

Abstract
A large body of evidence indicates that polybrominated diphenyl ether (PBDE) flame retardants have become widespread environmental pollutants. Body burden is particularly high in infants and toddlers, due to exposure through maternal milk and house dust. Animal studies suggest that PBDEs may exert developmental neurotoxicity, via mechanisms that are still elusive. PBDEs have been reported to cause oxidative stress and apoptotic cell death in neurons in vitro, when tested in mono-cultures. Here we report the results of experiments in which mouse cerebellar granule neurons (CGNs) were co-cultured with cerebellar astrocytes. Astrocytes were found to protect neurons against the toxicity of the PBDE mixture DE-71. Astrocytes from Gclm (−/−) mice, which lack the modifier subunit of glutamate cysteine ligase and, as a consequence, have very low GSH levels, were much less effective at protecting CGNs from DE-71 toxicity. The protective effects was mostly due to the ability of Gclm (+/+) astrocytes to increase GSH levels in neurons. By increasing GSH, GSH ethylester provided a similar protective effect. In vivo, where both neurons and astrocytes would be either Gclm (+/+) or Gclm (−/−), the toxicity of DE-71 to CGNs is predicted to vary 16.8-fold, depending on genotype. Hence, in addition to being intrinsically more susceptible to DE-71 toxicity because of their low GSH content, CGNs in Gclm (−/−) mice would also lack the full protective effect provided by astrocytes. Since several polymorphisms, including some in the Gclm gene, cause very low levels of GSH, it may be speculated that such individuals might display a higher susceptibility to the neurotoxic effects of PBDEs.
Introduction
Polybrominated diphenyl ethers (PBDEs) are a class of flame retardants that have been extensively used in the past thirty years, particularly in textiles, carpets, television sets, computers and small appliances. Since they are not fixed to the polymer product through chemical binding, PBDEs can leak into the environment, and have become persistent environmental pollutants (deWit, 2002; Hale et al. 2003; Law et al. 2006). PBDEs have been found in a wide variety of species, including humans, where the highest body burden is found in infants and toddlers, because of their exposure through maternal milk and house dust (McDonald, 2005; Fischer et al. 2006; Zuurbier et al. 2006; Lorber, 2008). PBDEs can cross the placenta, and similar concentrations are found in maternal and fetal blood (Mazdai et al. 2003; Antignac et al. 2008).
The high exposure to PBDEs during development has raised concerns regarding their potential developmental toxicity. Recent evidence suggests that PBDEs may be developmental neurotoxicants (Branchi et al. 2003; Birnbaum and Staskal, 2004; McDonald, 2005; Costa and Giordano, 2007), as indicated by animal studies in which pre- or post-natal exposure to various PBDEs was found to cause behavioral alterations particularly in the domains of locomotor activity and cognition (Eriksson et al. 2001; Branchi et al. 2002; Viberg et al. 2003; 2006; Dufault et al. 2005; Rice et al. 2007, Gee and Moser, 2008; Onos et al. 2008).
Two modes of action, that are not necessarily mutually exclusive, are being suggested as possible mechanisms underlying the developmental neurotoxicity of PBDEs, one related to an impairment of thyroid hormone homeostasis, the other involving direct effects of PBDEs on neuronal and/or glial cells (Zhou et al. 2002; Costa and Giordano, 2007). Some in vitro studies have shown that PBDEs can affect signal transduction pathways, such as protein kinase C or calcium homeostasis (e.g. Kodavanti and Ward, 2005; Coburn et al. 2008; Dingemans et al. 2008), while others have indicated that these compounds may cause apoptotic cell death of neurons, by mechanisms that involve oxidative stress (e.g. Reistad et al. 2006; He et al. 2008a;b).
We recently reported that the PBDE mixture DE-71 caused oxidative stress and apoptosis in mouse neurons and astrocytes, and that these effects were modulated by intracellular glutathione (GSH) levels (Giordano et al. 2008). GSH is found at higher levels in astrocytes than in neurons (Rice and Russo-Menna, 1998; Giordano et al. 2008), and indeed DE-71 toxicity is greater in neurons than astrocytes (Giordano et al. 2008). In the brain, however, astrocytes are in close proximity to neurons. Since neurotoxicity of PBDEs has been so far investigated in neurons or astrocytes in mono-culture, the present study aimed at determining the neurotoxicity of DE-71 in co-cultures of mouse cerebellar astrocytes and cerebellar granule neurons (CGNs).
