

Abstract
All photosynthetic reaction centers share a common structural theme. Two related, integral membrane polypeptides sequester electron transfer cofactors into two quasi-symmetrical branches, each of which incorporates a quinone. In type II reaction centers [photosystem (PS) II and proteobacterial reaction centers], electron transfer proceeds down only one of the branches, and the mobile quinone on the other branch is used as a terminal acceptor. PS I uses iron-sulfur clusters as terminal acceptors, and the quinone serves only as an intermediary in electron transfer. Much effort has been devoted to understanding the unidirectionality of electron transport in type II reaction centers, and it was widely thought that PS I would share this feature. We have tested this idea by examining in vivo kinetics of electron transfer from the quinone in mutant PS I reaction centers. This transfer is associated with two kinetic components, and we show that mutation of a residue near the quinone in one branch specifically affects the faster component, while the corresponding mutation in the other branch specifically affects the slower component. We conclude that both electron transfer branches in PS I are active.
Upon excitation of the
photosystem (PS) I primary donor (P700), an
electron is transferred to the primary acceptor,
A0 (a chlorophyll a molecule) and in
less than 100 ps to a secondary acceptor, A1,
identified as a phylloquinone. From A1, the
electrons are transferred to an iron-sulfur cluster,
FX, and then to the terminal iron-sulfur
acceptors, FA and FB, which
are bound by the extrinsic PsaC polypeptide (1) (see Fig.
1a). The structure of PS I at
4-Å resolution (2) revealed the existence of two sets of redox
cofactors between P700 and
FX, bound with an almost perfect symmetry by the
two largest subunits, PsaA and PsaB. Due to this symmetry, the obvious
similarity of the PsaA and PsaB polypeptides (≈45–50% identical
throughout their length; ref. 3) and the fact that the bacterial type I
reaction centers are homodimers (4, 5), the possibility of two parallel
electron transfer pathways up to FX should be
considered. However, based on the structural analogies of the core of
PS I to the purple bacteria reaction center, which suggested a common
evolutionary origin for all photosynthetic reaction centers (6), it has
been generally thought that electron transport in PS I is
unidirectional, as it is in the type II reaction centers. Most data
supporting this view comes from electron paramagnetic resonance (EPR)
experiments, in which only a single quinone anion radical can be
accumulated during strong illumination under reducing conditions
sufficient to reduce the terminal acceptors (7, 8) (although see ref.
9). However, these extreme conditions may well bias the results.
Transient EPR experiments under conditions of forward electron
transport would be preferable (10, 11), but this technique may lack the
time resolution required to observe very fast events. Using visible
spectroscopy both forward electron transfer from A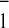 to FX (12, 13), and charge recombination between
P
to FX (12, 13), and charge recombination between
P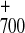 and A
and A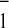 (14) have been shown to
be biphasic. The biphasic oxidation of A
(14) have been shown to
be biphasic. The biphasic oxidation of A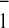 in isolated
PS I centers initially was interpreted as reflecting a low equilibrium
constant between A1 and FX
(Fig. 1b), leading to a fast (≈25 ns) redox equilibration
of A1 and FX and a slower
(≈150 ns) decay of this quasi-equilibrium state associated with
electron transfer from FX to
FA/FB (12). Based on the
lack of sensitivity of these kinetics to the membrane potential, this
model was recently challenged and alternative hypotheses were proposed
(13): if there is only one active branch, then there must be two
populations of PS I in equilibrium that differ structurally to give
rise to different kinetics (Fig. 1c). Alternatively, the
existence of two active branches would explain the biphasic kinetics as
transfer from the two different quinones, one of which is primarily
bound by PsaA and the other by PsaB (Fig. 1d).
in isolated
PS I centers initially was interpreted as reflecting a low equilibrium
constant between A1 and FX
(Fig. 1b), leading to a fast (≈25 ns) redox equilibration
of A1 and FX and a slower
(≈150 ns) decay of this quasi-equilibrium state associated with
electron transfer from FX to
FA/FB (12). Based on the
lack of sensitivity of these kinetics to the membrane potential, this
model was recently challenged and alternative hypotheses were proposed
(13): if there is only one active branch, then there must be two
populations of PS I in equilibrium that differ structurally to give
rise to different kinetics (Fig. 1c). Alternatively, the
existence of two active branches would explain the biphasic kinetics as
transfer from the two different quinones, one of which is primarily
bound by PsaA and the other by PsaB (Fig. 1d).
Figure 1.

(a) Scheme of cofactor arrangement in PS I.
P700 is a pair of chlorophylls that serves as the primary
electron donor. The accessory (“acc”) and A0
chlorophylls are both single chlorophyll molecules; A0 is
known to serve as an intermediate in electron transfer, but the role of
the accessory chlorophyll in this transfer is still not clear (1, 2).
A1 is a phylloquinone. FX, FA, and
FB are 4Fe-4S clusters. Trp represents the tryptophan near
the phylloquinone that has been mutated in this study.
(b–d) Different models to explain the
biphasic kinetics of A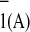 reoxidation.
(b) Only one branch is active. A rapid equilibrium is
established between A1 and FX, which is
depleted by forward electron transfer to
FA/FB. (c) Only one branch is
active, but the phylloquinone can exist in one of two possible
conformations, which may be in equilibrium (slow on the time scale of
electron transfer). (d) Both branches are active, but
the two phylloquinones are in slightly different environments, giving
rise to different kinetics of electron transfer to FX.
reoxidation.
(b) Only one branch is active. A rapid equilibrium is
established between A1 and FX, which is
depleted by forward electron transfer to
FA/FB. (c) Only one branch is
active, but the phylloquinone can exist in one of two possible
conformations, which may be in equilibrium (slow on the time scale of
electron transfer). (d) Both branches are active, but
the two phylloquinones are in slightly different environments, giving
rise to different kinetics of electron transfer to FX.
Because the understanding of the origin of the biphasic kinetics could
give new insights in the electron transport mechanism in PS I, we
examined the quinone reoxidation in site-directed mutants of PsaA and
PsaB in the genetically tractable alga Chlamydomonas
reinhardtii. The data presented here show that mutations
introduced in the environment of each phylloquinone specifically alter
the two phases of electron transfer from A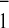 . These
observations lead to the conclusion that, unlike the situation in the
purple bacteria reaction center, both electron transfer branches in PS
I are active.
