

Abstract
We show that laser-tweezers Raman spectroscopy of eukaryotic cells with a significantly larger diameter than the tight focus of a single beam laser trap leads to optical trapping of the cell by its optically densest part, i.e. typically the cell’s nucleus. Raman spectra of individual optically trapped monocytes are compared with location-specific Raman spectra of monocytes adhered to a substrate. When the cell’s nucleus is stained with a fluorescent live cell stain, the Raman spectrum of the DNA-specific stain is observed only in the nucleus of individual monocytes. Optically trapped monocytes display the same behavior. We also show that the Raman spectra of individual monocytes exhibit the characteristic Raman signature of cells that have not yet fully differentiated and that individual primary monocytes can be distinguished from transformed monocytes based on their Raman spectra. This work provides further evidence that laser tweezers Raman spectroscopy of individual cells provides meaningful biochemical information in an entirely nondestructive fashion that permits discerning differences between cell types and cellular activity.
Keywords: Monocytes, Raman spectroscopy, laser tweezers, single cell microscopy
Introduction
Monocytes are mononuclear immune cells that circulate in the blood. Human monocytes are 14 to 20 μm in diameter with a kidney-shaped nucleus that occupies about one quarter to one third of the total cell volume. Unlike other white blood cells, however, monocytes are considered to be immature cells [1]. They circulate in blood for an average of about 3 days before entering tissue through the space between endothelial cells. In tissue they mature into macrophages that recognize and digest cellular debris and pathogens by phagocytosis [1]. Monocytes, while still circulating in the blood, also act as sentinels for pathogens and become activated after interactions with typical bacterial compounds, such as lipopolysaccharide or mannose [2]. Interestingly, monocytes also play a key role in the development of atherosclerosis [3, 4]. This, together with their important role as part of the innate immune system makes monocytes particularly interesting cells for cell-based assays. Here, we show that the label-free chemical analysis of individual, living monocytes by Raman spectroscopy enables us to rapidly probe robust Raman signatures of individual cells that are sensitive to cellular activation and also allows us to distinguish primary cells from transformed, cultured cells.
Raman spectroscopy enables the label-free, non-destructive, and dynamic chemical analysis of living cells based on the inelastic scattering of photons by molecular bond vibrations [5, 6]. When combined with confocal microscopy it allows for the nondestructive compositional analysis of the endogenous biochemical signature of a cell with diffraction-limited spatial resolution [7, 8].
The recent combination of micro-Raman spectroscopy with laser tweezers using near-infrared laser wavelengths permits the convenient analysis of living cells in suspension [8-14]. Individual cells can now be probed in their natural environment, and the optical trapping process facilitates easy sample handling, and optimized Raman spectral data acquisition of non-adherent cells. Here, we show on the example of individual monocytes that optical trapping of cells that are significantly larger than the tight focus of a single beam laser trap leads to trapping of the cell by its optically densest part, i.e. typically the cell nucleus. We demonstrate that monocyte Raman spectra exhibit signatures that reflect their immature nature, and we show that individual primary monocytes can readily be distinguished from transformed monocytes based on their Raman spectra.
Materials and Methods
Raman spectroscopy of single cells
Laser-tweezers Raman spectroscopy of single cells in suspension was achieved based on a custom-built, inverted Raman microscope as schematically depicted in Figure 1a. The main microscope platform consists of an Olympus IX-71 inverted optical microscope equipped with a 60x, NA 1.2, water immersion objective optimized for near-IR operation (MO - Olympus America, Center Valley, PA). The laser source (L) is an 80 mW, 785 nm diode-pumped solid-state laser (Crystalaser, Reno, NV). The microscope is also equipped with a mercury arc lamp (LA) for white-light fluorescence excitation through appropriate filters (using a DAPI filter set to excite the DNA-binding dye Hoechst 33342), as well as transmitted light differential interference contrast (DIC) for visualizing cells. Cells can be viewed both in transmitted light as well as fluorescence mode through an analog CCD camera (Edmunds Optics), which is digitized and displayed on a personal computer through a frame grabber board (Data Translation, Inc.). In order to visualize cells by fluorescence and subsequently take Raman spectra within distinct parts of the cell, cells were also allowed to adhere to fused silica cover slips (170 μm thickness). Cells in suspension are placed in a 35mm diameter culture dish formed by a stainless steel cell chamber (AttoChamber, Invitrogen, Eugene, OR) and a glass cover slip (BK7 glass, #1 cover slip, Fisher Scientific). Raman spectra are excited by routing the expanded and collimated 785 nm laser beam through a narrow bandpass (BP) filter (Chroma Technology, Brattleboro, VT) into the microscope, reflecting it off of a dichroic mirror (DM - 810DRL, Chroma Optical) into the objective lens. Due to losses in the clean up filter, dichroic filter, and the objective, the laser power in the optical focus is reduced to ~ 30 mW. The Raman-scattered light is collected by the same microscope lens, passes the dichroic mirror, and any remaining laser light is filtered by a 785 nm long pass (LP) filter (Razoredge, Semrock, Inc., Rochester, NY). After passing a 150 μm diameter pinhole (SF) acting as a confocal aperture, the light is re-collimated and then sent into a 300 mm imaging spectrograph (SP - SP2300i, Roper Scientific, Trenton, NJ) equipped with a back-illuminated thermoelectrically-cooled deep-depletion charge-coupled device (CCD) camera with 1340×100 pixels (PIXIS 100, Roper Scientific, Trenton, NJ). Cells are trapped manually by manipulating the xy translation stage of the microscope and adjusting the focus. They are then moved to 30 μm height above the glass cover slip prior to acquiring Raman spectra. The spectral integration time for all Raman spectra is 120s.
