

Abstract
Obesity, diabetes, and metabolic syndromes are increasingly recognized as health concerns worldwide. Overnutrition and insulin resistance are the major causes of diabetic hyperglycemia and hyperlipidemia in humans. Studies in the past decade provide evidence that bile acids are not just biological detergents facilitating gut nutrient absorption, but also important metabolic regulators of glucose and lipid homeostasis. Pharmacological alteration of bile acid metabolism or bile acid signaling pathways such as using bile acid receptor agonists or bile acid binding resins may be a promising therapeutic strategy for the treatment of obesity and diabetes. On the other hand, bile acid signaling is complex, and the molecular mechanisms mediating the bile acid effects are still not completely understood. This paper will summarize recent advances in our understanding of bile acid signaling in regulation of glucose and lipid metabolism, and the potentials of developing novel therapeutic strategies that target bile acid metabolism for the treatment of metabolic disorders.
1. Introduction
Bile acids are produced only in the liver as the end products of cholesterol catabolism [1, 2]. In addition to the classic function of bile acids in facilitating hepatobiliary secretion of endogenous metabolites and xenobiotics and intestine absorption of lipophilic nutrients, bile acids also play equally important roles in controlling the metabolism of glucose and lipids in the enterohepatic system, and energy expenditure in peripheral tissues [3, 4]. Because of such a close association between bile acid signaling and metabolic homeostasis, targeting bile acid metabolism by using bile acid receptor agonists or bile acid-binding resins have proven to be effective in improving lipid and glucose homeostasis in obesity and diabetes [5]. Furthermore, stimulating de novo bile acid synthesis prevented, whereas, disruption of bile acid signaling caused insulin resistance and dyslipidemia in mice, indicating that impaired bile acid homeostasis may likely contribute to the pathogenesis of metabolic disorders [6–9]. This paper will summarize recent advances in our understanding of bile acid signaling regulation of glucose and lipid metabolism and the potentials of developing novel therapeutic strategies that target bile acid metabolism for the treatment of metabolic disorders.
2. Bile Acid Synthesis
Bile acids are the end product of cholesterol catabolism in the liver [1, 10–12]. In humans, the bile acid pool consists of primary bile acids cholic acid (CA), chenodeoxycholic acid (CDCA), secondary bile acids deoxycholic acid (DCA), and lithocholic acid (LCA). Primary bile acids are synthesized from cholesterol through two general pathways, the classic pathway and the alternative pathway. Secondary bile acids are derived from primary bile acids in the intestine by bacterial enzymes. Enzymes that catalyze these multistep reactions are located in the endoplasmic reticulum, mitochondria, cytosol, and peroxisomes. In humans, the classic pathway is considered as the major bile acid synthetic pathway (accounts for more than 90% of total bile acid production) and produces CA and CDCA in approximately equal amounts. Cholesterol 7α-hydroxylase (CYP7A1), a microsomal cytochrome p450 enzyme, catalyzes the first and rate-limiting step in the classic pathway [13]. The intermediate product in the classic pathway 7α-hydroxy-4-cholestene-3-one serves as the common precursor for both CA and CDCA. 7α-hydroxy-4-cholestene-3-one can be hydroxylated at C-12 position by microsomal sterol 12α-hydroxylase (CYP8B1), followed up by several reactions including mitochondrial 27-hydroxylase (CYP27A1) to cleave a 3-carbon unit and eventually converted to CA. Without 12α-hydroxylation, 7α-hydroxy-4-cholestene-3-one is converted to CDCA. Thus, CYP7A1 controls the overall rate of bile acid production, while CYP8B1 controls the CA: CDCA ratio in the bile acid pool. The alternative pathway (also known as acidic pathway), which is thought to account for less than 10% of the total bile acid synthesis in humans, mainly produces CDCA. CYP27A1 catalyzes hydroxylation of cholesterol to 27-hydroxycholesterol and then 3β-dihydroxy-5-cholestionic acid [14]. Oxysterol 7α-hydroxylase (CYP7B1) then catalyzes the hydroxylation reaction at C-7 position of these two intermediates, which are subsequently converted to CDCA by the same enzymes in the classic pathway.
3. Bile Acid Transport and Enterohepatic Circulation
Bile acids, once produced in the liver, are transported across the canalicular membrane of the hepatocytes into the bile and stored in the gallbladder. After each meal, gallbladder bile acids are released into the intestinal tract, efficiently reabsorbed in the ileum, and transported back to the liver via portal blood for reexcretion into the bile. This process is referred to as enterohepatic circulation of bile acids [10] (Figure 1). Bile acid transporters play important roles in this transport process [15]. The biliary excretion of bile acids is the major driving force of bile flow. The bile acid pool size is defined as the total amount of bile acids circulating in the enterohepatic circulation. In humans, bile acid pool consists of CA, CDCA, and DCA in an approximate 40 : 40 : 20 ratio. In mice, the majority of the CDCA is converted into muricholic acids (MCAs), which are highly soluble and less toxic.
Figure 1.
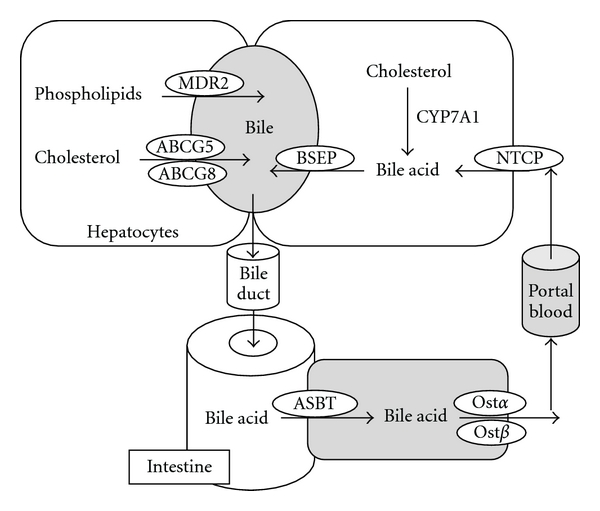
Enterohepatic circulation of the bile. Bile acids are synthesized from cholesterol in the hepatocytes. CYP7A1 regulates the rate-limiting step in the classic bile acid biosynthetic pathway. Bile acids are secreted into the gallbladder via BSEP. Phospholipids are transported via MDR2, and cholesterol is transported by the ABCG5/G8 transporters into the bile. In the gallbladder, bile acids, phospholipids, and cholesterol form mixed micelles to solubilize cholesterol and to reduce bile acid toxicity. After meal intake, gallbladder releases bile into the small intestine where bile acids facilitate the absorption of dietary lipids and vitamins. At the terminal ileum, most of the bile acids are reabsorbed by ASBT into the enterocytes, and secreted into the portal circulation via basolateral bile acid transporters Ostα/Ostβ. At the basolateral membrane of the hepatocytes, bile acids are taken up by the NTCP transporter for resecretion into the gallbladder.