. These
observations lead to the conclusion that, unlike the situation in the
purple bacteria reaction center, both electron transfer branches in PS
I are active.
Materials and Methods
Mutant Strains of C. reinhardtii.
The single mutants were created by bio-ballistic transformation of appropriate gene deletion strains with plasmids containing the point mutations in psaA or psaB, followed by selection for the linked antibiotic-resistance marker (15). The plasmids in which the mutations were constructed contain the entire psaB gene or psaA third exon along with flanking sequences to direct recombination and a coinserted aadA gene, which serves as a selectable marker. The PsaA–W693F mutation converts codon 693 from TGG (Trp) to TTC (Phe) and was made in plasmid pKR154 (15). The PsaB–W673F mutation was made in plasmid pKR164 (15) and converts codon 673 from TGG to TTC. It also changes codon 674 from CAA (Gln) to CAG (Gln); this silent change introduces a BstNI site used to detect the mutation during cloning.
For kinetic analysis, the recipients were KRC51–3A (cbn1–48 FUD7 psaAΔ) or KRC52–8A (cbn1–48 FUD7 psaBΔ). The FUD7 genotype corresponds to a psbA deletion that prevents accumulation of PS II (16) whereas the cbn1–48 mutation leads to a chlorophyll b-less mutant with a reduced amount of light-harvesting complexes (17). The control strain was made by transformation of KRC51–3A with plasmid pKR154, encoding wild-type psaA-3. Double mutants were made by cotransformation with both psaA and psaB plasmids, followed by a genetic cross with KRC62–16A (mt− cbn1–48 nac2–26 psaAΔpsaBΔ) to generate a cbn1 nac2 strain (the nac2 mutation prevents PS II accumulation; ref. 18) harboring both mutations.
Optical Spectroscopy.
Spectroscopic measurements on whole cells of C. reinhardtii were performed by using a home-built spectrophotometer where absorption changes were probed by using short monochromatic flashes. In the 425- to 500-nm spectral range, the pulses were produced by a Nd:Yag pumped optical parametric oscillator Quanta-Ray MOPO-710 (Spectra-Physics) and in the 375- to 420-nm range by a frequency doubler accessory device FDO-900 (Spectra-Physics). The sample was excited at 700 nm by a tunable dye-laser pumped by the second harmonic of a Nd:Yag laser. The high signal-to-noise ratio of the spectrophotometer (105) when using samples with high optical densities such as cell suspensions and the time resolution (5 ns) of the technique have been described (19). C. reinhardtii cells were grown in Tris-acetate-phosphate medium (20) at 25°C under low light (6 μE⋅m−2⋅s−1). For spectroscopic measurements the cells were resuspended in 20 mM Hepes (pH 7.2) containing 20% (wt/vol) Ficoll, as well as 5 μM carbonyl cyanide (4-(trifluoromethoxy)phenyl)hydrazone to collapse the permanent transmembrane potential. The dependence of the flash-induced absorption changes at 430 and 380 nm upon the energy of the actinic flash was measured for each strain. Kinetics measurements were performed with light energies just sufficient to saturate the signal at 380 nm. When significantly higher actinic energies were used, we observed an additional fast decay component (τ < 5 ns) at 430 nm in all strains. The decay-associated spectra of the kinetic phases were obtained from a global analysis of the individual kinetics obtained at each wavelength, using the mexfit program (21).
Results
The mutations in PsaA and PsaB were designed by using the crystal
structures of PS I and the related purple bacterial reaction center as
a guide (2, 6, 22). The structural data on PS I suggested that the most
likely region for binding the phylloquinones was the stretch of highly
conserved residues in PsaA and PsaB forming the stromal α-helices n
and n′ (2). Additionally, magnetic resonance data indicated that an
aromatic nitrogen-containing amino acid was close to the phylloquinone
A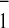 radical (23). We have introduced site-directed
mutations in the tryptophan and histidine residues conserved between
PsaA and PsaB in this region (Fig. 2) and
found that mutation of Trp-693 in the PsaA (PsaA–W693F) polypeptide
specifically disturbed the A
radical (23). We have introduced site-directed
mutations in the tryptophan and histidine residues conserved between
PsaA and PsaB in this region (Fig. 2) and
found that mutation of Trp-693 in the PsaA (PsaA–W693F) polypeptide
specifically disturbed the A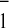 EPR spectrum (B.B., F.
MacMillan, C. Teutloff, K. Brettel, F. Gu, S. Grimaldi, R. Bittl, and
K.R., unpublished work). Here we examine the effect of the conversion
of these tryptophans (PsaA–W693 and PsaB–W673) to phenylalanines upon
the in vivo kinetics of A
EPR spectrum (B.B., F.
MacMillan, C. Teutloff, K. Brettel, F. Gu, S. Grimaldi, R. Bittl, and
K.R., unpublished work). Here we examine the effect of the conversion
of these tryptophans (PsaA–W693 and PsaB–W673) to phenylalanines upon
the in vivo kinetics of A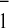 reoxidation. We
also constructed a double mutant that contains both point mutations
(PsaA–W693F/PsaB–W673F). Three additional mutants were examined:
two single substitutions of the conserved glutamates 695 and 698 in
PsaA for glutamines (mutants PsaA–E695Q and PsaA–E698Q) and a double
mutant in which the two tryptophans further along the n/n′ helices
(Fig. 2) were modified to phenylalanines (PsaA–W702F/PsaB–W682F).
All single mutations as well as the double mutations did not prevent
photoautotrophic growth of the mutant strains under either aerobic or
anaerobic conditions (data not shown). To simplify the spectroscopic
and kinetic analysis, we introduced these mutations into a control
strain lacking PS II and chlorophyll b.
reoxidation. We
also constructed a double mutant that contains both point mutations
(PsaA–W693F/PsaB–W673F). Three additional mutants were examined:
two single substitutions of the conserved glutamates 695 and 698 in
PsaA for glutamines (mutants PsaA–E695Q and PsaA–E698Q) and a double
mutant in which the two tryptophans further along the n/n′ helices
(Fig. 2) were modified to phenylalanines (PsaA–W702F/PsaB–W682F).