Figure 1.
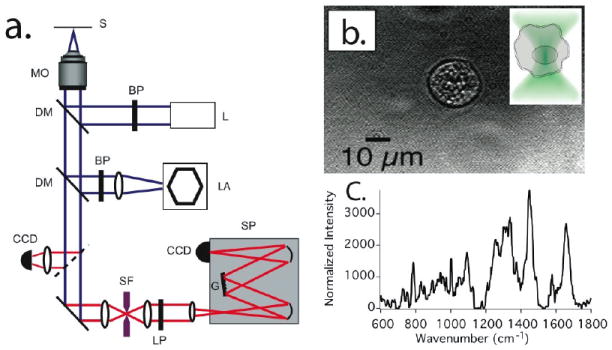
a) Schematic representation of the combined laser tweezers Raman spectroscopy and fluorescence microscopy system (Abbreviations: S: Sample, MO: microscope objective, DM: dichroic mirror, BP: bandpass filter, L: laser, LA: xenon lamp, CCD: charge coupled device camera, SF: spatial filter, LP: longpass filter, G: grating, SP: spectrometer). b)Transmitted light micrograph of an optically trapped living primary monocyte in PBS solution. The cell was trapped by using 30mW of 785 nm continuous wave laser light focused to a tight spot by a 1.2 NA water immersion objective. Insert: Diagram illustrating a mononuclear cell suspended in the focus of an optical trap and showing trapping by its nuclear compartment. c) Background-subtracted Raman spectrum obtained from the optically trapped cell after 60s signal integration time.
Acquisition of Raman spectra from Hoechst stained monocyte nuclei and cytoplasm
THP-1 monocyte cells stained with Hoechst 33342 dye, as described below, were allowed to adhere to a fused silica coverslip. The cells, their cytoplasm and nuclei are distinguished by viewing them through a combination of mercury-arc lamp excited fluorescence and DIC optics, based on the selective Hoechst-labeling of the nucleus. The excitation laser spot was positioned in either the nucleus or cytoplasm of the cells by using a manual positioning stage, the fluorescence and DIC channels were blocked, and Raman spectra acquired in each location within 60 s signal integration time.
Hoechst-Dye stained monocyte time course experiment
A single Hoechst dye stained THP-1 monocyte was held in the optical trap for ~ 1 hour and Raman spectra (120 s integration time) were acquired every 5 minutes.
Isolation of primary human T-cells and monocytes
Normal human T-cells and monocytes were obtained from peripheral blood mononuclear cells (PBMC) after Ficoll-Hypaque density gradient separation of heparinized venous blood obtained from healthy volunteers. The cells were washed 3 times in RPMI 1640 medium prior to purification. Normal T-cells were separated by rosette formation with activated sheep red blood cells followed by repeat Ficoll-Hypaque density gradient centrifugation. T-cells were recovered from the rosette pellet after twice lysing the sheep red blood cells with ammonium chloride lysis buffer (ACK Lysing Buffer, Cambrex Bio Science, Walkersville, MD) and thrice washing with RPMI 1640 medium. T-cell populations were suspended in RPMI 1640 medium supplemented with 10% fetal calf serum at a density of approximately 106 cells/ml. Monocytes were isolated from purified PBMC populations using a commercially available human monocyte isolation kit (Miltenyi Biotec, Inc. Auburn, CA).
T-lymphocyte and monocyte PBMC subpopulations were routinely >98% lymphoid or myeloid, respectively, as assessed by Wright-Giemsa stain of cytospin cell preparations. All cell preparations were >99% viable as assessed by trypan blue dye exclusion. Immediately prior to the acquisition of Raman spectra the cells were washed and suspended in phosphate buffered saline (PBS). The use of fresh human samples for this study was in accordance with the University of California Institutional Review Board practice guidelines.
Preparation of transformed monocytes
Transformed human monocytes (THP-1 monocytes, American Type Culture Collection [ATCC], Manassas, VA) are grown in suspension (37 deg. C and 5% CO2 atmosphere) in either T-25 or T-75 tissue culture flasks, and in RPMI 1640 medium (ATCC) supplemented with 10% fetal bovine serum (ATCC), and containing Gentamicin/Amphotericin B antibiotic (Cascade Biologics, Portland, OR) and 0.05 mM 2-mercaptoethanol (Sigma-Aldrich). Before performing laser tweezers Raman experiments, the cells, typically at a concentration of 5×10 5 cells/mL, are washed and transferred to 5 – 10 times the volume of phosphate buffered saline (Dulbecco’s PBS, Ca and Mg free, Gibco-Invitrogen, Carlsbad, CA) to avoid trapping more than one cell at a time. For experiments using Hoechst stained nuclei, THP-1 cells were incubated in medium with 10 ug/mL Hoechst at 37° C for 30 minutes, then subsequently washed and re-suspended in PBS.