Material and methods
Materials
DE-71 (Lot # 05500F16P) was purchased from Wellington Laboratories (Guelph, ON, Canada). The composition of DE-71 is reported as follows: BDE-99, 44%; BDE-47, 32%; BDE-100, 9%; BDE-153, 4%; other PBDEs, 11%. Other DE-71 mixtures have been reported to contain detectable amounts of polybrominated dibenzofurans and polybrominated dibenzodioxins (Hanari et al. 2006; Sanders et al. 2005). None were reported by the vendor, and no chemical analysis of the DE-71 used in this study was carried out. Dimethylsulfoxide (DMSO) and 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Neurobasal-A medium, fetal bovine serum (FBS) and gentamycin were from Invitrogen (Carlsbad, CA, USA).
Generation of Gclm-null mice and genotyping
All procedures for animal use were in accordance with the National Institute of Health Guide for the Use and Care of Laboratory Animals, and were approved by the University of Washington Animal Care and Use Committee. Gclm-null [Gclm (−/−)] mice were derived by homologous recombination techniques in mouse embryonic stem cells, as previously described in detail (Giordano et al. 2006; McConnachie et al. 2007). Pups were genotyped as described by Giordano et al. (2006).
Cultures of cerebellar granule neurons and cerebellar astrocytes
Cultures of cerebellar granule neurons (CGN) were prepared from 7 day-old mice, as described by Giordano et al. (2006). Neurons were grown for 10–12 days before treatments. Primary mouse astrocytes were obtained from PND 7 cerebellum, as previously described by Giordano et al. (2006). After 10 days in culture, cells were plated in 24-well plates for the experiments at the density of 5×104 astrocytes/well.
Co-cultures of neurons and astrocytes, cell treatments and cytotoxicity assay
Astrocytes were cultured as described above. After ten days, astrocytes were dissociated with 0.25% trypsin and 0.1% DNase in Hanks’ balanced salt solution and subcultured on permeable membranes (inserts) for 5 days. To maintain the correct hydrostatic pressure across the insert membrane, volumes in the co-cultures were regulated such that 15% of the total incubation volume was inside the insert and 85% in the well. DE-71 was dissolved in DMSO to obtain a stock solution of 25 mM, which was diluted appropriately in medium at the time of use. Three to five different concentrations of DE-71, in triplicate, were used to allow determination of IC50 values. Co-culture treatment was performed as described by Morken et al. (2005) with minor modifications. Briefly, the inserts containing cultured astrocytes were transferred to the neuronal plates, containing cultured neurons, 24 h prior to incubation with DE-71. At time of insert transfer, half of the neuronal medium was replaced with astrocyte medium. The ratio of astrocytes to neurons was 2.5–3.0 is all experiments. Cell viability was quantified by a colorimetric method utilizing the metabolic dye 3-(4,5- dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT), as previously described (Giordano et al. 2006; 2008).
Measurement of GSH levels
Intracellular GSH levels were measured spectrophotometrically, as described by Giordano et al. (2006).
Statistical Analysis
Data are expressed as the mean ± SD of at least three independent experiments. IC50 values were calculated from a concentration-response curve with 3–5 concentrations of DE-71 (GraphPad Prism Software), using a non-linear regression with a sigmoidal fit model. Statistical analysis was carried out by one way ANOVA followed by Bonferroni’s multiple comparison test.
Results and discussion
In the present study we utilized CGNs and cerebellar astrocytes from Gclm (+/+) and Gclm (−/−) mice. The latter lack the modifier subunit of glutamate cysteine ligase (GCL) the first and rate-limiting step in the synthesis of GSH (Dringen, 2000). In the absence of GCLM, the efficiency of the catalytic subunit GCLC to synthesize GSH is drastically impaired, resulting in GSH levels that are 9–20% of those found in Gclm (+/+) animals (Yang et al. 2002; Giordano et al. 2006; 2008; McConnachie et al. 2007). We had previously found that the toxicity of DE-71 was enhanced in neurons (CGNs and others) from Gclm (−/−) mice, due to their low GSH content (Giordano et al. 2008). In the co-culturing system utilized in the present study, astrocytes were plated on poly-D-lysine coated, permeable membranes, and allowed to attach. Inserts were then transferred onto a neuron monolayer. The co-culture was then treated with different concentrations of DE-71. Cytotoxicity in neurons and astrocytes can be assessed separately in this system, thus allowing determination of the possible protective role of astrocytes toward neurons.
