

Abstract
The authors report a very rare case of adreno-cortical carcinoma arising in a giant adrenal pseudocyst. A 64-year-old woman presented to the emergency department with a 6 week history of progressively worsening severe left abdominal pain, anorexia, anergia and constipation. On examination, she was cachectic with tenderness over the left abdomen and flank. Medical history was significant for gastritis and anaemia. During her investigation, a well-defined para-renal 12×6 centimetre multi-loculated cyst, of uncertain origin was identified on CT. Ultrasound-guided biopsy was not diagnostic. MRI showed the cyst to be likely adrenal in origin. Serum and urinary catecholamines were unremarkable. At laparotomy an unresectable large, tense, fixed, cystic mass was seen to occupy the left side of the abdomen. The cyst was de-roofed. Pathology showed a high-grade poorly differentiated adreno-cortical carcinoma with a pseudo-capsule. She died 2 months postoperatively.
Background
Adrenal pseudocysts are cystic lesions arising within the adrenal gland surrounded by a fibrous tissue wall devoid of a recognisable lining layer. Adreno-cortical carcinoma is a rare endocrine malignancy causing up to 0.2% of all cancer deaths. Only approximately 7% of pseudocysts are associated with malignancy.
Case presentation
A 64-year-old woman presented to the emergency department with a 6 week history of progressively worsening severe left abdominal pain, anorexia, anergia and constipation. On examination, she was cachectic with tenderness over the left abdomen and flank. One month prior to presentation, she had undergone an oesophogastroduodenoscopy and full colonoscopy, which were unremarkable. Medical history was significant for osteoporosis, gastro-oesphageal reflux disease and chronic anaemia. There was no surgical history. Socially she lived with her husband. She smoked 10–25 cigarettes a day for the preceding 45 years. She did not consume alcohol. Family history was positive for cardiovascular disease, lung and pancreatic cancer. Her father aged 70 years and her brother aged 44 years died from myocardial infarcts. A sister died aged 65 years from lung cancer, and a brother aged 63 years from pancreatic cancer.
Investigations
During her investigation, a well-defined para-renal 12×6 centimetre multi-loculated cyst, of uncertain origin was identified on CT (figure 1). MRI showed the cyst to be separate from both kidney and pancreas, though very closely adjacent to both (figures 2 and 3). The left adrenal gland was not separately identified from the mass, suggesting adrenal origin. The walls of the mass were irregular and thickened, showing uniform enhancement. No lymphadenopathy was seen. The radiological appearance was consistent with an adrenal pseudocyst. Ultrasound-guided biopsy was not diagnostic. Serum and urinary catecholamines and metanephrines were unremarkable.
Figure 1.

(A, B) Transverse sections of a CT abdomen showing a large multi-loculated cystic fluid collection in the left side of the abdomen in the retroperitoneum and the perinephric space.
Figure 2.
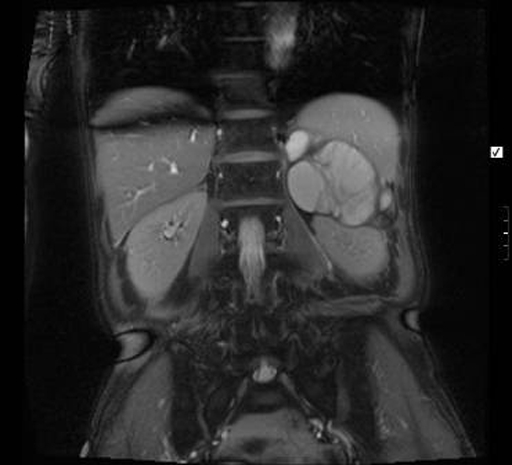
Frontal section of a MRI abdomen showing a well-defined left-sided para-renal multi-loculated cystic mass seen to be separate from the left kidney and pancreas. The left adrenal gland is not separately identified.
Figure 3.
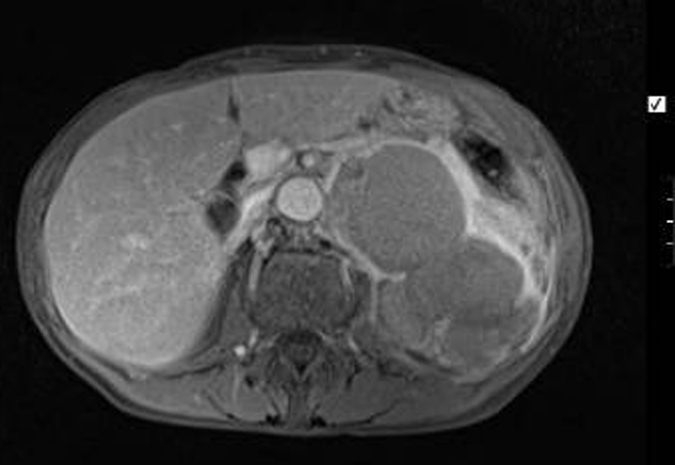
Transverse section of a MRI abdomen showing a well-defined left-sided para-renal multi-loculated cystic mass seen to be separate from the left kidney and pancreas. The left adrenal gland is not separately identified.