All single mutations as well as the double mutations did not prevent
photoautotrophic growth of the mutant strains under either aerobic or
anaerobic conditions (data not shown). To simplify the spectroscopic
and kinetic analysis, we introduced these mutations into a control
strain lacking PS II and chlorophyll b.
Figure 2.

Sequence alignment of PsaA and PsaB in the region of the n/n′ helix. Asterisks indicate the position of the residues mutated in this work. Conservation profile is color coded for identity (black, 100%; dark gray, 80%; light gray, 60%).
Transient absorption signals in the nanosecond-microsecond time scale
were recorded at discrete wavelengths in the 370- to 500-nm spectral
range after selective laser flash excitation (700 nm) of PS I in the
control strain. Global analysis of the data resolved four components:
three exponential decays (two in the nanosecond and one in the
microsecond time range) and a component that did not decay on the
detection time scale (30 μs). The spectrum of the nondecaying
component was associated with the electrochromic shifts due to the
flash-induced delocalized membrane potential. The spectrum had a
positive peak at 475 nm and negative peaks at 490, 460, and 420 nm,
characteristic for the pigment electrochromic changes in mutants
lacking chlorophyll b (24). The spectra of the three
exponential decays are shown in Fig. 3.
The microsecond component was assigned to P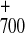 reduction, based on the typical bleaching at 430 nm (25) and a
half-time of 4 μs characteristic of its reduction by plastocyanin in
intact algae (26). The spectra of the two nanosecond phases were
attributed to reoxidation of A
reduction, based on the typical bleaching at 430 nm (25) and a
half-time of 4 μs characteristic of its reduction by plastocyanin in
intact algae (26). The spectra of the two nanosecond phases were
attributed to reoxidation of A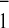 . As previously
observed in cyanobacteria (27) and Chlorella (13), the
nanosecond phases displayed positive absorption in the 370- to 400-nm
range, characteristic for semiphylloquinone, and contributions from the
electrochromic band shifts induced by the presence of a charge on
A1. The positive band at 480 nm has been
attributed to a shift in the absorption spectrum of a carotenoid
molecule in the close vicinity of the phylloquinone. It should be noted
that the two decay-associated spectra exhibited subtle but significant
differences in shape in the near-UV region (371–410 nm) and more
obvious differences above 410 nm. The half-times (13 and 140 ns) and
spectra of the two nanosecond decays were similar to those seen in
Chlorella (13). In the case of Chlorella, two
differences should be noted, a slightly slower half-time of the fast
phase (20 ns compared with 13 ns) and almost equivalent absorption at
380 nm for the two nanosecond decays. Thus, biphasic kinetics of
electron transfer from A1 to
FX are also present in Chlamydomonas
PS I and may well be a universal feature.
. As previously
observed in cyanobacteria (27) and Chlorella (13), the
nanosecond phases displayed positive absorption in the 370- to 400-nm
range, characteristic for semiphylloquinone, and contributions from the
electrochromic band shifts induced by the presence of a charge on
A1. The positive band at 480 nm has been
attributed to a shift in the absorption spectrum of a carotenoid
molecule in the close vicinity of the phylloquinone. It should be noted
that the two decay-associated spectra exhibited subtle but significant
differences in shape in the near-UV region (371–410 nm) and more
obvious differences above 410 nm. The half-times (13 and 140 ns) and
spectra of the two nanosecond decays were similar to those seen in
Chlorella (13). In the case of Chlorella, two
differences should be noted, a slightly slower half-time of the fast
phase (20 ns compared with 13 ns) and almost equivalent absorption at
380 nm for the two nanosecond decays. Thus, biphasic kinetics of
electron transfer from A1 to
FX are also present in Chlamydomonas
PS I and may well be a universal feature.
Figure 3.

Decay associated spectra of the 13 ns (○), 140 ns (●), and 4 μs (▴) components, obtained from global analysis of transient absorption data in whole cells of the C. reinhardtii control strain. Flash-induced kinetics in the 5 ns-30 μs time range were measured at each wavelength to obtain the three exponential components. Deconvolution with more exponential components did not improve the results.
The P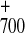 re-reduction was followed at 430 nm in all
mutant strains, and the kinetics could be well fit by a single
exponential with a half-time of ≈4 μs (Table
1). We did not observe absorption changes
at this wavelength in the tens of nanoseconds time range, where charge
recombination in the primary pair P
re-reduction was followed at 430 nm in all
mutant strains, and the kinetics could be well fit by a single
exponential with a half-time of ≈4 μs (Table
1). We did not observe absorption changes
at this wavelength in the tens of nanoseconds time range, where charge
recombination in the primary pair P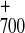 A
A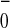 would occur (1). This indicated that the mutations
did not prevent electron transfer up to A1.
would occur (1). This indicated that the mutations
did not prevent electron transfer up to A1.
Table 1.
Kinetic parameters of electron transfer in the mutant strains
| Strain | A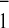 oxidation
oxidation
|
Amplitude* (% of total) | P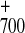 reduction,
t430 nm (μs) reduction,
t430 nm (μs) |
|||
|---|---|---|---|---|---|---|
| Fast
phase
|
Slow phase
|
|||||
| t380 nm (ns) | t480–457 nm (ns) | t380 nm (ns) | t480–457 nm (ns) | |||
| Control | 13 ± 2 | 13 ± 3 | 143 ± 10 | 160 ± 11 | 34 ± 3 | 4.0 ± 0.2 |
| PsaA–W693F | 15 ± 2 | 12 ± 3 | 490 ± 21 | 355 ± 120 | 34 ± 2 | 4.0 ± 0.2 |
| PsaB–W673F | 73† | 62† | 143† | 160† | 33 ± 10 | 4.2 ± 0.3 |
| W693F/W673F | 73 ± 17 | 62 ± 28 | 485 ± 70 | 350 ± 100 | 49 ± 9 | 4.1 ± 0.2 |
| PsaA–E695Q | 11 ± 5 | n.d. | 277 ± 33 | n.d. | 44 ± 11 | 4.4 ± 0.3 |
| PsaA–E698Q | 11 ± 4 | n.d. | 222 ± 30 | n.d. | 36 ± 10 | 3.9 ± 0.2 |
| W702F/W682F | 19 ± 12 | n.d. | 187 ± 28 | n.d. | n.d. | 3.9 ± 0.2 |
The half-times (t) were estimated from the data using fits to one or two exponential decay components. The measurement at 380 nm is in the spectral region of the semiphylloquinone absorption, the difference 480–457 nm is in the electrochromic band shift induced by the presence of charge on A1. The half-time of P700 re-reduction was calculated from the decay of the absorption change at 430 nm. n.d., not determined.