As shown in Table 1, DE-71 decreased the viability of mouse CGNs from Gclm (+/+) mice (in mono-culture), with an IC50 of 7.2 μM. When CGNs were co-cultured with cerebellar astrocytes from Gclm (+/+) mice, the toxicity of DE-71 was decreased by fivefold, resulting in an IC50 of 38.7 μM. In contrast, cerebellar astrocytes from GClm (−/−) mice were less effective at protecting Gclm (+/+) CGNs, as the IC50 of DE-71 increased by only two-fold to 13.8 μM (Table 1).
Table 1.
Neurotoxicity of DE-71 in CGNs co-cultured with cerebellar astrocytes
| Astrocytes | CGNs | IC50 (uM) |
|---|---|---|
| None | Gclm (+/+) | 7.2 ± 0.8 |
| Gclm (+/+) | Gclm (+/+) | 38.7 ± 3.6**## |
| Gclm (−/−) | Gclm (+/+) | 13.8 ± 1.6** |
| None | Gclm (−/−) | 1.1 ± 0.4 |
| Gclm (+/+) | Gclm (−/−) | 8.1 ± 0.7* # |
| Gclm (−/−) | Gclm (−/−) | 2.3 ± 0.6 |
Values represent IC50 (uM) in the MTT assay, derived from concentration-response curves obtained with 3–4 concentrations of DE-71, and are the means (± SD) of three separate determinations.
Significantly different from the respective CGNs alone, p<0.01;
p<0.001.
Significantly different from the respective co-cultures with Gclm (−/−) astrocytes, p<0.01;
p<0.001.
As previously reported (Giordano et al. 2008), CGNs from Gclm (−/−) mice were significantly more susceptible to DE-71 toxicity (IC50 = 1.1 μM). Cerebellar astrocytes from Gclm (+/+) mice, co-cultured with Gclm (−/−) CGNs, provided a high degree of protection (almost eight-fold). In contrast, astrocytes from Gclm (−/−) mice were much less protective, and the IC50 of DE-71 increased only by two-fold to 2.3 μM (Table 1).
The toxicity of DE-71 toward cerebellar astrocytes was lower than in neurons, and was higher in astrocytes from Gclm (−/−) animals than in astrocytes from Gclm (+/+) animals (Table 2). The presence of CGNs of either genotype did not have any effect on the susceptibility of cerebellar astrocytes to DE-71 (Table 2).
Table 2.
Neurotoxicity of DE-71 in cerebellar astrocytes co-cultured with CGNs
| CGNs | Astrocytes | IC50 (uM) |
|---|---|---|
| None | Gclm (+/+) | 41.1 ± 3.0 |
| Gclm (+/+) | Gclm (+/+) | 42.9 ± 3.6 |
| Gclm (−/−) | Gclm (+/+) | 38.0 ± 2.5 |
| None | Gclm (−/−) | 12.7 ± 2.6 |
| Gclm (+/+) | Gclm (−/−) | 14.8 ± 1.4 |
| Gclm (−/−) | Gclm (−/−) | 12.2 ± 2.3 |
Values represent IC50 (uM) in the MTT assay, derived from concentration-response curves obtained with 3–4 concentrations of DE-71, and are the means (± SD) of three separate determinations. All results in Gclm (−/−) astrocytes were significantly different from those in Gclm (+/+) astrocytes (p<0.01). There were no significant differences within astrocyte genotypes.
These findings indicate that astrocytes can protect neurons toward DE-71 toxicity, and that the degree of protection is dependent upon the GSH content of the astrocytes. A mechanism involved in this protective effect may be represented by a lower accumulation of DE-71 in CGNs as a consequence of the presence of astrocytes, as has been shown for example in case of methyl mercury (Morken et al. 2005). However, this would not explain why astrocytes from Gclm (−/−) mice are less protective than those of Gclm (+/+) mice.