Processing and Analysis of Raman spectra
Despite using near-infrared laser excitation, Raman spectra from single cells typically contain some residual background from fluorescence and substrate scattering. To remove these background contributions, an automated background subtraction routine following the procedure developed by Lieber et al. was utilized [15]. This method first fits the original Raman spectrum with a 5th order polynomial. All the sharp spectral peaks above the polynomial are replaced with the polynomial, while areas below the polynomial are retained from the original spectrum. Then, the resulting spectrum is subjected to fitting with another 5th order polynomial and treated similarly. This process is repeated, typically 20 times for each spectrum. The last fit-polynomial obtained by this procedure is then used to subtract a broad, feature-less background spectrum from the original spectrum. Regions where this background subtraction scheme results in negative spectral values are replaced by zero, which might result in isolated spectrally flat areas in some of the spectra, typically observed in the 1150 – 1200 cm-1 region of some of the spectra shown in this paper. After background subtraction all spectra are normalized to their total area under the spectrum in order to avoid biasing of specific peaks and enable a fair comparison of spectral differences between different cells. All spectral processing was conducted utilizing custom-written routines in MATLAB (Mathworks, Natick, MA, USA).
Results
Raman spectra of individual monocytes and their comparison to mature leukocytes
All monocytes studied in this paper are living cells probed in saline buffer at room temperature. In order to minimize background contributions from media, all cells are washed in PBS, resuspended and probed in PBS immediately before the experiment. The cells in suspension are kept in polyethylene micro-centrifuge tubes and small aliquots (~20 μl) are placed on a glass coverslip. Without further surface treatment cells will begin sticking to the glass coverslip within about 10-15 minutes after application. This allows for sufficient time to probe approximately 10 cells. Once the majority of cells begin adhering to the coverslip surface a new cell suspension has to be prepared. In order to minimize variations in background contributions, all cells are consistently probed at the same height of 30 μm above the coverslip surface by translating the microscope focus and adjusting it to 30 μm for every cell after capturing the cell with the laser trap. Figure 1b shows a transmitted light micrograph of an optically trapped primary monocyte in PBS solution. The inset to Fig. 1b depicts schematically the relative size of the laser spot compared to the optically trapped cell. All monocytes are trapped by exposing them to approximately 30 mW of laser power from a 785 nm continuous wave laser focused to a diffraction-limited spot by a 1.2 NA water immersion objective. Figure 1c shows a typical background-subtracted Raman spectrum obtained by averaging the spectra of ~ 10 optically trapped monocytes, where each cell was probed for 120s.
To determine the difference between primary monocyte, cultured monocytes, and other, more mature hematopoietic cells, we obtained the spectra of typically ~ 20 cells from each group. Figure 2 compares the average background-subtracted Raman spectra for the different cell types. Figure 2a compares the averaged Raman spectrum obtained from isolated primary monocytes to that of cultured transformed THP-1 monocytes. The difference spectrum from these cells is shown in Figure 2b and is obtained by subtracting the primary monocyte averaged spectrum from that of the THP-1 cells. To demonstrate differences between Raman spectra obtained from monocytes and those obtained from other, mature white blood cells, Figure 2c depicts the average Raman spectrum of optically trapped primary T-cells in comparison to that of the same primary monocytes shown in Fig. 2a. In order to highlight differences between the different cell types shown in Figure 2c, the difference spectrum of the cells is shown below the spectra in Figure 2d.
Figure 2.
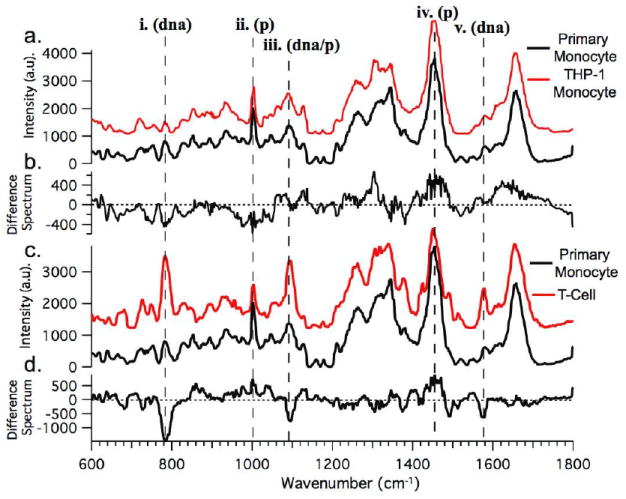
a) Averaged Raman spectra obtained from primary monocytes (black trace) and THP-1 transformed cultured monocytes (red trace). b) Difference spectrum between averaged spectra shown in a. (Difference = Primary − THP-1). c) Averaged Raman spectrum obtained from optically trapped primary T cells (red trace) shown with same averaged monocyte spectrum from a. (black trace.) d) The difference spectrum between the averaged spectra shown in c). (Difference = Monocyte − T-cell.)
Comparison of the spectral signature of monocytes and transformed THP-1 cells
To demonstrate that monocytes isolated from the blood can be distinguished from transformed THP-1 monocytes based on their Raman spectra, a comparison of two independent data sets from each cell type is shown in Figure 3a. Peaks that correspond to contributions from DNA (dna) and protein (p) are indicated by the dashed lines i.-iv. The averaged spectra from the sets of isolated primary monocytes are shown in the figure as red and black traces. Each set was obtained from a different healthy volunteer. The averaged THP-1 Raman spectra are shown in the figure as green and blue traces. These data were acquired from two different cells cultures started from different frozen stocks (both stocks, however, came from the original ATCC THP-1 cell line). Additionally, the plus and minus standard deviation from each data set is shown along with each averaged spectrum (black traces) as gray traces in Figure 3b-e. Differences between the primary and THP-1 monocyte Raman spectra are obvious by visual comparison of the spectra, especially at the Raman shifts indicated in the figure (dashed lines i.-iv.).
Figure 3.