Differential diagnosis
-
▶
Adrenal pseudocyst
-
▶
Pancreatic pseudocyst
-
▶
Complex renal cyst
-
▶
Adreno-cortical cancer.
Treatment
Due to the malignant potential of the pseudocyst we proceeded to laparotomy. At laparotomy an unresectable large, tense, fixed, cystic mass was seen to occupy the left side of the abdomen. The cyst was de-roofed.
Outcome and follow-up
Pathology showed a high-grade poorly differentiated adreno-cortical carcinoma with a pseudo-capsule. She died 2 months postoperatively.
Discussion
Cysts of the adrenal gland are usually benign lesions with an estimated incidence at autopsy of 0.064%–0.18%, with adrenal pseudocysts accounting for an extremely small amount of this percentage.1 Adrenal pseudocysts most commonly arise from haemorrhage within the adrenal gland, secondary to extreme stress, birth, trauma, surgery or malignancy. They have long been known to be associated with malignancy, with an estimated incidence of approximately 7%.1 Among the malignancies found in adrenal pseudocysts; adreno-cortical carcinoma (ACC) is by far the most common.1
ACC has an overall incidence of only one to two persons per one million population.2 It is primarily an adult disease with the average age of onset being 44 years. Overall the prevalence of ACC is slightly higher in females.1 Many studies have shown a left prevalence of ACC with the tumour being bilateral in approximately 2%–6% of cases.2
Affected patients usually present with abdominal pain or with symptoms related to the mass effect or hormonal activity of the tumour. Adrenal cancers are thus classified as either being functional or non-functional. ACCs are classed functional when their hormonal secretion results in clinical manifestations such as Cushing’s syndrome, virilisation syndrome, feminisation syndrome or a mixed Cushing-virilising syndrome. Approximately 60% of patients with ACC present with evidence of adrenal steroid hormone excess, with excessive cortisol secretion seen in 30% of functioning tumours, androgen hypersecretion in 20%, oestrogen hypersecretion in 10% and aldosterone hypersecretion in 2%.1 2 Thirty-five percent of functioning ACC’s secrete multiple hormones.2 This figure is actually likely to be higher than 35% because some non-functional tumours may initially present as non-functional but later become functional. The presenting complaint with regard to non-functioning tumours is usually related to tumour size, with typical presenting symptoms of abdominal pain or pressure secondary to mass effect. Other clinical features include weight loss, weakness, fever, anorexia, nausea and myalgia.2
The majority of ACCs arises sporadically and have an idiopathic aetiology. However, ACC does develop as part of a constellation of tumours in inherited familial cancer syndromes: Li-Fraumeni Syndrome, Beckwidth-Wiedemann Syndrome, Gardener Syndrome and Multiple Endocrine Neoplasia type 1 being among the most common of these syndromes.3 While genes and DNA ploidy certainly have a role in inheritance, the part they play as prognostic indicators is somewhat controversial, some studies, such as ECOG-1879,4 for example, shows correlation between aneuploidy and prognosis, and other studies,5 show no correlation.
Initial evaluation of a potential ACC should include, in addition to appropriate endocrine studies, CT and/or MRI of the abdomen. MR is much more useful in the diagnosis of adrenal cysts and tumours. The diagnostic accuracy rates of MRI, CT and B-mode ultrasonography are estimated to be 100%, 87% and 78%, respectively.6 While MRI has a superior diagnostic accuracy for ACC, CT is considered the diagnostic study of choice for evaluating adrenal masses. On thin section, CT nodules as small as 3–5 mm can be identified. CT provides information on size, homogenicity, calcification, the area of necrosis and the extent of local invasion.6 Several studies have looked to differentiate benign versus malignant disease via imaging. Belldegrun et al noted that 92% of adrenal carcinomas were greater than 6 cm, while most other reviews indicate that malignant lesions tend to be 5 cm or greater, have an irregular shape and will show heterogenous contrast enhancement.7 Patients found to have large ACC, particularly those of the right adrenal gland should undergo evaluation of the vena cava for thrombus.6
Angiography and adrenal venography can have a role to play in identifying small tumours and for distinguishing tumours of the adrenal gland from those of the upper pole of the kidney. Positron emission tomography may have a useful role in identifying unsuspected sites of metastases; however, its role in staging is currently unclear.8
Retrospective studies have highlighted two important prognostic factors: completeness of resection and stage of disease. The staging system for ACC depends on tumour size, adjacent organ invasion, lymph node involvement and distant metastases. Patients without evidence of invasion into local tissues or spread to lymph nodes have an improved prognosis.9 However, it is estimated that only 30% of ACC is confined to the adrenal gland at the time of presentation, meaning that ACC is only curable when it presents at a very early stage.10
As radical excision is the treatment of choice for those patients with localised malignancies, ACCs are typically not biopsied prior to surgery as was the situation in our case. Diagnosis is thus confirmed with histo-pathological assessment. Grossly, ACCs are often very large, with a tan-yellow cut surface, and areas of haemorrhage and necrosis. On microscopic examination, the tumour usually displays sheets of atypical cells with some resemblance to the cells of the normal adrenal cortex. The presence of invasion and mitotic activity help differentiate small cancers from ACCs.11
Complete surgical excision is the only means by which long-term disease-free survival may be achieved. Overall 5 year survival for tumours resected for cure is approximately 40%. The choice of open versus laproscopic has long been controversial. Laproscopic adrenalectomy has been proven to be the standard of care for managing small benign adrenal masses.12 Recent prospective studies have also shown that laproscopic adrenalectomy is safe and feasible for large functioning adrenal masses as well as non-functioning adrenal masses.13 Studies have shown that large adrenal tumours suspected of harbouring malignancy can be managed laproscopically, provided there is no peri-adrenal involvement or capsular disruption.13
Mitotane was the mainstay of medical management in patients with unresectable disease and in those in whom recurrent or metastatic disease could not be treated with reoperation. It works as an adrenolytic agent by causing alterations in mitochondrial function and altering the extra-adrenal metabolism of cortisol and androgens.14 However, many studies have questioned the improved survival benefit of such treatment with mitotane and other trialed chemotherapeutic agents and the role they will have in the future is now uncertain.