As previously reported (Giordano et al. 2008), DE-71 toxicity in CGNs is primarily represented by oxidative stress-mediated apoptosis. Astrocytes have been shown to protect neurons against oxidative stress by providing GSH. Cystine is taken up by astrocytes and converted to cysteine, which serves for the synthesis of GSH; GSH is then released from astrocytes, and metabolized by γ-glutamyl transpeptidase (GGT) to cysteinylglycine, from which cysteine is released by an endopeptidase located on the neuronal membrane surface. Cysteine is then taken up by neurons through the EAAC1 transporter (Aoyama et al. 2006) and utilized for GSH synthesis. Astrocytes have also been shown to induce transcriptional up-regulation of neuronal GSH through the release of still unidentified factors (Iwata-Ichikawa et al. 1999). Furthermore, GGT has also been shown to transfer the γ-glutamyl moiety of GSH to extracellular cystine to form γ-glutamylcystine, which can be taken up into cells, reduced to γ-glutamylcysteine, and used by GSH synthethase to synthesize GSH, thus bypassing glutamate-cysteine ligase (GCL; Anderson and Meister, 1983; Chinta et al. 2006).
To test the hypothesis that astrocytes may protect neurons by increasing their GSH content, we measured GSH levels in CGNs cultured alone or in the presence of astrocytes. As shown in Table 3, Gclm (+/+) astrocytes were able to significantly increase GSH levels in CGNs of both genotypes. In contrast, Gclm (−/−) astrocytes were much less effective in this regard. The astrocyte-induced increase in CGNs’ GSH content was also observed when the co-cultures were exposed to DE-71, which, alone, did not alter GSH content in CGNs (Table 3). To further probe the hypothesis that a major mechanism of CGN protection by astrocytes is the ability of the latter to increase GSH levels in neurons, CGNs alone or co-cultured with astrocytes were treated with DE-71 in the presence of GSH ethylester (GSHee). We had previously shown that GSHee (at 2.5 mM) significantly increases GSH levels in CGNs of both mouse genotypes (Giordano et al. 2006), and this was confirmed in the present study (not shown). Fig.1 shows that treatment of CGNs, cultured alone or with astrocytes, with GSHee, protected them from the cytotoxicity of DE-71.
Table 3.
Total intracellular GSH levels in CGNs cultured alone or with cerebellar astrocytes
| Astrocytes | CGNs | Control | +DE-71 |
|---|---|---|---|
| None | Gclm (+/+) | 13.5 ± 1.0 | 11.7 ± 1.5 |
| Gclm (+/+) | Gclm (+/+) | 17.9 ± 1.1 * | 18.6 ± 1.9* |
| Gclm (−/−) | Gclm (+/+) | 15.2 ± 1.4 | 14.6 ± 1.4 |
| None | Gclm (−/−) | 2.6 ± 0.7 | 3.1 ± 0.9 |
| Gclm (+/+) | Gclm (−/−) | 8.3 ± 1.1** | 6.7 ± 0.8* |
| Gclm (−/−) | Gclm (−/−) | 3.8 ± 0.8 | 3.5 ± 0.7 |
Astrocytes and neurons were co-cultured as described in Methods and treated for 24 hr with DE-71 (10 μM). Total intracellular GSH levels were measured as described by Giordano et al. (2006). Values are expressed as nmol/mg protein, and results represent the mean (± SD) of three separate experiments.
Significantly different from CGNs alone (without astrocytes), p<0.01;
p<0.001.
Fig. 1.
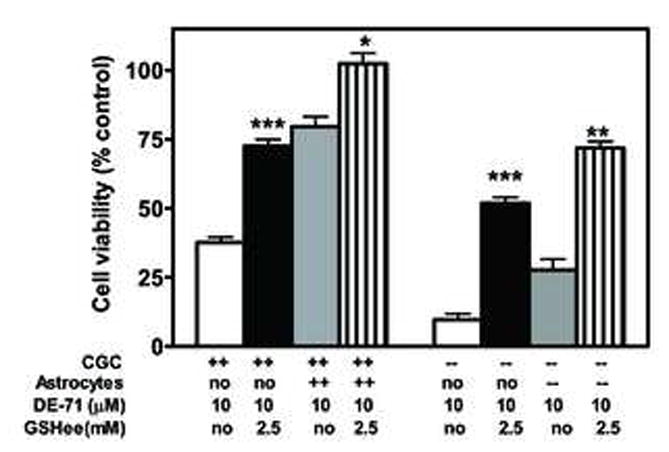
Glutathione ethyl ester (GSHee) protects CGNs in monoculture and in co-culture from neurotoxicity induced by DE-71. Astrocytes and neurons were co-cultured as described in methods and treated for 24 hr with DE-71 (10 μM) alone, or after a 30 min pre-incubation with GSHee (2.5mM). Citotoxicity was assessed by the MTT assay. Results represent the mean (± SD) of three separate determinations. *Significantly different from DE-71 in the absence of GSHee, p<0.05; **, p<0.01; ***, p<0.001.