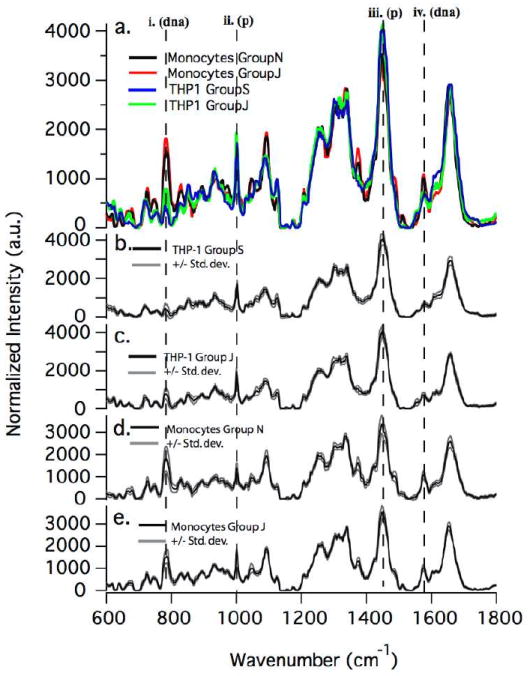
Comparison of spectra from two different data sets of primary monocytes and transformed THP-1 monocytes, respectively. Data sets from each cell type were acquired during different sample preparations and on different days to demonstrate repeatability of method. Standard deviations (+/-) are also shown as gray lines. Key Raman modes corresponding to DNA (dna) and protein (p) content are indicated with dashed lines i. –iv.
Single beam laser traps hold large cells by their nucleus
To determine whether the Raman spectra obtained from individual optically trapped cells with nucleus-to-cytoplasm volume ratios of about 1:1 reflect average Raman spectra of the entire cell or a specific location within the cell, we have compared the spectra obtained from optically trapped cells with those taken from specific locations within the cell after allowing cells to adhere to a fused silica coverslip substrate. This was also done to test the reproducibility of the Raman spectra obtained from individual optically trapped cells. In order to highlight the nuclei of single living cells and provide specific markers for the nucleus, we have also infused some of the cells with the fluorescent stain Hoechst 33342. This stain, when probed with 785 nm excitation, provides a characteristic Raman response that positively identifies high local concentrations of the dye. In Figure 4 (inset) we show a combined fluorescence and transmitted light micrograph of a single transformed monocyte treated with 10 μg/mL Hoechst 33342. This image was obtained by illuminating the cells in epifluorescence mode with a mercury arc lamp through a DAPI filter set, while at the same time providing some contrast by trans-illuminating the sample with white light from a Halogen lamp. The Hoechst 33342 dye preferentially stains the nucleus of living cells, which serves as a means of identifying the nucleus and the cytoplasm of the cells. In Figure 4, we present Raman spectra obtained from optically trapped THP-1 cells before and after treatment with 10 μg/mL Hoechst stain, as well as their difference spectra. The Hoechst dye has a well-defined Raman signature with distinct peaks at 1606, 1588, 1543, 1454, 1267, 1285, and 980 cm-1. The difference spectrum shows more clearly the contribution from the Hoechst Stain to the overall combined Raman spectrum obtained from an optically trapped Hoechst-stained THP-1 monocyte. The two peaks at 1606 and 1543 cm -1 are used most frequently in our analysis and comparison of data acquired from Hoechst-stained cells as these peaks are both the most intense and the least convoluted with other peaks of interest in the fingerprint region. Interestingly, it can be seen from the difference spectrum that the Raman modes generally assigned to protein vibrations, e.g. 1655 cm-1, experience a decrease in intensity upon Hoechst-staining.
Figure 4.
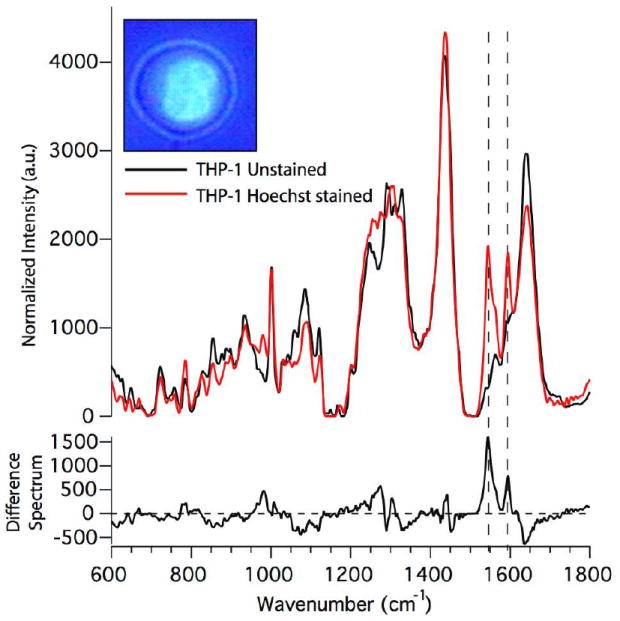
Raman spectra obtained from optically trapped monocytes without Hoechst stain and after treatment with 10 μg/mL Hoechst stain and their difference spectra. Inset shows a mixed fluorescence and transmitted light micrograph of a THP-1 transformed monocytes treated with Hoechst stain. (Hoechst dye preferentially stains the nucleus of living cells.). The dashed lines indicate those Raman modes that are most notably affected by loading the cell with the Hoechst dye.