Hepatocytes are polarized epithelial cells with basolateral (sinusoidal) and apical (canalicular) membrane domains. Hepatocytes take up bile acids through the basolateral membrane, which is in direct contact with the portal blood plasma, and excrete bile acid at the canalicular membrane into the bile [16]. Since the biliary bile acids concentration is about 100- to 1000-fold higher in the bile than in the hepatocytes, canalicular bile acid transport represents the rate-limiting step in bile formation. Several members of the ATP-binding cassette (ABC) transporter family are responsible for transporting bile acids and other organic compounds across the canalicular membrane against their concentration gradients. The bile salt export pump (BSEP, ABCB11), originally identified as the sister of P-glycoprotein (SPGP), is mainly responsible for bile acid transport at the canalicular membrane [17] (Figure 1). Mutations in BSEP were first identified in patients with progressive familial intrahepatic cholestasis subtype 2 (PFIC-2). The absence of functional BSEP in the hepatic canalicular membrane and less than 1% of normal biliary bile acid concentration found in these patients suggested that BSEP is the major canalicular bile acid transport system [18]. Phospholipids are excreted via the phospholipid flippase MDR2 (ABCB4) (Figure 1), and the major phospholipid in the bile is phosphatidylcholine [19, 20]. Biliary-free cholesterol secretion mediated by ABCG5/G8 transporters is an important route for hepatic cholesterol elimination. Bile acids, phospholipids, and cholesterol are three major organic solutes of the bile and once secreted, they form mixed micelles to increase cholesterol solubility and reduce their toxicity to the bile duct. Bile acids are conjugated with taurine or glycine in the peroxisomes and present as bile salts. They cannot cross the hepatocyte membrane and need active transport mechanisms for cellular uptake [21]. Two bile acid transporters, Na+-dependent taurocholate transporter (NTCP) (Figure 1) and organic anion transporters (OATPs) are responsible for basolateral bile acid transport into the hepatocytes.
In the intestine, bile salts are deconjugated, and bacterial 7α-dehydroxylase removed a hydroxy group from C-7 and converts CA to DCA and CDCA to LCA. These secondary bile acids are highly toxic. In the intestine lumen, bile acids are reabsorbed mostly at the terminal ileum. Like the hepatic basolateral uptake system, intestinal bile acid uptake is also mainly mediated by the apical sodium-dependent bile salt transporter (ASBT) (Figure 1) [22]. Once absorbed into the enterocytes, bile acids bind the intestinal bile acid binding protein (I-BABP) and are transported to the basolateral membrane for secretion [23]. Recently identified heterodimeric organic solute transporters OSTα and OSTβ appeared to be the major basolateral bile acid transport system in the intestine and many other epithelial cells (Figure 1) [24]. This is supported by studies showing that overexpression of OSTα and OSTβ in mice enhanced basolateral efflux of taurocholate, while mice lacking ostα showed marked decreases in intestinal bile acid absorption, serum bile acid concentration, and bile acid pool size.
4. Regulation of Bile Acid Synthesis
4.1. Bile Acid Feedback Regulation of Its Own Synthesis
It is clear that bile acid synthesis is mainly controlled via the transcriptional regulation of the CYP7A1 gene [11]. It is well established that the CYP7A1 gene is repressed by bile acids. This bile acid feedback repression mechanism allows the liver to efficiently increase or decrease bile acid synthesis in response to changes in bile acid levels and thus to maintain a constant bile acid pool. It is thought that bile acid feedback inhibition of CYP7A1 is mediated by the bile acid-activated nuclear receptor farnesoid X receptor (FXR) in the hepatocytes and the enterocytes (Table 1). It was first discovered that hepatic FXR induced a negative nuclear receptor small heterodimer partner (SHP), which interacts with and represses the transcriptional activator liver-related homologue-1 (LRH-1) that binds to the CYP7A1 gene promoter and thus inhibit CYP7A1 transcription [25]. However, the repression of CYP7A1 by bile acids and FXR agonist in SHP-deficient mice implies that FXR-SHP-LRH-1 cascade is not the only pathway mediating bile acid feedback inhibition of CYP7A1 and redundant pathways also exist [26, 27]. More recently, FXR was shown to induce intestine fibroblast growth factor 15 (FGF15) which may act as an endocrine hormone to repress CYP7A1 gene transcription via ERK signaling activation [28]. Direct infusion of recombinant FGF15 into mouse blood circulation or overexpression of FGF15 in mouse livers via an adenovirus expression vector caused marked repression of CYP7A1 mRNA expression. The identification of an intestine-initiated endocrine mechanism in mediating bile acid feedback regulation is consistent with the fact of intestine being the major organ for bile acid reabsorption and retention. Such finding also provides an explanation to a long observed phenomenon that intraduodenal, but not intravenous, infusion of taurocholic acid repressed CYP7A1 mRNA expression in rats [29]. In mice lacking functional Ostα/Ostβ, bile acid transport to the liver was reduced and bile acids accumulated in the intestine. Interestingly, these mice showed increased intestine FGF15 expression and reduced liver CYP7A1 mRNA and total bile acid pool [30]. Furthermore, intestine-specific FXR knockout, but not liver-specific FXR knockout, prevented GW4064 repression of liver CYP7A1 gene expression in mice [31]. These studies collectively suggest that intestine FXR plays a dominant role in mediating bile acid feedback repression of bile acid synthesis. Unfortunately, data on detection of the presence of FGF15 protein in the mouse circulation is lacking, and such evidence is needed in order to eventually establish the endocrine mechanism of feedback regulation of bile acid synthesis. Human FGF19 shares ~51% amino acid sequence identity with mouse FGF15 and is considered as the mouse FGF15 orthologue. FGF19 has been shown to repress CYP7A1 in human hepatocytes [32]. In contrast to FGF15 that is not detectable in mouse livers and circulation, FGF19 mRNA is detectable in human livers and human hepatocytes [32, 33]. FGF19 protein is present in human circulation [34]. In human hepatocytes, FGF19 is highly inducible by bile acids or FXR agonists [32]. Since adenovirus-mediated overexpression of FGF15 in mouse liver has been shown to repress CYP7A1, it is likely that bile acid accumulation in human liver may induce FXR/FGF19 pathway to repress CYP7A1 in an autocrine manner [32]. Previous studies from us and others showed that bile acids were able to activate FXR-independent cell signaling pathways to repress the CYP7A1 gene [35, 36]. We recently showed that feeding FXR knockout mice a cholic acid-containing diet still repressed CYP7A1 despite the absence of FGF15 or SHP induction [6]. These results indicate that in response to super-physiological concentrations of bile acids, redundant pathways are stimulated to repress bile acid synthesis. These pathways ensure prompt repression of de novo bile acid synthesis in response to elevated bile acid levels in the liver and/or the intestine.