Altogether, these results confirm that intracellular GSH content is a most important determinant of CGN susceptibility to DE-71 neurotoxicity. By providing GSH, astrocytes protect neurons against the oxidative stress-mediated toxicity of DE-71. However, additional minor mechanisms may also be involved in the protective effect, as suggested, for example, by the finding that Gclm (−/−) astrocytes provided a 2-fold protection of Gclm (−/−) CGNs, without significantly increasing their GSH levels (Tables 1 and 3).
In vivo, both neurons and astrocytes would be either Gclm (+/+) or Gclm (−/−). Thus, in an in vitro system expected to mimic the in vivo situation (where both neurons and astrocytes are present) the toxicity of DE-71 to CGNs would vary from 38.7 to 2.3 μM (ratio = 16.8), depending on genotype. Hence, in addition to being intrinsically more susceptible to DE-71 toxicity because of their low GSH content, CGNs in Gclm (−/−) mice would also lack a full protective effect provided by astrocytes. This may be of relevance for individuals with genetic predispositions causing low GSH levels, as they may display a higher susceptibility to PBDE developmental neurotoxicity. Indeed, several polymorphisms in GCL have been described (Dalton et al. 2004; Botta et al. 2008), including some in the Gclm gene, that are associated with low levels of GSH (Nakamura et al. 2002).
Acknowledgments
This study was supported in part by grant P30ES07033 from the National Institute for Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson ME, Meister A. Transport and direct utilization of γ-glutamylcyst(e)ine for glutathione synthesis. Proc Natl Acad Sci USA. 1983;80:707–711. doi: 10.1073/pnas.80.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antignac JP, Cariou R, Maume D, Marchand P, Monteau F, Zalko D, Berrebi A, Cravedi JP, Andre F, Le Bizec B. Exposure assessment of fetus and newborn to brominated flame retardants in France: preliminary data. Mol. Nutr. Food Res. 2008;52:258–265. doi: 10.1002/mnfr.200700077. [DOI] [PubMed] [Google Scholar]
- Ayoama K, Suh SW, Hamby AM, Liu J, Chan J, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nature Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS, Staskal DF. Brominated flame retardants: cause for concern? Environ Health Perspect. 2004;112:9–17. doi: 10.1289/ehp.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta D, White CC, Vliet-Gregg P, Mohar I, Shi S, McGrath MB, McConnachie LA, Kavanagh TJ. Modulating GSH synthesis using glutamate cysteine ligase transgenic and gene-targeted mice. Drug Metab Rev. 2008;40:465–477. doi: 10.1080/03602530802186587. [DOI] [PubMed] [Google Scholar]
- Branchi I, Alleva E, Costa LG. Effects of perinatal exposure to a polybrominated diphenyl ether (PBDE 99) on mouse neurobehavioral development. Neurotoxicology. 2002;23:375–384. doi: 10.1016/s0161-813x(02)00078-5. [DOI] [PubMed] [Google Scholar]
- Branchi I, Capone F, Alleva E, Costa LG. Polybrominated diphenyl ethers: neurobehavioral effects following developmental exposure. Neurotoxicology. 2003;24:449–462. doi: 10.1016/S0161-813X(03)00020-2. [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Kumar JM, Zhang H, Forman HJ, Andersen JK. Up-regulation of gamma-glutamyl transpeptidase activity following glutathione depletion has a compensatory rather than an inhibitory effect on mitochondrial complex I activity: implications for Parkinson’s disease. Free Rad Biol Med. 2006;40:1557–1563. doi: 10.1016/j.freeradbiomed.2005.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn CG, Curraz-Collazo MC, Kodavanti PRS. In vitro effects of environmentally relevant polybrominated diphenyl ether (PBDE) congeners on calcium buffering mechanisms in rat brain. Neurochem Res. 2008;33:355–364. doi: 10.1007/s11064-007-9430-x. [DOI] [PubMed] [Google Scholar]