The relative amplitude of the fast nanosecond phase expressed as percentage of the total amplitude was estimated from the kinetics at 380 nm. Values that are significantly different from those of the control strain are in bold.
The half-times of the two phases used to fit the data for PsaB–W673F strain were fixed parameters with values taken from the double mutant for the fast phase and from the control strain for the slow phase, as described in the text.
To examine the effects of the mutations on electron transfer from
A1 to FX, we measured the
kinetics of the absorption decay at 380 nm, in the semiphylloquinone
absorption band, where contributions from P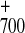 reduction and electrochromic shifts are small. The results for the two
symmetric mutants PsaA–W693F and PsaB–W673F and the double mutant
containing both mutations (PsaA–W693F/PsaB–W673F) are shown in Fig.
4. The mutation of the tryptophan in PsaA
affected specifically the kinetics of the slow nanosecond component,
increasing its half-time from ≈140 ns to ≈490 ns. The rate constant
of the fast component and the ratio between the amplitudes of the two
phases were the same as in the control strain (Fig. 4a;
Table 1). Mutation of the corresponding tryptophan in PsaB
(PsaB–W673F) slowed the initial part of the kinetics but did not
modify the later part, which is clearly seen in the conformity of the
latter part of the traces (Fig. 4b). In the double mutant
(PsaA–W693F/PsaB–W673F), the overall kinetics were retarded and two
distinct phases were well resolved (Fig. 4c), a slow one
with the same half-time as in the single PsaA–W693F mutant (≈490 ns)
and a faster one with a half-time of ≈73 ns (Table 1). The kinetics
in the PsaB–W673F mutant could be fit poorly with a single exponential
component (t1/2 = 110 ns). However,
the fit was significantly better if we assumed that two components were
still present in the kinetics of A
reduction and electrochromic shifts are small. The results for the two
symmetric mutants PsaA–W693F and PsaB–W673F and the double mutant
containing both mutations (PsaA–W693F/PsaB–W673F) are shown in Fig.
4. The mutation of the tryptophan in PsaA
affected specifically the kinetics of the slow nanosecond component,
increasing its half-time from ≈140 ns to ≈490 ns. The rate constant
of the fast component and the ratio between the amplitudes of the two
phases were the same as in the control strain (Fig. 4a;
Table 1). Mutation of the corresponding tryptophan in PsaB
(PsaB–W673F) slowed the initial part of the kinetics but did not
modify the later part, which is clearly seen in the conformity of the
latter part of the traces (Fig. 4b). In the double mutant
(PsaA–W693F/PsaB–W673F), the overall kinetics were retarded and two
distinct phases were well resolved (Fig. 4c), a slow one
with the same half-time as in the single PsaA–W693F mutant (≈490 ns)
and a faster one with a half-time of ≈73 ns (Table 1). The kinetics
in the PsaB–W673F mutant could be fit poorly with a single exponential
component (t1/2 = 110 ns). However,
the fit was significantly better if we assumed that two components were
still present in the kinetics of A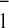 reoxidation and
constrained the half-times of the two components to match the modified
fast phase found in the double mutant and the slow phase of the wild
type (73 ns and 140 ns, Table 1). The combined results of the kinetics
deconvolution with all parameters free in the double mutant and the
constrained fit for the single mutant PsaB–W673F showed that the fast
phase (≈13 ns) of A
reoxidation and
constrained the half-times of the two components to match the modified
fast phase found in the double mutant and the slow phase of the wild
type (73 ns and 140 ns, Table 1). The combined results of the kinetics
deconvolution with all parameters free in the double mutant and the
constrained fit for the single mutant PsaB–W673F showed that the fast
phase (≈13 ns) of A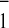 reoxidation was specifically
affected by mutation of the tryptophan W673 in PsaB polypeptide.
Similar values also were obtained when the electrochromic band shift
(480–457 nm) was used as a reporter of A
reoxidation was specifically
affected by mutation of the tryptophan W673 in PsaB polypeptide.
Similar values also were obtained when the electrochromic band shift
(480–457 nm) was used as a reporter of A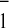 reoxidation (Table 1).
reoxidation (Table 1).
Figure 4.

Kinetics of A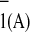 oxidation measured at 380 nm for
the control strain (■), PsaA–W693F mutant
(●), PsaB–W673F mutant (▴), and the
PsaA–W693F/PsaB–W673F double mutant (*). The solid lines are
theoretical fits with the parameters shown in Table 1. The dotted lines
are the extrapolation of the slow phase to show its amplitude in the
control strain and the PsaA–W693F mutant strain.
oxidation measured at 380 nm for
the control strain (■), PsaA–W693F mutant
(●), PsaB–W673F mutant (▴), and the
PsaA–W693F/PsaB–W673F double mutant (*). The solid lines are
theoretical fits with the parameters shown in Table 1. The dotted lines
are the extrapolation of the slow phase to show its amplitude in the
control strain and the PsaA–W693F mutant strain.
Mutation of the residues Glu-695 and Glu-698 in PsaA affected
specifically the kinetics of the slow phase although to a much smaller
extent than in the PsaA–W693F mutant (Table 1). Both glutamates are
located on the stromal helix a few residues from Trp-693, and it has
been suggested that their negatively charged side chains could
contribute to lowering the redox potential of the phylloquinone (23).