Table 1.
FXR target genes and their function and lipid and glucose metabolism.
| Gene | Tissue | Regulation | Function | |
|---|---|---|---|---|
| Bile acid metabolism | CYP7A1 | liver | down | Encodes the rate-limiting enzyme in classic bile acid synthetic pathway |
| BSEP | liver | up | Rate-limiting step in canalicular bile acid transport into the gallbladder | |
| NTCP | liver | down | Basolateral bile acid uptake into the hepatocytes | |
| OSTα/β | intestine | up | Enterocyte basolateral bile acid secretion into the portal blood | |
| I-BABP | intestine | up | Intracellular bile acid transport | |
| FGF15/19 | intestine | up | Bile acid synthesis inhibition | |
| SHP | liver | up | Bile acid synthesis inhibition | |
|
| ||||
| Glucose metabolism | PEPCK | liver | up | Gluconeogenesis |
| FGF15/19 | intestine | up | Stimulates glycogen synthesis, repress gluconeogenesis | |
| Insulin | pancreas | up | Glucose metabolism | |
|
| ||||
| Cholesterol metabolism | ApoA1 | liver | down | HDL metabolism |
| LDLR | liver | down | LDL uptake | |
| ABCG5/G8 | liver | up | Biliary-free cholesterol secretion | |
| SRB1 | liver | up | Hepatic HDL uptake, biliary cholesterol secretion | |
| PCSK9 | liver | down | Induces LDL receptor degradation | |
|
| ||||
| Fatty acid metabolism | ApoC II | liver | up | LPL activator |
| ApoC III | liver | down | LPL inhibitor | |
| SREBP1 | liver | down | Lipogenesis | |
4.2. Nutrient Effects on Hepatic Bile Acid Synthesis
Despite the majority of studies being focused on the regulatory roles of bile acids in nutrient metabolism, there is also evidence that nutrition could directly regulate bile acid synthesis. In humans, CYP7A1 activity, as determined by a serum surrogate 7alpha-hydroxycholest-4-en-3-one (C4), increases during postprandial periods during the day and decreases during fasting and at night [37]. A metabolomic study also identified bile acids as the most markedly elevated metabolites in human sera after an oral glucose challenge in patients with normal glucose tolerance, but this response was blunted in patients with impaired glucose tolerance [38]. Since postprandial period is the highly active in metabolism and humans undergo fasting-to-feeding cycles a few times a day, these observations could indicate an important link between bile acid synthesis and postprandial nutrient absorption and metabolism. Nutrient-activated signaling such as those by glucose or insulin is by far the most important signaling that regulates postprandial metabolism. Using primary human hepatocytes, we have demonstrated that insulin rapidly induced while glucagon repressed CYP7A1 mRNA [39, 40]. We also reported that glucose induced CYP7A1 gene transcription via inducing histone hyperacetylation in CYP7A1 gene chromatin [41]. Although our in vitro study provide mechanistic support for human observations, studies using in vivo mouse models yielded controversial results. It has been shown that PGC-1α acts as a coactivator of HNF4α and induces CYP7A1 during fasting in mice [42]. Furthermore, CYP7A1 mRNA was induced in STZ-treated type-I diabetic rats lacking insulin secretion [43], which has led to the speculation that insulin may repress CYP7A1 gene in rats. On the other hand, more recent studies seem to be contradictory to these early observations. First, mouse CYP7A1 mRNA expression peaked during the early dark cycle when food intake was the most active [44, 45]. Furthermore, restricted feeding during light cycles shifted the peak of CYP7A1 mRNA expression from dark cycles to light cycles [44]. Such evidence seems to imply that liver bile acid synthesis and liver metabolism are coordinately controlled. Further studies are necessary to determine if nutritional regulation of bile acid synthesis may play a role in metabolic homeostasis during fasting to refeeding cycles.
