relocalization took place. Possibly, as the prion protein got expelled from the membrane, a limited initial sequential drift occurred, mostly affecting the terminal regions. These changes, however, did not have to fully reshape the protein as the environmental change was not, at least from a functional point of view, that severe. As the protein moved from the membrane to its extracellular side only, the former extracellular domain that corresponds to today's structured part between residues 135–231 (cf. Fig. 1 and refs. 41 and 43) could carry on virtually unimpeded with the same structure and function it possessed. Such function(s), possibly in cellular communication, recognition, and signal transduction (52, 53), could survive relocalization by the lipid anchor tethering the protein close to its previous localization and orientation. As a matter of fact, flexibility and adaptability of the overall structure may have endowed this domain with such capabilities that extended way beyond its original functional limits. The extracellular functional domain, therefore, could serve as a seed for PrP in its pursuit of structural and functional survival, and eventual revival.
Conclusions
In a structural sense, the prion protein links foldable proteins that have a single low energy conformation and nonfoldable polypeptides that possess a practically unlimited number of three-dimensional structures of comparable energies. In our view, the emergence of this structural peculiarity can be explained by assuming that a radical change in the cellular environment of this protein occurred some time during evolution. Fig. 2 recapitulates our suggestion on the possible course and aftermath of this evolutionary event.
Acknowledgments
This work was supported by Grants T 30566, T 22069, T 29059, and T 32360 from the Hungarian Scientific Research Fund (OTKA) and AKP 98–45 3,3 from the Hungarian Academy of Sciences. I.S. and M.C. acknowledge the support of the Hungarian–British intergovernment science and technology fund. G.E.T. is supported by a Magyary Zoltán Postdoctoral Fellowship.
Abbreviations
- PrP
prion protein
- PrPC
cellular form of prion protein
- PrPSc
scrapie form of prion protein
- ER
endoplasmic reticulum
- STE
stop transfer effector
- TM
transmembrane
Footnotes
URL localization of prediction methods used in predicting transmembrane topology of proteins: TOPPRED, http://www.sbc.su.se/∼erikw/toppred2; DAS, http://www.sbc.su.se/∼miklos/DAS; PHDhtm, http://www.embl-heidelberg.de/predictprotein/; HMMTOP, http://www.enzim.hu/hmmtop.
References
- 1.Weissmann C. FEBS Lett. 1996;389:3–11. doi: 10.1016/0014-5793(96)00610-2. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S B. Science. 1997;278:245–251. doi: 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner S B. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Telling G C, Parchi P, DeArmond S J, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner S B. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 5.Nymeyer H, Garcia A E, Onuchic J N. Proc Natl Acad Sci USA. 1998;95:5921–5928. doi: 10.1073/pnas.95.11.5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobson C M. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 7.Prusiner S B, McKinley M P, Bowman K A, Bolton D C, Bendheim P E, Groth D F, Glenner G G. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 8.Kitamoto T, Tateishi J, Tashima T, Takeshita I, Barry R A, DeArmond S J, Prusiner S B. Ann Neurol. 1986;20:204–208. doi: 10.1002/ana.410200205. [DOI] [PubMed] [Google Scholar]
- 9.Bamborough P, Wille H, Telling G C, Yehiely F, Prusiner S B, Cohen F E. Cold Spring Harbor Symp Quant Biol. 1996;61:495–509. [PubMed] [Google Scholar]
- 10.Gabizon R, McKinley M P, Prusiner S B. Proc Natl Acad Sci USA. 1987;84:4017–4021. doi: 10.1073/pnas.84.12.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellinger-Kawahara C G, Kempner E, Groth D, Gabizon R, Prusiner S B. Virology. 1988;164:537–541. doi: 10.1016/0042-6822(88)90569-7. [DOI] [PubMed] [Google Scholar]
- 12.McKinley M P, Meyer R K, Kenaga L, Rahbar F, Cotter R, Serban A, Prusiner S B. J Virol. 1991;65:1340–1351. doi: 10.1128/jvi.65.3.1340-1351.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Heijne G. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 14.Cserző M, Bernassau J M, Simon I, Maigret B. J Mol Biol. 1994;243:388–396. doi: 10.1006/jmbi.1994.1666. [DOI] [PubMed] [Google Scholar]
- 15.Cserző M, Wallin E, Simon I, von Heijne G, Elofsson A. Protein Eng. 1997;10:673–676. doi: 10.1093/protein/10.6.673. [DOI] [PubMed] [Google Scholar]
- 16.Rost B, Fariselli P, Casadio R. Protein Sci. 1996;5:1704–1718. doi: 10.1002/pro.5560050824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tusnády G E, Simon I. J Mol Biol. 1998;283:489–506. doi: 10.1006/jmbi.1998.2107. [DOI] [PubMed] [Google Scholar]