The kinetics of A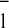 reoxidation in the double mutant
PsaA–W702F/PsaB–W682F, where the conserved tryptophans located
further along the stromal helix n/n′ were modified to phenylalanines,
were not significantly different from the control strain (Table 1).
This indicated that the effects observed upon mutation of the
Trp-693/Trp-673 pair are related to their proximity to the
phylloquinones and their specific interaction.
reoxidation in the double mutant
PsaA–W702F/PsaB–W682F, where the conserved tryptophans located
further along the stromal helix n/n′ were modified to phenylalanines,
were not significantly different from the control strain (Table 1).
This indicated that the effects observed upon mutation of the
Trp-693/Trp-673 pair are related to their proximity to the
phylloquinones and their specific interaction.
Fig. 5 compares the spectral features of
the two nanosecond decay components collected in the semiphylloquinone
absorption region for the control strain, PsaA–W693F, and the double
mutant PsaA–W693F/PsaB–W673F. Globally the fast component in the
control strain had relatively flat spectrum between 380 and 405 nm
whereas the spectrum of the slow component presented two bands with
different intensities, one at ≈380 nm and the other at ≈400 nm, the
ratio between them ≈1.4. In the PsaA–W693F mutant the spectrum and
the half-time of the fast component (17 ns) were similar to the control
strain. Interestingly, in the spectrum of the slow component the
position of the two bands were not modified but the ratio between them
decreased to ≈1.1. Thus the mutation in PsaA not only slowed down the
slow phase of A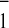 reoxidation but modified its
spectral properties. This modified spectrum, with a ratio
ΔA380/ΔA400 of ≈1.1
is seen again in the double mutant. Moreover, the spectrum of the
retarded fast decay is also changed with a net absorption increase at
400 nm. We could therefore attribute the similar spectral modification
(decrease of the ratio
ΔA380/ΔA400) to the
mutation of PsaB–Trp-673 to phenylalanine.
reoxidation but modified its
spectral properties. This modified spectrum, with a ratio
ΔA380/ΔA400 of ≈1.1
is seen again in the double mutant. Moreover, the spectrum of the
retarded fast decay is also changed with a net absorption increase at
400 nm. We could therefore attribute the similar spectral modification
(decrease of the ratio
ΔA380/ΔA400) to the
mutation of PsaB–Trp-673 to phenylalanine.
Figure 5.

Comparison of the spectra of the two nanosecond decay components in the region of semiphylloquinone absorption in the control strain (Left), PsaA–W693F (Center), and PsaA–W693F/PsaB–W673F (Right) mutants. The amplitudes of the components at the probe wavelengths, where the kinetics were measured, are shown as symbols. Closed symbols are the spectra of the fast phase, open symbols are the spectra of the slow phase.
Discussion
In this work we have studied the reoxidation kinetics of
A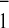 in whole cells of the green algae C.
reinhardtii. The spectra and kinetics associated with
phylloquinone reoxidation in C. reinhardtii were similar to
other green algae (13). We found that the two kinetic components of
A
in whole cells of the green algae C.
reinhardtii. The spectra and kinetics associated with
phylloquinone reoxidation in C. reinhardtii were similar to
other green algae (13). We found that the two kinetic components of
A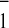 reoxidation can be modulated independently and
specifically by mutating Trp-693 in PsaA or Trp-673 in PsaB. The fact
that mutation of the tryptophans PsaA–W702 and PsaB–W682, which are
located at the end of the stromal helices n/n′, had no effect upon
phylloquinone reoxidation indicates that they are not in the proximal
environment of the phylloquinones. This is consistent with recent
high-resolution crystallographic data showing that both PsaA–W693 and
the symmetry-related PsaB–W673 are in contact with the phylloquinones
on their respective side (P. Jordan, P. Fromme, and N. Krauß, personal
communication). Therefore, we conclude that both quinones can be
reduced by PS I photochemistry and that both are reoxidized by
FX. Furthermore, we can assign the fast (≈13
ns) component to the reoxidation of the phylloquinone on the PsaB side
and the slow (≈140 ns) component to the reoxidation of the
phylloquinone on the PsaA side. The implication of these results is
that electron transfer in PS I, unlike type II reaction centers, is
bi-directional.
reoxidation can be modulated independently and
specifically by mutating Trp-693 in PsaA or Trp-673 in PsaB. The fact
that mutation of the tryptophans PsaA–W702 and PsaB–W682, which are
located at the end of the stromal helices n/n′, had no effect upon
phylloquinone reoxidation indicates that they are not in the proximal
environment of the phylloquinones. This is consistent with recent
high-resolution crystallographic data showing that both PsaA–W693 and
the symmetry-related PsaB–W673 are in contact with the phylloquinones
on their respective side (P. Jordan, P. Fromme, and N. Krauß, personal
communication). Therefore, we conclude that both quinones can be
reduced by PS I photochemistry and that both are reoxidized by
FX. Furthermore, we can assign the fast (≈13
ns) component to the reoxidation of the phylloquinone on the PsaB side
and the slow (≈140 ns) component to the reoxidation of the
phylloquinone on the PsaA side. The implication of these results is
that electron transfer in PS I, unlike type II reaction centers, is
bi-directional.
Our results strongly argue against both of the models that use a single
active branch. In the equilibrium model (Fig. 1b), it is not
possible to change the rate of only one of the phases while keeping the
other rate and the ratio between the phases constant. Likewise, the
structural heterogeneity model (Fig. 1c) would predict that
mutation of one of the tryptophans (the one next to the “inactive
quinone”) would have no effect on either phase. Mutations can exert
effects over relatively long distances, and one might invoke such
indirect effects to explain the phenotype of the mutation near the
inactive quinone. However, this would require a complex reasoning: a
mutation near the active quinone would have a direct effect upon it
(modifying the spectrum of the semiquinone and slowing its rate of
oxidation) only when the reaction center is in one conformation but not
in the second; at the same time, a mutation on the other side would act
over a relatively long distance to exert the exact same kind of effect
on the active quinone, but only when in the second conformation.
Therefore, we favor the simplest interpretation of our data, which is
that charge separation can proceed down either branch (Fig.