5. Bile Acid Regulation of Glucose Metabolism
5.1. FXR and Glucose Metabolism
Diabetes is associated with impaired peripheral glucose clearance and increased hepatic glucose production during fasting, which lead to postprandial and fasting hyperglycemia. Initial evidence that bile acids may regulate glucose metabolism came from studies showing that FXR agonist induced phosphoenolpyruvate Carboxykinase (PEPCK) mRNA expression (Table 1) and glucose output in human and rat hepatocytes [46]. Treating mice with an FXR agonist also induced hepatic PEPCK mRNA expression in mice in vivo [46]. A FXR binding site has been identified in the promoter of PEPCK gene. In contrast, later studies carried out in fxr knockout mice revealed that FXR-deficient mice had insulin resistance and hyperglycemia phenotypes. Administration of a FXR agonist GW4064 decreased serum glucose, increased liver glycogen, and improved insulin sensitivity in diabetic db/db mice [7, 8]. A number of recent studies showed that bile acids and FXR repressed hepatic PEPCK and G6Pase gene expression and thus liver gluconeogenesis. In this case, it is shown that bile acids may induce the repressor SHP, which inhibits PEPCK via inhibiting C/EBP [47], FoxO1 [48], and Glucocorticoid receptor [49]. Although these liver effects of FXR activation may prevent fasting hyperglycemia, it does not sufficiently explain the increased insulin sensitivity and glucose disposal in FXR agonist-treated mice as determined by glucose and insulin tolerance tests. In a similar study, Cariou et al. used hyperinsulinemic euglycemic clamp and demonstrated that FXR-deficiency is associated with decreased whole-body glucose disposal, suggesting a role of FXR in regulating peripheral glucose metabolism [50]. FXR is not expressed in muscle but is expressed in white adipose at a very low level. It is noticed that fxr−/− mice had smaller adipocytes, and FXR agonist GW4064 treatment increased adipose differentiation and insulin-dependent glucose uptake in 3T3-L1 cells in vitro. Another study suggests that FXR agonist INT747 induced adipose differentiation via inducing the expression of adipocyte-related genes including C/EBPα and PPARγ [51]. In addition to a role of FXR in adipose, two recent studies provided additional mechanism by which FXR may regulate peripheral glucose homeostasis. These studies revealed that FXR is also expressed in pancreatic β cells and positively regulates glucose-dependent insulin secretion [52]. It is suggested that FXR activation stimulates insulin gene transcription. On the other hand, FXR activation is associated with increased AKT phosphorylation and GLUT2 translocation to the cell membrane and thus enhances glucose uptake into pancreatic β cells and glucose-dependent insulin secretion. Intestine is another major FXR expressing tissue. A recent study showed that FGF15/19, expressed in the intestine and secreted into the blood circulation, acts as a postprandial factor that promotes glycogen synthesis, which may be an important mechanism controlling postprandial glucose metabolism [53]. It has been shown that serum FGF19 increases during postprandial period in humans, presumably due to increased bile acid signaling [34]. Therefore, identification of the regulatory role of FGF15/19 in postprandial glycogen synthesis provides a novel link between bile acid signaling and glucose metabolism. In addition to the nuclear receptor FXR-mediated effects, bile acids have been shown to directly activate hepatic AKT via a Gα i protein coupled receptor signaling pathway, which stimulated hepatic glycogen synthesis [54]. Recently, it was further demonstrated that bile acid activation of the Gα i-AKT signaling cascade was involved in the bile acid induction of FXR and SHP and downregulation of gluconeogenic gene expressions in the liver [55]. In summary, these studies suggest that bile acid regulation of hepatic glucose metabolism involves complex crosstalk between FXR-dependent pathways and FXR-independent signaling pathways.
5.2. TGR5 and Glucose Metabolism
Bile acids also activate a cell surface G-protein coupled receptor TGR5, which is mainly expressed in the intestine, brown adipose, white adipose, and gallbladder. Low levels of TGR5 expression has also been detected in liver and skeletal muscle. Upon activation, TGR5 leads to intracellular cAMP production and PKA activation. Based on the ability to induce cellular cAMP production, taurolithocholic acid (TLCA) and LCA show highest potency in activating TGR5 with EC50 of 0.33 and 0.53 μM, respectively, while DCA, CDCA, and CA (in rank order) activate TGR5 at higher 1–8 μM concentrations [56]. It is suggested that in brown adipocytes, bile acid activation of TGR5-cAMP-PKA cascade results in induction of downstream deiodinase, fatty acid oxidation genes, and uncoupling proteins, which increase energy expenditure and promote weight loss [57]. As increased free fatty acid release and cytokine production associated with obesity clearly contribute to the development of insulin resistance, bile acids/TGR5 regulation of weight loss certainly could play a role in regulating glucose homeostasis. However, studies in tgr5−/− mice showed that neither under chow condition nor under high fat diet feeding condition did tgr5−/− develop obesity or hyperglycemia [58]. The high fat diet feeding effect on insulin sensitivity, as determined by insulin tolerance tests, also seemed to be gender-specific in tgr5−/− mice, with male showing impaired, but female showing improved insulin sensitivity [58]. The most potent endogenous ligand for TGR5 is TLCA. TLCA is highly toxic and once synthesized, is rapidly metabolized in the intestine and the liver. Under physiological conditions, liver efficiently extracts bile acids from the portal circulation, and bile acid concentration in the systemic circulation is very low. Because these primary and secondary bile acids activate TGR5 at a higher EC50, it is possible that TGR5 is not activated by physiological concentration of circulating bile acids outside of the entero-hepatic system. Thus, opposing to the clear pharmacological benefits of TGR5 activation, the physiological role of TGR5 in mediating bile acid signaling control of metabolic homeostasis needs to be further investigated. In addition to brown adipose, intestine is another major TGR5 expressing tissue. Using an enteroendocrine cell line STC-1, Katsuma et al. first demonstrated that bile acids stimulate glucagon like peptide-1 (GLP-1) production via TGR5 activation [59]. The pharmacological significance of this pathway was then demonstrated by a detailed study carried out by Thomas et al. [60]. These authors showed that administration of a potent TGR5 agonist INT777 raised intracellular ATP/ADP ratio and calcium influx, which leads to enhanced GLP-1 secretion from the intestine. GLP-1 is known to promote insulin secretion and thus regulate glucose homeostasis. Because GLP-1 mimetics and receptor agonists are currently under clinical development and have shown promise in improving glucose homeostasis in diabetes, bile acid-based TGR5 agonists may be a potential therapeutic to stimulate GLP-1 secretion in diabetic patients [61].
In contrast to these studies, bile acid sequestrants, which remove bile acids from the body by binding to bile acids in the intestine and prevent bile acids from being reabsorbed, have been shown to improve insulin sensitivity and lower fasting glucose in both men and several different experimental models [62]. Two studies conducted in rats have so far suggested that bile acid sequestrants may improve insulin sensitivity by increasing GLP-1 release [63, 64]. Although the molecular mechanism is still not clear, both studies suggested that such effect is likely bile acid receptor-independent. This is because both studies showed that administration of bile acid sequestrants significantly lowered serum bile acid levels, which was associated with decreased FXR activation in the liver and the intestine. Furthermore, it is shown that blocking intestine bile acid transport using SC-435, an apical sodium-dependent bile acid transport inhibitor, also lowered serum bile acid levels, but did not modulate insulin sensitivity or GLP-1 secretion. Thus, it is likely that bile acid sequestrants exert its effect by directly modulating cellular signaling in the intestine rather than by altering circulating bile acid levels or modulating bile acid pool.