The prognosis for patients with ACC is generally poor with approximate 5 year survivals for stages I–IV: 30% to 45%, 12.5% to 57%, 5% to 18% and 0%, respectively.15 Median survival in patients with unresectable tumours is in the order of 3–9 months, whereas after complete resection median survival is estimated to be 13–28 months.
Learning points.
-
▶
Adrenal pseudocysts are cystic lesions arising within the adrenal gland surrounded by a fibrous tissue wall devoid of a recognisable lining layer.
-
▶
Only approximately 7% of pseudocysts are associated with malignancy.
-
▶
Adreno-cortical carcinoma is a rare endocrine malignancy causing up to 0.2% of all cancer deaths.
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Castillo OA, Litvak JP, Kerkebe M, et al. Laproscopic management of symptomatic and large adrenal cysts. J Urol 2006;173:915–7 [DOI] [PubMed] [Google Scholar]
- 2.Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab 2006;91:2027–37 [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009;27:1250–6 [DOI] [PubMed] [Google Scholar]
- 4.Camuto P, Schinella R, Gilchrist K, et al. Adrenal cortical carcinoma: flow cytometric study of 22 cases, an ECOG study. Urology 1991;37:380–4 [DOI] [PubMed] [Google Scholar]
- 5.Haak HR, Cornelisse CJ, Hermans J, et al. Nuclear DNA content and morphological characteristics in the prognosis of adrenocortical carcinoma. Br J Cancer 1993;68:151–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin GB, Xu M, Wang H. [Diagnosis and treatment of adrenal cyst: report of 15 cases]. Zhonghua Yi Xue Za Zhi 2009;89:409–11 [PubMed] [Google Scholar]
- 7.Schultz CL, Haaga JR, Fletcher BD, et al. Magnetic resonance imaging of the adrenal glands: a comparison with computed tomography. AJR Am J Roentgenol 1984;143:1235–40 [DOI] [PubMed] [Google Scholar]
- 8.Becherer A, Vierhapper H, Pötzi C, et al. FDG-PET in adrenocortical carcinoma. Cancer Biother Radiopharm 2001;16:289–95 [DOI] [PubMed] [Google Scholar]
- 9.Lee JE, Berger DH, el-Naggar AK, et al. Surgical management, DNA content, and patient survival in adrenal cortical carcinoma. Surgery 1995;118:1090–8 [DOI] [PubMed] [Google Scholar]
- 10.Norton JA. Adrenal tumors. In: DeVita VT, Jr, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 7th edition Philadelphia, Pa: Lippincott Williams & Wilkins; 2005;1528–39 [Google Scholar]
- 11.Cote R, Suster S, Weiss L, Weidner N. Modern Surgical Pathology. 2 Volume Set London: W B Saunders; 2002:ISBN 0-7216-7253-1 [Google Scholar]
- 12.Gagner M, Lacroix A, Bolté E. Laparoscopic adrenalectomy in Cushing’s syndrome and pheochromocytoma. N Engl J Med 1992;327:1033. [DOI] [PubMed] [Google Scholar]
- 13.Sharma R, Ganpule A, Veeramani M, et al. Laparoscopic management of adrenal lesions larger than 5 cm in diameter. Urol J 2009;6:254–9 [PubMed] [Google Scholar]
- 14.Terzolo M, Angeli A, Fassnacht M, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007;356:2372–80 [DOI] [PubMed] [Google Scholar]
- 15.Wajchenberg BL, Albergaria Pereira MA, Medonca BB, et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer 2000;88:711–36 [PubMed] [Google Scholar]