1d). We should make clear that our data do not directly
examine initial charge separation. However, if one posits that this
event were biased to a unique branch to produce a specific
P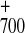 A
A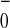 state, then one must explain
the reduction of the phylloquinone on the other branch.
A1 is reduced faster than the resolution time of
our instrument (5 ns), consistent with previous reports for
A0 reoxidation with a half time of a few tens of
picoseconds (28). Electron transfer between the two phylloquinones,
based on the distance between them (22.3 Å; ref. 2) and the
“Moser-Dutton ruler” (29), would be slower than 10 ms, which is
several orders of magnitude slower than A1
reoxidation. In a similar fashion we can rule out electron transfer
across branches at the level of A0 based on the
distances. The distance between eC3 and eC3′ in the PS I structure is
26.5 Å (2) and the distance from A0 to the
phylloquinone on the other branch is even longer.
state, then one must explain
the reduction of the phylloquinone on the other branch.
A1 is reduced faster than the resolution time of
our instrument (5 ns), consistent with previous reports for
A0 reoxidation with a half time of a few tens of
picoseconds (28). Electron transfer between the two phylloquinones,
based on the distance between them (22.3 Å; ref. 2) and the
“Moser-Dutton ruler” (29), would be slower than 10 ms, which is
several orders of magnitude slower than A1
reoxidation. In a similar fashion we can rule out electron transfer
across branches at the level of A0 based on the
distances. The distance between eC3 and eC3′ in the PS I structure is
26.5 Å (2) and the distance from A0 to the
phylloquinone on the other branch is even longer.
Thus, we conclude that the initial excited state
P*700 can evolve into either
P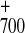 A
A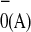 or
P
or
P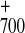 A
A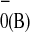 states, with
A0(A) and A0(B)
representing the analogous chlorophyll of the A or B branch,
respectively. Secondary electron transfer events (in the picosecond
time scale) then would lead to the states
P
states, with
A0(A) and A0(B)
representing the analogous chlorophyll of the A or B branch,
respectively. Secondary electron transfer events (in the picosecond
time scale) then would lead to the states
P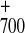 A
A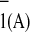 or
P
or
P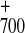 A
A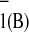 . The next stage, which is
the one that we observe in this study, is the tertiary electron
transfer leading to the singular state
P
. The next stage, which is
the one that we observe in this study, is the tertiary electron
transfer leading to the singular state
P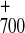 F
F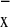 . Theoretically, we could
estimate the contribution of electron transfer down each branch by the
relative amplitude of each kinetic phase. If the amplitude of the
spectral changes associated with the reoxidation of the two
phylloquinones were identical, then we would estimate that electrons
are partitioned between each branch with a preference (55–66%) for
the one leading to the quinone on the PsaA side. However, these numbers
should not be considered rigorous, as the spectra displayed some
dissimilarity that could be interpreted as differences in the local
environment of each phylloquinone and/or different contributions of
the electrochromic band shifts. Some of these differences may be
explained by new crystallographic data, which reveal that the proximity
and orientations of the carotene(s) nearest to the phylloquinone on
either side are significantly different (P. Jordan, P. Fromme, and N.
Krauß, personal communication).
. Theoretically, we could
estimate the contribution of electron transfer down each branch by the
relative amplitude of each kinetic phase. If the amplitude of the
spectral changes associated with the reoxidation of the two
phylloquinones were identical, then we would estimate that electrons
are partitioned between each branch with a preference (55–66%) for
the one leading to the quinone on the PsaA side. However, these numbers
should not be considered rigorous, as the spectra displayed some
dissimilarity that could be interpreted as differences in the local
environment of each phylloquinone and/or different contributions of
the electrochromic band shifts. Some of these differences may be
explained by new crystallographic data, which reveal that the proximity
and orientations of the carotene(s) nearest to the phylloquinone on
either side are significantly different (P. Jordan, P. Fromme, and N.
Krauß, personal communication).
It is worth noting that mutation of each tryptophan had a similar effect on the quinone close to it. In both cases, conversion to a phenylalanine had two effects on one of the kinetic phases: a modification of the decay-associated spectrum and a 3- to 5-fold decrease in the rate of its decay. The spectral change was also similar in both cases, manifesting itself as an increase in absorbance at ≈400 nm. Two possible explanations that are not mutually exclusive could be given for this change: (i) Because the Trp residues (PsaA–Trp-693 and PsaB–Trp-673) are involved in a π-stacking interaction with the phylloquinones, as previously suggested based on electron spin echo envelope modulation spectroscopy (23), their mutation to Phe will likely perturb the electron coupling between the quinone ring and the aromatic side chain leading to a different absorption properties of A1. (ii) The mutation could lead to a change in the local environment of the phylloquinone, and thus to slightly different interactions with the neighboring chromophores, most probably chlorophylls, modifying the electrochromic bandshifts.