6. Bile Acids and Lipid Metabolism
6.1. Bile Acids and Cholesterol Metabolism
It has been known for a long time that preventing bile acid reabsorption in the intestine by bile acid sequestration increases hepatic CYP7A1 and bile acid synthesis [65]. The resulting increase in hepatic cholesterol catabolism caused compensatory induction of LDL receptor (LDLR) and LDL cholesterol (LDL-C) uptake. Because of the activation of this liver pathway, cholestyramine has been used to effectively lowering serum cholesterol in human patients. Paradoxically, activation of FXR by its potent agonists, which repress hepatic bile acid synthesis, also decreased serum cholesterol in animal models [7]. In wild-type mice, activation of FXR is mainly associated with a reduction of HDL-C, while in hypercholesterolemic animal models, activation of FXR decreases both LDL-C and HDL-C. In vitro, FXR was shown to induce LDLR expression and repress PCSK9, an LDLR inhibitor [66]. However, activation of FXR still significantly decreased serum non-HDL cholesterol in ldlr−/− mice [67]. Furthermore, CDCA administration has been shown to raise serum LDL-C levels in humans. It remains to be determined whether activation of FXR will provide benefits in lowering LDL-C in men.
FXR agonists have been shown to prevent atherosclerosis in various experimental models [68]. Serum HDL transports cholesterol from peripheral tissues to the liver for elimination and thus plays a critical role in reverse cholesterol transport and the development of atherosclerosis. However, the role of FXR in regulating HDL metabolism is still under debate because FXR inhibits the hepatic production of apolipoprotein A1 (ApoAI), a key structural component of HDL, and activation of FXR is associated with decreased serum HDL [69]. Nevertheless, a recent study showed that activation of FXR promotes reverse cholesterol transport in mice by inducing hepatic expression of scavenger receptor B1 (SR-B1) [70], which is suggested to play a role in both hepatic uptake of HDL-C and biliary secretion of free cholesterol [71]. A FXR binding site has been identified in the SR-B1 gene promoter [72]. In a recent study, we demonstrated that stimulating de novo bile acid synthesis by transgenic expression of a CYP7A1 gene in mouse liver prevented diet-induced hypercholesterolemia [6]. Different from CA feeding or FXR agonist administration, Cyp7a1-tg mice showed both increased hepatic cholesterol catabolism and bile acid signaling. Using this model, we demonstrated that bile acid activation of FXR induces hepatic expression of ABCG5 and ABCG8 through a common FXRE, which promoted biliary-free cholesterol secretion and fecal cholesterol loss. It is well known that cholesterol activation of LXR only induces mouse, but not human CYP7A1 gene expression [73]. We also showed that cholesterol/LXR signaling only induced ABCG5 and ABCG8 in mice, but not in primary human hepatocytes [6]. These studies suggest that upon hepatic cholesterol accumulation, LXR may stimulate cholesterol catabolism or biliary cholesterol secretion in mouse livers, but not human livers. Thus, it is possible that bile acid/FXR/ABCG5/G8 pathway plays a more important role in maintaining hepatic cholesterol homeostasis in response to increased cholesterol levels in humans.
6.2. Bile Acids and Fatty Acid Metabolism
It has been known for a long time that serum bile acid and serum triglycerides are inversely correlated, suggesting that bile acid negatively regulates serum triglycerides [74, 75]. Current studies suggest that bile acids may lower serum triglycerides by repressing both hepatic triglyceride production/secretion and stimulating serum triglyceride clearance. In the liver, it is shown that bile acid activation of FXR repressed LXR-induction of SREBP-1 and its target genes in de novo lipogenesis by inducing the repressor SHP, which not only decreased hepatic fat accumulation, but also led to reduced hepatic VLDL secretion [76]. One study showing both LXR-dependent and FXR-dependent induction of hepatic lipogenesis by bile acid sequestrants administration supported this conclusion [68]. The finding that FXR represses microsomal triglyceride transfer protein (MTP) and thus hepatic VLDL secretion seems to provide additional support that the FXR/SHP pathway reduces hepatic triglyceride output [77]. Genetic knockout of shp in ob/ob mice increased MTP and VLDL secretion [78]. Diabetes and obesity are associated with increased hepatic VLDL output. Both increased fatty acid supply to the liver and hepatic insulin resistance may be involved. On the other hand, there are also studies showing that hepatic VLDL secretion was impaired in diabetic mouse models despite increased triglyceride output [79]. Furthermore, increased VLDL secretion in shp knockout mice seemed to be beneficial in reducing hepatic fat accumulation in ob/ob mice [78]. Thus, the bile acid regulation of hepatic VLDL secretion and its in vivo significance seem to be complex and may depend on the experimental conditions. Serum triglyceride is cleared after VLDL-triglyceride is hydrolyzed by lipoprotein lipase (LPL) and subsequently taken up by the peripheral tissues. It has been reported that obesity and diabetes are also associated with impaired peripheral triglyceride clearance, contributing to diabetic hypertriglyceridemia. Activation of FXR has been shown to induce apolipoprotein CII (ApoCII), which is an LPL activator, and repress apolipoprotein CIII (ApoCIII), which is an LPL inhibitor in the liver [80, 81]. Increasing ApoCII or decreasing ApoCIII stimulates LPL to hydrolyze triglycerides carried by VLDL, thus accelerates serum VLDL clearance upon FXR activation.
7. Conclusion
Extensive investigations conducted in the past decade have shown that bile acids are important regulators of glucose and lipid metabolism. The identification of bile acid-activated nuclear receptor FXR and cell surface G protein coupled receptor TGR5 has significantly advanced our understanding on how bile acid signaling regulates cellular metabolism in physiological and diseased conditions. The identification of these regulatory mechanisms also provided molecular basis for developing bile acid receptor agonists and receptor antagonists for treating human metabolic diseases. On the other hand, conflicting studies in the field are present, which not only reflects the complex nature of the bile acid signaling in regulation of whole body metabolism, but also implies the difference between physiological role and pharmacological role of bile acid signaling in metabolic control. Furthermore, studies that focus on the regulation of bile acid metabolism in diseased conditions, especially obesity and diabetes, are still insufficient. Future advances in the field are needed to improve our understanding in the bile acid control of metabolism, which is also critical in developing better drug therapies for the treatment of metabolic disorders.