To compare the Raman spectrum from optically trapped Hoechst 33342 stained monocytes to that specifically originating from the nuclear region vs. the cytoplasm of the cell, we also obtained location-specific spectra from the same Hoechst stained THP-1 monocytes which were immobilized by allowing them to adhere to a fused silica coverslip. In this way, by using the Hoechst 33342 fluorescence as an identifier for nuclear vs. cytoplasmic compartments of the cells, we were able to position the excitation laser in either region, respectively. These results are shown in Figure 5. The spectra shown in Fig. 5a were averaged over approximately 10 different cells in each case, and corresponding standard deviations are shown in Fig. 5b. It should also be pointed out that both the nucleus and cytoplasm spectra were obtained for each individual cell. The average spectrum from the nuclear region clearly shows the presence of the Hoechst Raman contributions at 1606 and 1543 cm-1. The difference spectrum obtained from subtracting the averaged cytoplasm spectrum from the averaged nucleus spectrum is shown in Fig. 5c.
Figure 5.
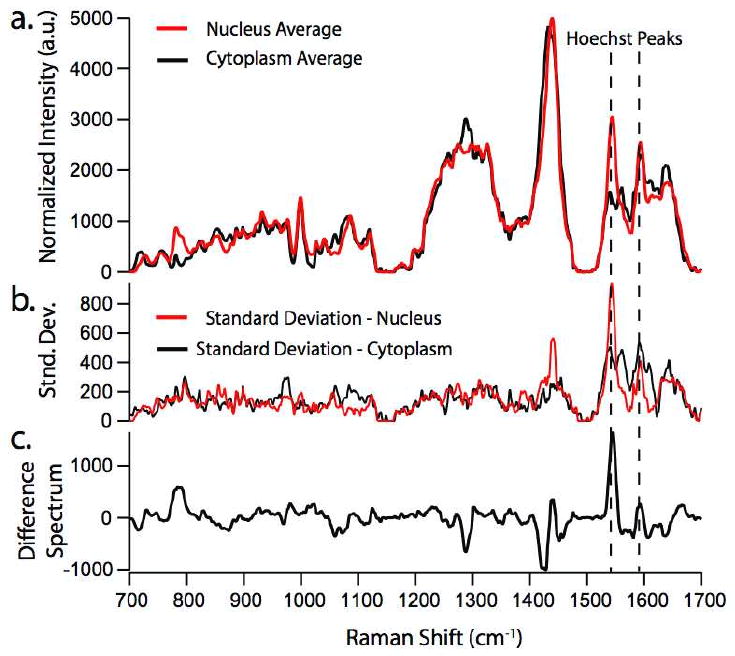
a) Raman spectra obtained from monocytes treated with 10μM Hoechst 33342 adhered to a fused silica coverslip. This allows the isolation of spectra from the cytoplasm of the cell (black trace) and the nucleus (red trace) of the cell. b.) Standard deviation from the averaged spectra shown in a. c.) Difference spectra calculated by subtracting averaged spectrum of cytoplasm from that of the nucleus.
Nuclei of living THP-1 monocytes exhibit selective removal of Hoechst stain
As can be seen from the standard deviation of the nuclei spectra shown in Fig. 5b, the Hoechst Raman peaks at 1606 and 1543, and 1454 cm-1 exhibit relatively large standard deviations. We attribute this to the fact that after the washing step, over time, the nucleus of the THP-1 monocytes is selectively removing the Hoechst dye [16]. Thus, the spectra obtained at later times generally display less intense peaks at these Raman shifts. To test the hypothesis that the Hoechst stain is being pumped out of the cell’s nucleus, it was necessary to perform a time course measurement on a single cell. This was done in the laser-trapping mode, and spectra from a single trapped Hoechst stained THP-1 monocyte were obtained every 5 minutes for 1 hour. The results from this experiment are shown in Figure 6a, in which spectra are shown for the first (0 min. and black solid trace), the halfway (30 min. and black dashed trace), and the last (60 min. and red solid trace) time point. Figure 6b shows a plot illustrating the absolute reduction in the 1543 cm-1 peak when the t=0 min. spectrum is used to calculate difference spectra. It should be noted that this decrease in signal is clearly due to a change in dye concentration and not due to photobleaching, because we are probing the Raman spectra in a spectral range outside the electronic resonance of the dye. As can be seen, the Hoechst peaks at 1606 and 1543 cm-1 initially decrease with time, and then cease to decrease much further after the halfway point. Also notable is the increase in the peak at 1447 cm -1, generally assigned as a protein mode due to the C-H2 deformation mode, and the decrease in the peak at 785 cm-1, which is always assigned to DNA contributions.
Figure 6.
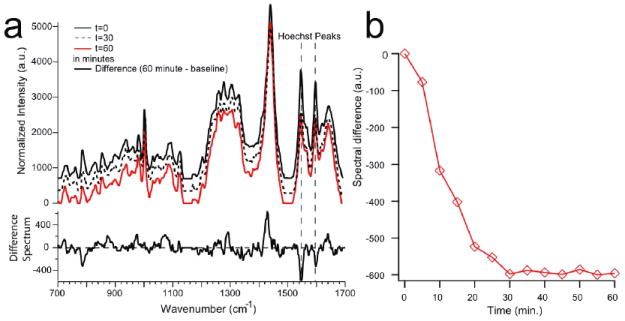
a) Raman spectra obtained from different time points of a single optically trapped Hoechst-stained THP-1 monocyte. A single cell was held in the optical trap for 60 minutes and spectra were obtained every five minutes. Shown here are select time points, i.e. baseline (or zero time point), 30 minutes, and 60 minutes. Also shown is the difference spectrum between the final (60 minute) and initial (zero time) point. Spectra are offset along the y-axis for better visibility. B) Plot showing the reduction in signal of the 1543 cm-1 Hoechst peak after the dye has been flushed out of the buffer solution surrounding the cells.