- Costa LG, Giordano G. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology. 2007;28:1047–1067. doi: 10.1016/j.neuro.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Rad Biol Med. 2004;37:1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- de Wit CA. An overview of brominated flame retardants in the environment. Chemosphere. 2002;46:583–624. doi: 10.1016/s0045-6535(01)00225-9. [DOI] [PubMed] [Google Scholar]
- Dingemans MML, de Groot A, van Kleef RGDM, Bergman A, van der Berg M, Vijverberg HPM, Westerink RHS. Hydroxylation increases the neurotoxic potential of BDE-47 to affect exocytosis and calcium homeostasis in PC12 cells. Environ Health Perspect. 2008;116:637–643. doi: 10.1289/ehp.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R. Metabolism and function of glutathione in brain. Progr Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Dufault C, Poles G, Driscoll LL. Brief postnatal PBDE exposure alters learning and the cholinergic modulation of attention in rats. Toxicol Sci. 2005;88:172–180. doi: 10.1093/toxsci/kfi285. [DOI] [PubMed] [Google Scholar]
- Eriksson P, Jakobson E, Fredriksson A. Brominated flame retardants: a novel class of developmental neurotoxicants in our environment? Environ Health Perspect. 2001;109:903–908. doi: 10.1289/ehp.01109903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Hooper K, Athanasiadou M, Athanassiadis I, Bergman A. Children show highest levels of polybrominated diphenyl ethers in a California family of four: a case study. Environ Health Perspect. 2006;114:1581–1584. doi: 10.1289/ehp.8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee JR, Moser VC. Acute postnatal exposure to brominated diphenylether 47 delays neuromotor ontogeny and alters motor activity in mice. Neurotoxicol Teratol. 2008;30:79–87. doi: 10.1016/j.ntt.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Giordano G, White CC, McConnachie LA, Fernandez C, Kavanagh TJ, Costa LG. Neurotoxicity of domoic acid in cerebellar granule neurons in a genetic model of glutathione deficiency. Mol Pharmacol. 2006;70:2116–2126. doi: 10.1124/mol.106.027748. [DOI] [PubMed] [Google Scholar]
- Giordano G, Kavanagh TJ, Costa LG. Neurotoxicity of a polybrominated diphenyl ether mixture (DE-71) in mouse neurons and astrocytes is modulated by intracellular glutathione levels. Toxicol Appl Pharmacol. 2008;232:161–168. doi: 10.1016/j.taap.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale RC, Alaee M, Manchester-Neesvig JB, Stapleton HM, Ikonomou MG. Polybrominated diphenyl ether flame retardants in the North American environment. Environ Int. 2003;29:771–779. doi: 10.1016/S0160-4120(03)00113-2. [DOI] [PubMed] [Google Scholar]
- Hanari N, Kannan K, Miyake Y, Okazawa T, Kodavanti PRS, Aldous KM, Yamashita N. Occurrence of polybrominated biphenyls, polybrominated dibenzo-p-dioxins, and polybrominated dibenzofurans as impurities in commercial polybrominated diphenl ether mixtures. Environ Sci Technol. 2006;40:4400–4405. doi: 10.1021/es060559k. [DOI] [PubMed] [Google Scholar]
- He P, He W, Wang A, Xia T, Xu B, Zhang M, Chen X. PBDE-47-induced oxidative stress, DNA damage and apoptosis in primary cultured hippocampal neurons. Neurotoxicology. 2008a;29:124–129. doi: 10.1016/j.neuro.2007.10.002. [DOI] [PubMed] [Google Scholar]
- He W, He P, Wang A, Xia T, Xu B, Chen X. Effects of BDE-47 on cytotoxicity and genotoxicity in human neuroblastoma cells in vitro. Mutat Res. 2008b;649:62–70. doi: 10.1016/j.mrgentox.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Iwata-Ichikawa E, Kondo Y, Miyazaki I, Asanuma M, Ogawa N. Glial cells protect neurons against oxidative stress via transcriptional up-regulation of glutathione synthesis. J Neurochem. 1999;72:2334–2344. doi: 10.1046/j.1471-4159.1999.0722334.x. [DOI] [PubMed] [Google Scholar]
- Kodavanti PRS, Ward TR. Differential effects of commercial polybrominated diphenyl ethers and polychlorinated biphenyl mixtures on intracellular signaling in rat brain in vitro. Toxicol Sci. 2005;85:952–962. doi: 10.1093/toxsci/kfi147. [DOI] [PubMed] [Google Scholar]