Acknowledgment
Supported by NIH grants DK44442 and DK58379.
Abbreviations
- CA:
Cholic acid
- CDCA:
Chenodeoxycholic acid
- DCA:
Deoxycholic acid
- LCA:
Lithocholic acid
- CYP7A1:
Cholesterol 7α-hydroxylase
- CYP8B1:
Microsomal sterol 12α-hydroxylase
- CYP27A1:
Mitochondrial 27-hydroxylase
- CYP7B1:
Oxysterol 7α-hydroxylase
- MCA:
Muricholic acids
- ABC:
ATP-binding cassette
- BSEP:
Bile salt export pump
- SPGP:
Sister of P-glycoprotein
- PFIC:
Progressive familial intrahepatic cholestasis
- MDR:
Multidrug resistance
- NTCP:
Na+-dependent taurocholate transporter
- OATP:
Organic anion transporter
- ASBT:
Apical sodium-dependent bile salt transporter
- I-BABP:
Intestinal bile acid binding protein
- OST:
Organic solute transporter
- FXR:
Farnesoid X receptor
- SHP:
Small heterodimer partner
- LRH-1:
Liver-related homologue-1
- FGF15:
Fibroblast growth factor 15
- PEPCK:
Phosphoenolpyruvate carboxykinase
- GLP-1:
Glucagon-like peptide-1
- LDL:
Low density lipoprotein
- ApoA1:
Apolipoprotein A1
- SR-B1:
Scavenger receptor B1
- MTP:
Microsomal triglyceride transfer protein
- LPL:
Lipoprotein lipase
- ApoCII:
Apolipoprotein CII
- ApoCIII:
Apolipoprotein CIII
- VLDL:
Very low density lipoprotein.
References
- 1.Russell DW, Setchell KDR. Bile acid biosynthesis. Biochemistry. 1992;31(20):4737–4749. doi: 10.1021/bi00135a001. [DOI] [PubMed] [Google Scholar]
- 2.Chiang JYL. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocrine Reviews. 2002;23(4):443–463. doi: 10.1210/er.2000-0035. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiological Reviews. 2009;89(1):147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cellular Signalling. 2008;20(12):2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Goldfine AB. Modulating LDL cholesterol and glucose in patients with type 2 diabetes mellitus: targeting the bile acid pathway. Current Opinion in Cardiology. 2008;23(5):502–511. doi: 10.1097/HCO.0b013e32830c6c37. [DOI] [PubMed] [Google Scholar]
- 6.Li T, Matozel M, Boehme S, et al. Overexpression of cholesterol 7α-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology. 2011;53(3):996–1006. doi: 10.1002/hep.24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Lee FY, Barrera G, et al. Activation of the nuclear FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(4):1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. The Journal of Clinical Investigation. 2006;116(4):1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li T, Owsley E, Matozel M, Hsu P, Novak CM, Chiang JYL. Transgenic expression of cholesterol 7α-hydroxylase in the liver prevents high-fat diet-induced obesity and insulin resistance in mice. Hepatology. 2010;52(2):678–690. doi: 10.1002/hep.23721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang JY. Regulation of bile acid synthesis. Frontiers in Bioscience. 1998;3:D176–D193. doi: 10.2741/a273. [DOI] [PubMed] [Google Scholar]
- 11.Chiang JYL. Bile acids: regulation of synthesis. Journal of Lipid Research. 2009;50(10):1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlahcevic ZR, Pandak WM, Heuman DM, Hylemon PB. Function and regulation of hydroxylases involved in the bile acid biosynthesis pathways. Seminars in Liver Disease. 1992;12(4):403–419. doi: 10.1055/s-2008-1040410. [DOI] [PubMed] [Google Scholar]
- 13.Myant NB, Mitropoulos KA. Cholesterol 7α hydroxylase. Journal of Lipid Research. 1977;18(2):135–153. [PubMed] [Google Scholar]
- 14.Pikuleva IA, Babiker A, Waterman MR, Björkhem I. Activities of recombinant human cytochrome P450c27 (CYP27) which produce intermediates of alternative bile acid biosynthetic pathways. The Journal of Biological Chemistry. 1998;273(29):18153–18160. doi: 10.1074/jbc.273.29.18153. [DOI] [PubMed] [Google Scholar]
- 15.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiological Reviews. 2003;83(2):633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 16.Boyer JL. New concepts of mechanisms of hepatocyte bile formation. Physiological Reviews. 1980;60(2):303–326. doi: 10.1152/physrev.1980.60.2.303. [DOI] [PubMed] [Google Scholar]
- 17.Childs S, Yeh RL, Georges E, Ling V. Identification of a sister gene to P-glycoprotein. Cancer Research. 1995;55(10):2029–2034. [PubMed] [Google Scholar]
- 18.Strautnieks SS, Kagalwalla AF, Tanner MS, et al. Identification of a locus for progressive familial intrahepatic cholestasis PFIC2 on chromosome 2q24. American Journal of Human Genetics. 1997;61(3):630–633. doi: 10.1086/515501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smit JJM, Schinkel AH, Elferink RPJO, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75(3):451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 20.Langheim S, Yu L, Von Bergmann K, et al. ABCG5 and ABCG8 require MDR2 for secretion of cholesterol into bile. Journal of Lipid Research. 2005;46(8):1732–1738. doi: 10.1194/jlr.M500115-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Meier PJ. Molecular mechanisms of hepatic bile salt transport from sinusoidal blood into bile. American Journal of Physiology. 1995;269(6, part 1):G801–G812. doi: 10.1152/ajpgi.1995.269.6.G801. [DOI] [PubMed] [Google Scholar]
- 22.Shneider BL, Dawson PA, Christie DM, Hardikar W, Wong MH, Suchy FJ. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. The Journal of Clinical Investigation. 1995;95(2):745–754. doi: 10.1172/JCI117722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong YZ, Everett ET, Schwartz DA, Norris JS, Wilson FA. Molecular cloning, tissue distribution, and expression of a 14-kDa bile acid-binding protein from rat ileal cytosol. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(11):4741–4745. doi: 10.1073/pnas.91.11.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dawson PA, Hubbert M, Haywood J, et al. The heteromeric organic solute transporter α-β, Ostα-Ostβ, is an ileal basolateral bile acid transporter. The Journal of Biological Chemistry. 2005;280(8):6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Molecular Cell. 2000;6(3):517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 26.Kerr TA, Saeki S, Schneider M, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Developmental Cell. 2002;2(6):713–720. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Lee YK, Bundman D, et al. Redundant pathways for negative feedback regulation of bile acid production. Developmental Cell. 2002;2(6):721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 28.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metabolism. 2005;2(4):217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Pandak WM, Li YC, Chiang JYL, et al. Regulation of cholesterol 7 α-hydroxylase mRNA and transcriptional activity by taurocholate and cholesterol in the chronic biliary diverted rat. The Journal of Biological Chemistry. 1991;266(6):3416–3421. [PubMed] [Google Scholar]