The change in the rate of electron transfer (ket) from the quinone to FX in the mutant strains can be easier to explain in the context of the empirical equation derived by Moser et al. (29)
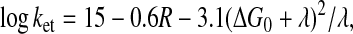 |
where R is the edge-to-edge distance between
A1 and FX in Å, and
ΔG0 and λ are the standard free energy and
the reorganization energy of the reaction, respectively. One or more of
the terms in this equation could have been modified in the mutant
strains. It is possible that the altered phylloquinone-binding site
causes a slight increase in the distance between the quinone and
FX and/or orientation of the quinone. A 3–5
time decrease in the rate of electron transfer would correspond to an
increase of ≈0.9 Å in R if all other parameters were
constant. Similarly the mutation can lead to an increase of either
ΔG0 or λ, and thus result in a lower rate of
electron transfer. The unusually low midpoint potential of
A1/A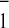 has been attributed to
its very hydrophobic environment and/or specific interactions with
the phylloquinone in PS I (30). Modification of either or both may
occur when the indole ring system of the Trp is exchanged for the
smaller phenyl side chain of Phe. The π-stacking interaction of the
side chain to the quinone could have been perturbed or the binding site
rendered more exposed to aqueous solvent. This would tend to stabilize
the semiquinone radical anion, thus decreasing driving force, and force
the movement of nearby polar molecules in response to the change in
charge during electron transfer, thus increasing the reorganization
energy. If the nearby conserved glutamates (695/675 and 698/678)
are deprotonated, then their negative charge might contribute to this
effect, and conversion to the uncharged glutamine would stabilize the
semiquinone anion and slow the rate of transfer to
FX. We did observe ≈2-fold and ≈1.5-fold
decreases in the rate of reoxidation of A
has been attributed to
its very hydrophobic environment and/or specific interactions with
the phylloquinone in PS I (30). Modification of either or both may
occur when the indole ring system of the Trp is exchanged for the
smaller phenyl side chain of Phe. The π-stacking interaction of the
side chain to the quinone could have been perturbed or the binding site
rendered more exposed to aqueous solvent. This would tend to stabilize
the semiquinone radical anion, thus decreasing driving force, and force
the movement of nearby polar molecules in response to the change in
charge during electron transfer, thus increasing the reorganization
energy. If the nearby conserved glutamates (695/675 and 698/678)
are deprotonated, then their negative charge might contribute to this
effect, and conversion to the uncharged glutamine would stabilize the
semiquinone anion and slow the rate of transfer to
FX. We did observe ≈2-fold and ≈1.5-fold
decreases in the rate of reoxidation of A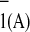 upon
mutation of the nearer (Glu-695) and farther (Glu-698) glutamic acid
residues of PsaA, lending support to this idea. However, these modest
changes may indicate that these carboxylates may be either relatively
far from the quinone or at least partially shielded by counter ions.
upon
mutation of the nearer (Glu-695) and farther (Glu-698) glutamic acid
residues of PsaA, lending support to this idea. However, these modest
changes may indicate that these carboxylates may be either relatively
far from the quinone or at least partially shielded by counter ions.
Reasoning similar to that above can be applied to explain the
difference in the rate of reoxidation of A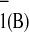 as
compared with that of A
as
compared with that of A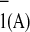 . However, a difference in
the distance between the two quinones and FX
seems less likely because an order of magnitude faster rate would
require 1.7-Å shorter distance on the PsaB branch. All available
structural data indicate equivalent positions for both quinones with
respect to the symmetry axis and thus to FX (2).
Therefore we would tend to attribute the difference in rate to a
difference in the amino acid compositions of the two phylloquinone
sites. To that end, we are now examining mutations in several conserved
amino acids (such as tryptophans and glutamates) that are not shared
between PsaA and PsaB.
. However, a difference in
the distance between the two quinones and FX
seems less likely because an order of magnitude faster rate would
require 1.7-Å shorter distance on the PsaB branch. All available
structural data indicate equivalent positions for both quinones with
respect to the symmetry axis and thus to FX (2).
Therefore we would tend to attribute the difference in rate to a
difference in the amino acid compositions of the two phylloquinone
sites. To that end, we are now examining mutations in several conserved
amino acids (such as tryptophans and glutamates) that are not shared
between PsaA and PsaB.
The presence of bi-directional electron transfer to quinones in
slightly different environments, leading to unequal rates of transfer
to the next cofactor immediately begs the question: what, if any, is
the functional significance of this arrangement? Because the primeval
reaction center was almost certainly homodimeric (discussed in refs. 3
and 31), and this situation persists in PS I's cousins found in green
sulfur bacteria (5) and heliobacteria (4), it is likely that the
original mode of electron transfer was bi-directional. The
heterodimeric type II reaction centers, which use one quinone
(QA) as a secondary acceptor (like
A1) and the other (QB) as a
terminal acceptor, have evolved to eliminate electron transfer directly
to QB. Thus, QA serves as a
“one-electron gate,” while QB is doubly
reduced, and electron transfer from QA to
QB is coupled to protonation of
QB to the quinol form, an essential part of its
function. In contrast, no such selection pressure was necessary in type
I reaction centers, where the semiquinone on one branch donates its
electron to a cofactor that sits in the symmetry axis, and it makes no
difference from which side it came. Reduction of ferredoxin by PS I is
limited by electron transfer from the iron-sulfur clusters, and
therefore the slower rate from A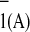 to
FX (t1/2
≈ 140 ns) would not restrict overall electron transfer. We also
should note that the PsaB–W673F mutant, in which the rates of electron
transfer from the two phylloquinones to FX are
almost equivalent, shows no observable defects in photosynthesis
in vivo and is indistinguishable from the wild type.
However, because PS I has a large antenna (100 chlorophyll molecules)
and the kinetics for
to
FX (t1/2
≈ 140 ns) would not restrict overall electron transfer. We also
should note that the PsaB–W673F mutant, in which the rates of electron
transfer from the two phylloquinones to FX are
almost equivalent, shows no observable defects in photosynthesis
in vivo and is indistinguishable from the wild type.
However, because PS I has a large antenna (100 chlorophyll molecules)
and the kinetics for 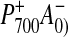 formation are on the same time scale as excitation energy transfer (1),
the presence of two branches for initial charge separation would double
the rate of exciton trapping and thus increase the efficiency of PS I
charge separation. Further studies in which the environment of
P700 and the primary electron acceptor(s) in PsaA
and PsaB is modified may help to answer the question of whether or not
the existence of two electron transfer pathways has functional
implications.
formation are on the same time scale as excitation energy transfer (1),
the presence of two branches for initial charge separation would double
the rate of exciton trapping and thus increase the efficiency of PS I
charge separation. Further studies in which the environment of
P700 and the primary electron acceptor(s) in PsaA
and PsaB is modified may help to answer the question of whether or not
the existence of two electron transfer pathways has functional
implications.
Acknowledgments
We thank F. Rappaport, R. Kuras, R. Metzger, and D. Oppenheimer for critical reading of the manuscript. K.R. acknowledges support from DuPont through a DuPont Young Professor Grant and the U.S. Department of Energy through an Energy Biosciences Grant. M.G.-K., A.J., and P.J. acknowledge support from the Centre National de la Recherche Scientifique and the Collège de France.