Discussion
Raman spectra of monocytes and comparison to T-cells
As was shown in the comparison of the Raman spectra from primary monocytes, transformed THP-1 monocytes, and T-cells (Figure 2a-d), there are distinct differences between the different cells types. Differences in the local biochemical composition and concentration of cellular constituents, e.g. proteins, DNA/RNA, and lipids, translate into characteristic differences in the cell’s Raman spectra, especially in the fingerprint region from ~600 – 1800 cm-1. Indeed, this has been shown previously in a comparison of different hematopoetic cells to their transformed cancerous analogs [12]. Here, we show that the main differences between monocytes and other hematopoetic cells are due to the local concentration of DNA and proteins present within the cell and the ratio of the two. These molecular differences can be quantified by comparing the intensities of specific Raman modes assigned to molecular bonds present in DNA and proteins, respectively. The physical basis leading to the modes present due to DNA contributions are nucleotide conformation (600 – 800 cm-1) and backbone geometry (800-1200 cm-1), as well as the electronic structure of the nucleotides (1200-1600 cm-1). Modes due mostly to C-C and C-H bonds contribute to the portion of the Raman spectrum originating from proteins and lipids. Also, proteins exhibit Raman modes due to amide vibrations, such as the amide I band (C=O stretching) and the amide III band (C-N stretching and N-H bending). In Figure 2c, the peaks that exhibit the most visually distinct differences between the two cell types are highlighted by the dashed lines (i. – v.), with their corresponding rough molecular assignment, i.e. either DNA or protein vibrations. Thus, it can be seen that primary monocytes have a locally lower protein to DNA ratio than T-cells (locally meaning: in their nucleus - as will be shown in more detail further below). This outcome is interesting in light of the fact that monocytes, in addition to being key cells of our immune system, have the potential to transform, or differentiate, into the more mature macrophage cells, which in some circumstances can continue to transform into foam cells, which are a histological hallmark of atherosclerotic plaques found in the arteries of individuals with heart disease [3, 4].
Comparison of primary monocytes to their transformed counterparts
A detailed analysis was conducted to highlight differences in the biochemical composition between monocytes isolated from the blood of healthy individuals and their transformed counterparts, the THP-1 cell line. Two independent data sets for each cell type were used to demonstrate the repeatability of LTRS measurements on our experimental system, and to test the validity, in general, of using LTRS to separate populations of cells based upon differences in their Raman spectra. As with the comparison between primary monocyte and T-cell Raman spectra, several visually distinct peaks differ significantly between the averaged spectra of the primary and THP-1 monocytes, as shown in Figure 3a. Most notable are the DNA peaks at 785, 1373, and 1575 cm-1, the protein peak at 1447 cm-1, and the combined DNA/protein peak at 1093 cm-1. Very similar to the earlier studies comparing T- and B- cells to their transformed counterparts, we observe a decrease in the local DNA concentration in the transformed THP-1 cells, and an overall higher protein to DNA ratio. It is evident that the averaged spectra from different groups of the same cell type are quite similar, and thus reproducible via the LTRS technique. There does exist some variability in the spectra from cell to cell within a given cell type, as can be seen from the standard deviation, shown along with the averaged spectra from each group in Figure 3b-e. The largest variations occur at the Raman shifts of 785 cm-1, 1093 cm-1, 1447 cm-1, and 1655 cm-1. These variations are not surprising since the concentration of DNA and protein will vary depending on cell cycle and cellular activation state.
The overall trend of lower DNA concentration and higher protein concentration can be attributed to the following: It is known that transformed cells exhibit increased RNA levels, a much larger nucleus, and a reduced cytoplasm. As we have indicated earlier, the LTRS technique leads to optical trapping by the nucleus of monocytes and a Raman spectrum stemming primarily from constituents within this region of the cell. Although other studies have shown a higher total DNA content relative to proteins in neoplastic cells, our studies indicate a decrease in the DNA concentration, and we attribute this to the increased size of the nuclei of the transformed cells. That is, the intensity of a given Raman mode is proportional to the concentration, and although the absolute DNA content in these cells might indeed be higher, its density is decreased inside the larger nucleus. Also, as transcription rates tend to be higher in neoplastic cells, this likely leads to a larger degree of decondensation of chromatin within the nucleus, and hence, a weaker Raman signal. These findings are consistent with our recent studies performed on T- and B-lymphocytes and their transformed analogs, as well as with reports from other groups investigating the biochemical differences between cancerous and normal cells using Raman spectroscopy [11, 12, 17-21]. These observations provide further evidence that in transformed tumors cells, such as THP-1 cells, chromatin is less densely packaged (thus, exhibits a locally lower DNA concentration) to allow for higher transcription rates, resulting in an apparent locally higher protein concentration most likely due to the presence of transcription factors, DNA helicases, etc. Of potential concern is that the actual culture medium might influence the separation between cultured cells and primary cells because of its compositional difference to human blood serum. This possibility is, however, quite low, since any such differences should not affect the composition of a cell’s chromatin.