- 30.Rao A, Haywood J, Craddock AL, Belinsky MG, Kruh GD, Dawson PA. The organic solute transporter α-β, Ostα-Ostβ, is essential for intestinal bile acid transport and homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(10):3891–3896. doi: 10.1073/pnas.0712328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim I, Ahn SH, Inagaki T, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. Journal of Lipid Research. 2007;48(12):2664–2672. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Song KH, Li T, Owsley E, Strom S, Chiang JYL. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7α-hydroxylase gene expression. Hepatology. 2009;49(1):297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaap FG, van der Gaag NA, Gouma DJ, Jansen PLM. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology. 2009;49(4):1228–1235. doi: 10.1002/hep.22771. [DOI] [PubMed] [Google Scholar]
- 34.Lundåsen T, Gälman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. Journal of Internal Medicine. 2006;260(6):530–536. doi: 10.1111/j.1365-2796.2006.01731.x. [DOI] [PubMed] [Google Scholar]
- 35.Li T, Jahan A, Chiang JYL. Bile acids and cytokines inhibit the human cholesterol 7α-hydroxylase gene via the JNK/c-Jun pathway in human liver cells. Hepatology. 2006;43(6):1202–1210. doi: 10.1002/hep.21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li T, Ma H, Chiang JYL. TGFβ1, TNFα, and insulin signaling crosstalk in regulation of the rat cholesterol 7α-hydroxylase gene expression. Journal of Lipid Research. 2008;49(9):1981–1989. doi: 10.1194/jlr.M800140-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gälman C, Angelin B, Rudling M. Bile acid synthesis in humans has a rapid diurnal variation that is asynchronous with cholesterol synthesis. Gastroenterology. 2005;129(5):1445–1453. doi: 10.1053/j.gastro.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 38.Shaham O, Wei R, Wang TJ, et al. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Molecular Systems Biology. 2008;4, article 214 doi: 10.1038/msb.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li T, Kong X, Owsley E, Ellis E, Strom S, Chiang JYL. Insulin regulation of cholesterol 7α-hydroxylase expression in human hepatocytes: roles of forkhead box O1 and sterol regulatory element-binding protein 1c. The Journal of Biological Chemistry. 2006;281(39):28745–28754. doi: 10.1074/jbc.M605815200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song KH, Chiang JYL. Glucagon and cAMP inhibit cholesterol 7α-hydroxylase (CYP7A1) gene expression in human hepatocytes: discordant regulation of bile acid synthesis and gluconeogenesis. Hepatology. 2006;43(1):117–125. doi: 10.1002/hep.20919. [DOI] [PubMed] [Google Scholar]
- 41.Li T, Chanda D, Zhang Y, -SikChoi H, Chiang JYL. Glucose stimulates cholesterol 7 α-hydroxylase gene transcription in human hepatocytes. Journal of Lipid Research. 2010;51(4):832–842. doi: 10.1194/jlr.M002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin DJ, Campos JA, Gil G, Osborne TF. PGC-1α activates CYP7A1 and bile acid biosynthesis. The Journal of Biological Chemistry. 2003;278(50):50047–50052. doi: 10.1074/jbc.M309736200. [DOI] [PubMed] [Google Scholar]
- 43.Subbiah MTR, Yunker RL. Cholesterol 7α-hydroxylase of rat liver: an insulin sensitive enzyme. Biochemical and Biophysical Research Communications. 1984;124(3):896–902. doi: 10.1016/0006-291x(84)91042-8. [DOI] [PubMed] [Google Scholar]
- 44.Ma K, Xiao R, Tseng HT, Shan L, Fu L, Moore DD. Circadian dysregulation disrupts bile acid homeostasis. PLoS One. 2009;4(8) doi: 10.1371/journal.pone.0006843. Article ID e6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Martelot G, Claudel T, Gatfield D, et al. REV-ERBα participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biology. 2009;7(9) doi: 10.1371/journal.pbio.1000181. Article ID e1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stayrook KR, Bramlett KS, Savkur RS, et al. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146(3):984–991. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- 47.Park MJ, Kong HJ, Kim HY, Kim HH, Kim JH, Cheong JH. Transcriptional repression of the gluconeogenic gene PEPCK by the orphan nuclear receptor SHP through inhibitory interaction with C/EBPα . Biochemical Journal. 2007;402(3):567–574. doi: 10.1042/BJ20061549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamagata K, Daitoku H, Shimamoto Y, et al. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. The Journal of Biological Chemistry. 2004;279(22):23158–23165. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 49.Borgius LJ, Steffensen KR, Gustafsson JÅ, Treuter E. Glucocorticoid signaling is perturbed by the atypical orphan receptor and corepressor SHP. The Journal of Biological Chemistry. 2002;277(51):49761–49766. doi: 10.1074/jbc.M205641200. [DOI] [PubMed] [Google Scholar]
- 50.Cariou B, Van Harmelen K, Duran-Sandoval D, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. The Journal of Biological Chemistry. 2006;281(16):11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 51.Rizzo G, Disante M, Mencarelli A, et al. The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Molecular Pharmacology. 2006;70(4):1164–1173. doi: 10.1124/mol.106.023820. [DOI] [PubMed] [Google Scholar]