- Law RJ, Allchin CR, deBoer J, Covaci A, Herzke D, Lepom P, Morris S, Tronczynski J, de Wit CA. Levels and trends of brominated flame retardants in the European environment. Chemosphere. 2006;64:187–208. doi: 10.1016/j.chemosphere.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Lorber M. Exposure of Americans to polybrominated diphenyl ethers. J Exp Sci Environ Epidemiol. 2008;8:2–19. doi: 10.1038/sj.jes.7500572. [DOI] [PubMed] [Google Scholar]
- Mazdai A, Dodder NG, Abernathy MP, Hites RA, Bigsby RM. Polybrominated diphenyl ethers in maternal and fetal blood samples. Environ Health Perspect. 2003;111:1249–1252. doi: 10.1289/ehp.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnachie LA, Mohar I, Hudson FN, Ware CB, Ladiges WC, Fernandez C, Chatterton-Kirchmeier S, White CC, Pierce RH, Kavanagh TJ. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetominophen-induced hepatotoxicity in mice. Toxicol Sci. 2007;99:628–636. doi: 10.1093/toxsci/kfm165. [DOI] [PubMed] [Google Scholar]
- McDonald TA. Polybrominated diphenylether levels among United States residents: daily intake and risk of harm to the developing brain and reproductive organs. Integr Environ Assess Manag. 2005;1:343–354. [PubMed] [Google Scholar]
- Morken TS, Sonnenwald U, Aschner M, Syversen T. Effects of methylmercury on primary brain cells in mono- and co-culture. Toxicol Sci. 2005;87:169–175. doi: 10.1093/toxsci/kfi227. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kugiyama K, Sugiyama S, Miyamoto S, Koide S, Fukushima H, Honda O, Yoshimura M, Ogawa H. Polymorphism in the 5’-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation. 2002;105:2968–2973. doi: 10.1161/01.cir.0000019739.66514.1e. [DOI] [PubMed] [Google Scholar]
- Onos KD, Kenny ER, Rice DC, Markowski VP. Long-term deficits following developmental exposure to the flame retardant decaBDE. Neurotoxicol Teratol. 2008 doi: 10.1016/j.ntt.2007.08.010. (in press) [DOI] [PubMed] [Google Scholar]
- Reistad T, Fonnum F, Mariussen E. Neurotoxicity of the pentabrominated diphenyl ether mixture, DE-71, and hexabromocyclododecane (HBCD) in rat cerebellar granule cells in vitro. Arch Toxicol. 2006;80:785–796. doi: 10.1007/s00204-006-0099-8. [DOI] [PubMed] [Google Scholar]
- Rice DC, Reeve EA, Herlihy A, Zoeller RT, Thompson WD, Markowski VP. Developmental delays and locomotor activity in the C57BL6/J mouse following neonatal exposure to the fully brominated PBDE, decabromodiphenyl ether. Neurotoxicol Teratol. 2007;29:511–520. doi: 10.1016/j.ntt.2007.03.061. [DOI] [PubMed] [Google Scholar]
- Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1998;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- Viberg H, Fredriksson A, Eriksson P. Neonatal exposure to polybrominated diphenyl ether (PBDE 153) disrupts spontaneous behavior, impairs learning and memory, and decreases hippocampal cholinergic receptors in adult mice. Toxicol Appl Pharmacol. 2003;192:95–106. doi: 10.1016/s0041-008x(03)00217-5. [DOI] [PubMed] [Google Scholar]
- Viberg H, Johansson N, Fredriksson A, Eriksson J, Marsh G, Eriksson P. Neonatal exposure to higher polybrominated diphenyl ethers, hepta-, octa- , or nonabromodiphenyl ether, impairs spontaneous behavior and learning and memory functions in adult mice. Toxicol Sci. 2006;92:211–218. doi: 10.1093/toxsci/kfj196. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dieter MZ, Che Y, Shetzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm (−/−) knockout mouse. Novel model system for compromised stress response. J Biol Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- Zhou T, Taylor MM, DeVito MJ, Crofton KM. Developmental exposure to polybrominated diphenyl ethers results in thyroid hormone disruption. Toxicol Sci. 2002;66:105–116. doi: 10.1093/toxsci/66.1.105. [DOI] [PubMed] [Google Scholar]
- Zuurbier M, Leijs M, Schoeteres G, ten Tusscher T, Koppe JG. Children’s exposure to polybrominated diphenyl ethers. Acta Pediatrica. 2006;95 (Suppl):65–70. doi: 10.1080/08035320600886299. [DOI] [PubMed] [Google Scholar]