Abbreviations
- EPR
electron paramagnetic resonance
- FX
FA, and FB, iron-sulfur clusters of photosystem I
- PS
photosystem
- P700
primary electron donor in PS I
- A0
primary electron acceptor in PS I (chlorophyll)
- A1
secondary electron acceptor in PS I (phylloquinone)
References
- 1.Brettel K. Biochim Biophys Acta. 1997;1318:322–373. [Google Scholar]
- 2.Klukas O, Schubert W D, Jordan P, Krauß N, Fromme P, Witt H T, Saenger W. J Biol Chem. 1999;274:7361–7367. doi: 10.1074/jbc.274.11.7361. [DOI] [PubMed] [Google Scholar]
- 3.Golbeck J H. Proc Natl Acad Sci USA. 1993;90:1642–1646. doi: 10.1073/pnas.90.5.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liebl U, Mockensturm-Wilson M, Trost J T, Brune D C, Blankenship R E, Vermaas W. Proc Natl Acad Sci USA. 1993;90:7124–7128. doi: 10.1073/pnas.90.15.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buttner M, Xie D L, Nelson H, Pinther W, Hauska G, Nelson N. Biochim Biophys Acta. 1992;1101:154–156. [PubMed] [Google Scholar]
- 6.Schubert W D, Klukas O, Saenger W, Witt H T, Fromme P, Krauß N. J Mol Biol. 1998;280:297–314. doi: 10.1006/jmbi.1998.1824. [DOI] [PubMed] [Google Scholar]
- 7.MacMillan F, Hanley J, van der Weerd L, Knupling M, Un S, Rutherford A W. Biochemistry. 1997;36:9297–9303. doi: 10.1021/bi971097d. [DOI] [PubMed] [Google Scholar]
- 8.Yang F, Shen G, Schlucter W M, Zybailov B L, Ganago A O, Vassiliev I R, Bryant D A, Golbeck J H. J Phys Chem B. 1998;102:8288–8299. [Google Scholar]
- 9.Heathcote P, Hanley J A, Evans M C W. Biochim Biophys Acta. 1993;1144:54–61. [Google Scholar]
- 10.Sieckmann I, van der Est A, Bottin H, Sétif P, Stehlik D. FEBS Lett. 1991;284:98–102. doi: 10.1016/0014-5793(91)80771-t. [DOI] [PubMed] [Google Scholar]
- 11.van der Est A, Bock C, Golbeck J, Brettel K, Setif P, Stehlik D. Biochemistry. 1994;33:11789–11797. doi: 10.1021/bi00205a015. [DOI] [PubMed] [Google Scholar]
- 12.Setif P, Brettel K. Biochemistry. 1993;32:7846–7854. doi: 10.1021/bi00082a002. [DOI] [PubMed] [Google Scholar]
- 13.Joliot P, Joliot A. Biochemistry. 1999;38:11130–11136. doi: 10.1021/bi990857c. [DOI] [PubMed] [Google Scholar]
- 14.Brettel K, Golbeck J H. Photosynth Res. 1995;45:183–193. doi: 10.1007/BF00015559. [DOI] [PubMed] [Google Scholar]
- 15.Redding K, MacMillan F, Leibl W, Brettel K, Rutherford A W, Breton J, Rochaix J-D. In: Photosynthesis: Mechanisms and Effects. Garab G, editor. Dordrecht, The Netherlands: Kluwer; 1998. pp. 591–594. [Google Scholar]
- 16.Bennoun P, Spierer-Herz M, Erickson J, Girard-Bascou J, Pierre Y, Delosme M, Rochaix J D. Plant Mol Biol. 1986;6:151–160. doi: 10.1007/BF00021484. [DOI] [PubMed] [Google Scholar]
- 17.Allen K D, Staehelin L A. Planta. 1994;194:42–54. [Google Scholar]
- 18.Kuchka M R, Goldschmidt-Clermont M, van Dillewijn J, Rochaix J D. Cell. 1989;58:869–876. doi: 10.1016/0092-8674(89)90939-2. [DOI] [PubMed] [Google Scholar]
- 19.Béal D, Rappaport F, Joliot P. Rev Sci Instrum. 1999;70:202–207. [Google Scholar]
- 20.Harris E H. The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use. San Diego: Academic; 1989. [DOI] [PubMed] [Google Scholar]
- 21.Muller K-H, Plesser T. Eur Biophys J. 1991;19:231–240. [Google Scholar]
- 22.Schubert W D, Klukas O, Krauß N, Saenger W, Fromme P, Witt H T. J Mol Biol. 1997;272:741–769. doi: 10.1006/jmbi.1997.1269. [DOI] [PubMed] [Google Scholar]
- 23.Hanley J, Deligiannakis Y, MacMillan F, Bottin H, Rutherford A W. Biochemistry. 1997;36:11543–11549. doi: 10.1021/bi971360a. [DOI] [PubMed] [Google Scholar]
- 24.Hildreth W W. Arch Biochem Biophys. 1970;139:1–8. doi: 10.1016/0003-9861(70)90038-x. [DOI] [PubMed] [Google Scholar]
- 25.Ke B. Arch Biochem Biophys. 1972;152:70–77. doi: 10.1016/0003-9861(72)90194-4. [DOI] [PubMed] [Google Scholar]
- 26.Delosme R. Photosynth Res. 1991;29:45–54. doi: 10.1007/BF00035205. [DOI] [PubMed] [Google Scholar]
- 27.Brettel K. FEBS Lett. 1988;239:93–98. [Google Scholar]
- 28.Hastings G, Kleinherenbrink F A, Lin S, McHugh T J, Blankenship R E. Biochemistry. 1994;33:3193–3200. doi: 10.1021/bi00177a008. [DOI] [PubMed] [Google Scholar]
- 29.Moser C C, Keske J M, Warncke K, Farid R S, Dutton P L. Nature (London) 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 30.Iwaki M, Itoh S. Biochemistry. 1991;30:5347–5352. doi: 10.1021/bi00236a004. [DOI] [PubMed] [Google Scholar]
- 31.Nitschke W, Matteoli T, Rutherford A W. In: Origin and Evolution of Biological Energy Conversion. Baltcheffsky M, editor. New York: VCH; 1996. pp. 177–203. [Google Scholar]