Raman Spectroscopy of Hoechst-Stained THP-1 cells
Another observation of interest from the analysis and comparison of all the cells discussed in the previous sections is the relatively good reproducibility of the spectra obtained from optically trapped cells. This can be seen by inspecting the standard deviations for each group, which are shown as grey outlines around the spectra in Figure 3. This number is less than 5% for most regions of the spectra, except for some of the peaks that also contribute the most to the variation between cells. For the cultured, transformed cells even the significant peaks exhibit standard deviations of at most 10%, which, again reflects the heterogeneity of primary circulating cells. Our hypothesis for explaining these low standard deviations is that large mononuclear cells with ~ 1:1 nucleus-to-cytoplasm ratios, are consistently optically trapped by their most optically dense compartment, i.e. the nucleus. To experimentally identify the specific part of large cells, such as monocytes, which is optically trapped during the application of LTRS, the nuclear stain Hoechst 33342 was used. This dye is typically used as a fluorescent marker for DNA in live cells and preferentially binds to A-T rich regions [16]. Thus, when applied to live THP-1 cells in culture, the majority of the stain is expected to be found within the nucleus. As pointed out earlier, Hoechst 33342 also has a readily identifiable Raman spectrum with distinct peaks within the fingerprint region. In the majority of our analysis of the Hoechst stained cell Raman spectra, we have focused on the two most intense peaks of the Hoechst Raman spectrum at 1606 and 1543 cm-1. Analysis of difference spectra acquired from the THP-1 cell Raman spectra before and after staining, shown in the bottom half of Figure 4, yields a few key outcomes. As the difference was calculated by subtracting the averaged Hoechst-stained THP-1 spectrum from the unlabeled THP-1 averaged spectrum, the contribution from the Hoechst Raman modes is expected to result in positive peaks in the difference spectrum at the corresponding Hoechst peak values, e.g. 1606, 1588, 1543 cm-1, etc. Indeed, this is observed, especially at the more intense Hoechst peaks (indicated in Figure 4 by the dashed vertical lines). Also observed is a decrease in the peaks at 1655 (21%), 1337 (20%), 785 (27%), as well as the peaks in the band between 1033 and 1133 cm-1 (with up to 40% change in peak intensity). It is difficult to say what is contributing to the change in these Raman peaks, as all of these modes, with the exception of that at 785 cm-1, have contributions from both DNA and protein modes, and in some cases, even lipids. However, it is well known that the Hoechst 33342 stain, even though used widely for live cell staining, is toxic to living cells at high concentrations, and thus, we speculate that these observed changes in the Raman spectrum might be indicative of modifications to the cell’s DNA and protein interactions that will ultimately lead to apoptosis. Finally, the observation of the intense Hoechst Raman peaks in the spectra obtained from optically trapped Hoechst-stained cells indicates that these cells are indeed being trapped by their optically most dense compartment; i.e. by their nucleus. To further substantiate this claim, a comparison of the Raman spectra obtained specifically from points within the nucleus and the cytoplasm of the same Hoechst-stained THP-1 is presented in Figure 5. It can be seen from the averaged spectra in Figure 5a that the Hoechst stain is only present at high concentration in the nucleus due to the observation of the Hoechst Raman peaks at 1606 and 1543 cm-1 in the nucleus spectrum and the lack of these peaks in the cytoplasmic spectrum. The standard deviations of the averaged spectra from Figure 5a are given in this example (Figure 5b) because a large variation in the Hoechst Raman peaks was observed in the nucleus Raman data set. We attribute this to the fact that after the cells have been washed subsequently to incubation with the Hoechst stain, the stain leaks out, or is actively pumped out of the cell over time. Thus, the concentration of Hoechst stain in the nucleus for cells whose spectra were obtained later after treatment with the stain is expected to be lower, yielding correspondingly less intense Hoechst Raman peaks. The time course study of a single optically trapped Hoechst-stained THP-1 cell (results shown in Figure 6) provides further evidence that the Hoechst stain is being actively removed from the nucleus over time. As was pointed out earlier, the intensity of the main Hoechst peaks at 1606 and 1543 cm-1 decreases initially and then ceases to decrease further after about 30 minutes. Again, a decrease in some of the peaks in the cell’s fingerprint Raman spectrum, as was observed in the data presented in Figure 4, is observed over time, possibly indicating cell death due to the toxicity of the Hoechst dye. Specifically, we see noticeable decreases in the 785 and 1447 cm-1 modes in the spectra from the initial to the final time points.
Conclusions
We have shown that there are significant differences in the Raman spectra, and hence the biochemical composition, between human monocytes and other cells from the hematopoetic lineage, such as T-cells. These differences were found primarily in the local DNA and protein concentration of the chromatin, and the ratio between the two. These differences might be indicative of the degree of maturity of these cells. Monocytes, once migrated into the tissue of arterial walls, are known to differentiate into macrophages. Furthermore, other cells are also known to be derived from monocytes, e.g. myeloid dendritic cells, osteoclasts, and Kupffer cells. T-cells, on the other hand, do not undergo transformation to further cell types. Differences between the Raman spectra from monocytes isolated from the peripheral blood of healthy donors and the leukemic transformed THP-1 analog are consistent with previous findings comparing other hematopoetic cell types to their cancerous equivalents. These cells can be separated and identified even by straight-forward spectral difference analysis. These results show promise for the use of Raman spectroscopy as a label-free means of identifying and separating normal healthy monocytes from cancerous cells in the blood of leukemia patients – similar to what has recently been demonstrated with T cells [11, 12]. We have also verified our working hypothesis that in a single beam optical trap large, mononuclear cells are optically trapped by their nuclei, and hence most of the Raman signal is originating from this cellular compartment. Furthermore, we have used the specific Raman spectroscopic peaks of the Hoechst dye to monitor its active removal from the nucleus of living THP-1 monocytes over time. These studies further confirm that Hoechst 33342 can act as a Raman probe for monitoring cellular activity, with the advantage that it does not undergo photobleaching, as is the case with fluorescence excitation.