- 18.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 19.Hobohm U, Sander C. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein F C, Koetzle T F, Williams G J, Meyer E E, Jr, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 21.Stahl N, Borchelt D R, Hsiao K, Prusiner S B. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 22.Hegde R S, Mastrianni J A, Scott M R, DeFea K A, Tremblay P, Torchia M, DeArmond S J, Prusiner S B, Lingappa V R. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 23.Lopez C D, Yost C S, Prusiner S B, Myers R M, Lingappa V R. Science. 1990;248:226–229. doi: 10.1126/science.1970195. [DOI] [PubMed] [Google Scholar]
- 24.Yost C S, Lopez C D, Prusiner S B, Myers R M, Lingappa V R. Nature (London) 1990;343:669–672. doi: 10.1038/343669a0. [DOI] [PubMed] [Google Scholar]
- 25.Hay B, Barry R A, Lieberburg I, Prusiner S B, Lingappa V R. Mol Cell Biol. 1987;7:914–920. doi: 10.1128/mcb.7.2.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartmann E, Rapoport T A, Lodish H F. Proc Natl Acad Sci USA. 1989;86:5786–5790. doi: 10.1073/pnas.86.15.5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hay B, Prusiner S B, Lingappa V R. Biochemistry. 1987;26:8110–8115. doi: 10.1021/bi00399a014. [DOI] [PubMed] [Google Scholar]
- 28.Gafvelin G, von Heijne G. Cell. 1994;77:401–412. doi: 10.1016/0092-8674(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 29.Prusiner S B. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 30.Levy D. Essays Biochem. 1996;31:49–60. [PubMed] [Google Scholar]
- 31.De Fea K A, Nakahara D H, Calayag M C, Yost C S, Mirels L F, Prusiner S B, Lingappa V R. J Biol Chem. 1994;269:16810–16820. [PubMed] [Google Scholar]
- 32.Krakauer D C, Zanotto P M, Pagel M. J Mol Evol. 1998;47:133–145. doi: 10.1007/pl00006370. [DOI] [PubMed] [Google Scholar]
- 33.Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp H P, DeArmond S J, Prusiner S B, Aguet M, Weissmann C. Nature (London) 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 34.Lledo P M, Tremblay P, DeArmond S J, Prusiner S B, Nicoll R A. Proc Natl Acad Sci USA. 1996;93:2403–2407. doi: 10.1073/pnas.93.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collinge J, Whittington M A, Sidle K C, Smith C J, Palmer M S, Clarke A R, Jefferys J G. Nature (London) 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- 36.Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, et al. Nature. 1996;380:528–531. doi: 10.1038/380528a0. [DOI] [PubMed] [Google Scholar]
- 37.Tobler I, Gaus S E, Deboer T, Achermann P, Fischer M, Rulicke T, Moser M, Oesch B, McBride P A, Manson J C. Nature (London) 1996;380:639–642. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 38.Peretz D, Williamson R A, Matsunaga Y, Serban H, Pinilla C, Bastidas R B, Rozenshteyn R, James T L, Houghten R A, Cohen F E, et al. J Mol Biol. 1997;273:614–622. doi: 10.1006/jmbi.1997.1328. [DOI] [PubMed] [Google Scholar]
- 39.Huang Z, Prusiner S B, Cohen F E. Fold Des. 1996;1:13–19. doi: 10.1016/S1359-0278(96)00007-7. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Stockel J, Mehlhorn I, Groth D, Baldwin M A, Prusiner S B, James T L, Cohen F E. Biochemistry. 1997;36:3543–3553. doi: 10.1021/bi961965r. [DOI] [PubMed] [Google Scholar]
- 41.Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 42.James T L, Liu H, Ulyanov N B, Farr-Jones S, Zhang H, Donne D G, Kaneko K, Groth D, Mehlhorn I, Prusiner S B, et al. Proc Natl Acad Sci USA. 1997;94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez Garcia F, Zahn R, Riek R, Wuthrich K. Proc Natl Acad Sci USA. 2000;97:8334–8339. doi: 10.1073/pnas.97.15.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muramoto T, Scott M, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1996;93:15457–15462. doi: 10.1073/pnas.93.26.15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muramoto T, DeArmond S J, Scott M, Telling G C, Cohen F E, Prusiner S B. Nat Med. 1997;3:750–755. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]
- 46.Tompa P, Friedrich P. Neuroscience. 1998;86:1037–1043. doi: 10.1016/s0306-4522(98)00148-1. [DOI] [PubMed] [Google Scholar]
- 47.Wickner R B. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 48.Wickner R B, Edskes H K, Maddelein M L, Taylor K L, Moriyama H. J Biol Chem. 1999;274:555–558. doi: 10.1074/jbc.274.2.555. [DOI] [PubMed] [Google Scholar]
- 49.True H L, Lindquist S L. Nature (London) 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- 50.Paushkin S V, Kushnirov V V, Smirnov V N, Ter-Avanesyan M D. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J J, Lindquist S. Nature (London) 1999;400:573–576. doi: 10.1038/23048. [DOI] [PubMed] [Google Scholar]
- 52.Westaway D, Carlson G A, Prusiner S B. Trends Neurosci. 1989;12:221–227. doi: 10.1016/0166-2236(89)90126-4. [DOI] [PubMed] [Google Scholar]
- 53.Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche J L, Lehmann S, Launay J M, Kellermann O. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]