- 52.Renga B, Mencarelli A, Vavassori P, Brancaleone V, Fiorucci S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochimica et Biophysica Acta. 2010;1802(3):363–372. doi: 10.1016/j.bbadis.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Kir S, Beddow SA, Samuel VT, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331(6024):1621–1624. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang Y, Studer E, Mitchell C, et al. Conjugated bile acids regulate hepatocyte glycogen synthase activity in vitro and in vivo via gαi signaling. Molecular Pharmacology. 2007;71(4):1122–1128. doi: 10.1124/mol.106.032060. [DOI] [PubMed] [Google Scholar]
- 55.Cao R, Cronk ZX, Zha W, et al. Bile acids regulate hepatic gluconeogenic genes and farnesoid X receptor via Gαi-protein-coupled receptors and the AKT pathway. Journal of Lipid Research. 2010;51(8):2234–2244. doi: 10.1194/jlr.M004929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. The Journal of Biological Chemistry. 2003;278(11):9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 58.Vassileva G, Hu W, Hoos L, et al. Gender-dependent effect of Gpbar1 genetic deletion on the metabolic profiles of diet-induced obese mice. Journal of Endocrinology. 2010;205(3):225–232. doi: 10.1677/JOE-10-0009. [DOI] [PubMed] [Google Scholar]
- 59.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochemical and Biophysical Research Communications. 2005;329(1):386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 60.Thomas C, Gioiello A, Noriega L, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metabolism. 2009;10(3):167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nauck MA. Incretin-based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. American Journal of Medicine. 2011;124(1):S3–S18. doi: 10.1016/j.amjmed.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 62.Kobayashi M, Ikegami H, Fujisawa T, et al. Prevention and treatment of obesity, insulin resistance, and diabetes by bile acid-binding resin. Diabetes. 2007;56(1):239–247. doi: 10.2337/db06-0353. [DOI] [PubMed] [Google Scholar]
- 63.Shang Q, Saumoy M, Holst JJ, Salen G, Xu G. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. American Journal of Physiology. 2010;298(3):G419–G424. doi: 10.1152/ajpgi.00362.2009. [DOI] [PubMed] [Google Scholar]
- 64.Chen L, McNulty J, Anderson D, et al. Cholestyramine reverses hyperglycemia and enhances glucose-stimulated glucagon-like peptide 1 release in Zucker diabetic fatty rats. Journal of Pharmacology and Experimental Therapeutics. 2010;334(1):164–170. doi: 10.1124/jpet.110.166892. [DOI] [PubMed] [Google Scholar]
- 65.Staels B, Kuipers F. Bile acid sequestrants and the treatment of type 2 diabetes mellitus. Drugs. 2007;67(10):1383–1392. doi: 10.2165/00003495-200767100-00001. [DOI] [PubMed] [Google Scholar]
- 66.Langhi C, Le May C, Kourimate S, et al. Activation of the farnesoid X receptor represses PCSK9 expression in human hepatocytes. FEBS Letters. 2008;582(6):949–955. doi: 10.1016/j.febslet.2008.02.038. [DOI] [PubMed] [Google Scholar]
- 67.Hartman HB, Gardell SJ, Petucci CJ, Wang S, Krueger JA, Evans MJ. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR-/- and apoE -/- mice. Journal of Lipid Research. 2009;50(6):1090–1100. doi: 10.1194/jlr.M800619-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hageman J, Herrema H, Groen AK, Kuipers F. A role of the bile salt receptor FXR in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(8):1519–1528. doi: 10.1161/ATVBAHA.109.197897. [DOI] [PubMed] [Google Scholar]
- 69.Claudel T, Sturm E, Duez H, et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. The Journal of Clinical Investigation. 2002;109(7):961–971. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Yin L, Anderson J, et al. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. The Journal of Biological Chemistry. 2010;285(5):3035–3043. doi: 10.1074/jbc.M109.083899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saddar S, Mineo C, Shaul PW. Signaling by the high-affinity HDL receptor scavenger receptor B type i. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(2):144–150. doi: 10.1161/ATVBAHA.109.196170. [DOI] [PubMed] [Google Scholar]
- 72.Chao F, Gong W, Zheng Y, et al. Upregulation of scavenger receptor class B type I expression by activation of FXR in hepatocyte. Atherosclerosis. 2010;213(2):443–448. doi: 10.1016/j.atherosclerosis.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 73.Chiang JYL. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. Journal of Hepatology. 2004;40(3):539–551. doi: 10.1016/j.jhep.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 74.Angelin B, Einarsson K, Hellstrom K, Leijd B. Effects of cholestyramine and chenodeoxycholic acid on the metabolism of endogenous triglyceride in hyperlipoproteinemia. Journal of Lipid Research. 1978;19(8):1017–1024. [PubMed] [Google Scholar]
- 75.Bateson MC, Maclean D, Evans JR, Bouchier IAD. Chenodeoxycholic acid therapy for hypertriglyceridaemia in men. British Journal of Clinical Pharmacology. 1978;5(3):249–254. doi: 10.1111/j.1365-2125.1978.tb01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. The Journal of Clinical Investigation. 2004;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hirokane H, Nakahara M, Tachibana S, Shimizu M, Sato R. Bile acid reduces the secretion of very low density lipoprotein by repressing microsomal triglyceride transfer protein gene expression mediated by hepatocyte nuclear factor-4. The Journal of Biological Chemistry. 2004;279(44):45685–45692. doi: 10.1074/jbc.M404255200. [DOI] [PubMed] [Google Scholar]
- 78.Huang J, Iqbal J, Saha PK, et al. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46(1):147–157. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- 79.Li X, Grundy SM, Patel SB. Obesity in db and ob animals leads to impaired hepatic very low density lipoprotein secretion and differential secretion of apolipoprotein B-48 and B-100. Journal of Lipid Research. 1997;38(7):1277–1288. [PubMed] [Google Scholar]
- 80.Claudel T, Inoue Y, Barbier O, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125(2):544–555. doi: 10.1016/s0016-5085(03)00896-5. [DOI] [PubMed] [Google Scholar]
- 81.Kast HR, Nguyen CM, Sinal CJ, et al. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Molecular Endocrinology. 2001;15(10):1720–1728. doi: 10.1210/mend.15.10.0712. [DOI] [PubMed] [Google Scholar]