Acknowledgments
This work was supported in part by funding from the National Science Foundation. The Center for Biophotonics Science and Technology is managed by the University of California, Davis, under Cooperative Agreement No. PHY 0120999. Additional funding was provided by a gift from the Keaton-Raphael Foundation for Childhood Cancer. T. Huser also acknowledges support by the Clinical Translational Science Center under grant number UL1 RR024146 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research. J.W. Chan acknowledges support from the LLNL Laboratory-directed Research and Development Program. Work at LLNL was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344.
References
- 1.Brown BA. Hematology: Principles and Procedures. 6. Philadelphia, PA: Lea & Febiger; 1993. [Google Scholar]
- 2.Alberts B, et al. Molecular Biology of the Cell. 4. New York, NY: Garland Science, Taylor and Francis Group; 2002. [Google Scholar]
- 3.den Hartigh LJ, et al. Fatty acids from Very Low-Density Lipoprotein Lipolysis Products Induce Lipid Droplet Accumulation in Human Monocytes. J Immunol. 2010;184(7):3927–3936. doi: 10.4049/jimmunol.0903475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 5.Chan JW, et al. Raman spectroscopy and microscopy of individual cells and cellular components. Laser & Photon Rev. 2008;2(5):325–349. [Google Scholar]
- 6.Wachsmann-Hogiu S, Weeks T, Huser T. Chemical analysis in vivo and in vitro by Raman spectroscopy – from single cells to humans. Curr Opin Biotechnol. 2009;20:63–73. doi: 10.1016/j.copbio.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puppels GJ, et al. Studying Single Living Cells and Chromosomes by Confocal Raman Microspectroscopy. Nature. 1990;V347(N6290):301–303. doi: 10.1038/347301a0. [DOI] [PubMed] [Google Scholar]
- 8.Xie CG, Dinno MA, Li YQ. Near-infrared Raman spectroscopy of single optically trapped biological cells. Optics Letters. 2002;27(4):249–251. doi: 10.1364/ol.27.000249. [DOI] [PubMed] [Google Scholar]
- 9.Xie CG, et al. Real-time Raman spectroscopy of optically trapped living cells and organelles. Optics Express. 2004;12(25):6208–6214. doi: 10.1364/opex.12.006208. [DOI] [PubMed] [Google Scholar]
- 10.Chan JW, et al. Reagentless identification of single bacterial spores in aqueous solution by confocal laser tweezers Raman spectroscopy. Analytical Chemistry. 2004;76(3):599–603. doi: 10.1021/ac0350155. [DOI] [PubMed] [Google Scholar]
- 11.Chan JW, et al. Non-destructive Identification of Individual Leukemia Cells by Laser Tweezers Raman Spectroscopy. Analytical Chemistry. 2008;80:2180–2187. doi: 10.1021/ac7022348. [DOI] [PubMed] [Google Scholar]
- 12.Chan JW, et al. Micro-Raman spectroscopy detects individual neoplastic and normal hematopoietic cells. Biophysical Journal. 2006;90(2):648–656. doi: 10.1529/biophysj.105.066761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan JW, et al. Monitoring dynamic protein expression in living E-coli. Bacterial Celts by laser tweezers raman spectroscopy. Cytometry Part A. 2007;71A(7):468–474. doi: 10.1002/cyto.a.20407. [DOI] [PubMed] [Google Scholar]
- 14.Chan J, et al. Label-free spectroscopic separation of human embryonic stem cells (hESCs) and their cardiac derivatives using Raman spectroscopy. Analytical Chemistry. 2009;81:1324–1331. doi: 10.1021/ac801665m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieber CA, Mahadevan-Jansen A. Automated method for subtraction of fluorescence from biological Raman spectra. Applied Spectroscopy. 2003;57:1363–1367. doi: 10.1366/000370203322554518. [DOI] [PubMed] [Google Scholar]
- 16.Petersen TW, et al. Chromatic shifts in the fluorescence emitted by murine thymocytes stained with Hoechst 33342. Cytometry Part A. 2004;60(2):173–181. doi: 10.1002/cyto.a.20058. [DOI] [PubMed] [Google Scholar]
- 17.Huang ZW, et al. Near-infrared Raman spectroscopy for optical diagnosis of lung cancer. International Journal of Cancer. 2003;107(6):1047–1052. doi: 10.1002/ijc.11500. [DOI] [PubMed] [Google Scholar]
- 18.Kaminaka S, et al. Near-infrared multichannel Raman spectroscopy toward real-time in vivo cancer diagnosis. Journal of Raman Spectroscopy. 2002;33(7):498–502. [Google Scholar]
- 19.Kaminaka S, et al. Near-infrared Raman spectroscopy of human lung tissues: possibility of molecular-level cancer diagnosis. Journal of Raman Spectroscopy. 2001;32(2):139–141. [Google Scholar]
- 20.Mahadevan-Jansen A, Richards-Kortum R. Raman Spectroscopy for the detection of cancers and precancers. Journal of Biomedical Optics. 1996;1(1):31–70. doi: 10.1117/12.227815. [DOI] [PubMed] [Google Scholar]
- 21.Utzinger U, et al. Near-infrared Raman spectroscopy for in vivo detection of cervical precancers. Applied Spectroscopy. 2001;55(8):955–959. [Google Scholar]