

Abstract
A 2-month-old male infant, born of second degree consanguineous parentage, presented with seizures not responding to phenytoin and phenobarbitone. His perinatal period had been uneventful and there was no family history of seizures. On examination, he had failure to thrive, perioral and perianal rash, alopecia with hypopigmented hair, seborrhoeic dermatitis, bilateral blepharitis, respiratory distress and stridor. Neurological examination revealed hypertonia of all the four limbs, exaggerated deep tendon reflexes and papilloedema. Biotinidase deficiency was suspected within 24 h of admission and empiric oral biotin 10 mg twice daily was started. The symptoms, especially seizures, dramatically improved within 48 h. Serum biotinidase levels revealed a profound deficiency (0.10 nmol/min/ml serum) and the parents were advised regarding the need for regular biotin supplementation. The child is presently 10 months old, thriving well, developmentally normal and is seizure free with total resolution of skin and hair lesions.
Background
Biotinidase deficiency is a rare, but easily treatable, inborn error of metabolism.1 This case illustrates the fact that the presentation of the disease may be highly varied and atypical, thereby requiring a high index of suspicion to diagnose this disorder. An excellent outcome can be achieved with prompt diagnosis and early treatment, which is inexpensive and prevents irreversible long-term complications like developmental delay, hearing loss and optic atrophy.2
Case presentation
A 58-day-old male infant, born of second degree consanguinity, presented with seizures for 2 weeks prior to presentation.
At admission, the child had multiple episodes of generalised tonic and myoclonic seizures not associated with fever. He also had respiratory distress and stridor. His perinatal period had been uneventful and the child had attained social smile. There was no family history of seizures.
The child had been unsuccessfully treated elsewhere for seizures with phenytoin and had been hospitalised there, around 1 week prior to presentation, for respiratory distress and stridor.
At the time of admission to our hospital, he was lethargic though afebrile. The skin showed erythematous maculopapular rash in the perioral and perianal regions (figure 1). He had alopecia, scanty eyebrows, blepharitis, conjunctivitis, balanitis and seborrhoeic dermatitis involving the scalp (figures 2 and 3). Hair over the scalp was hypopigmented. On neurological examination, the child was convulsing intermittently. Hypertonia was noted in all the four limbs with exaggerated deep tendon reflexes. Fundus examination revealed papilloedema. Rest of the systemic examination was normal.
Figure 1.
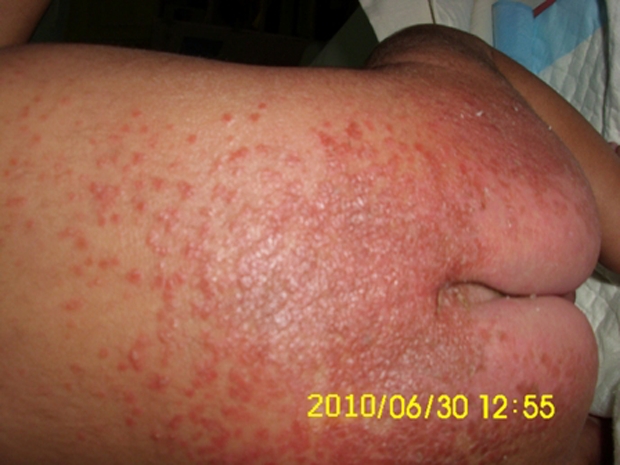
Erythematous maculopapular rash involving the back and perianal regions.
Figure 2.
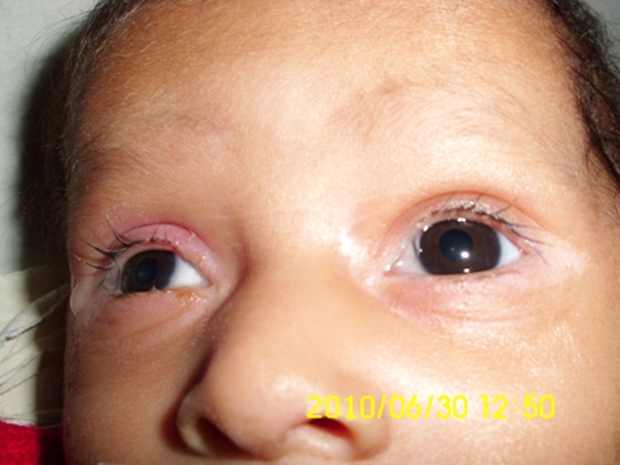
Blepharitis and conjunctivitis with scanty eyebrows.
Figure 3.
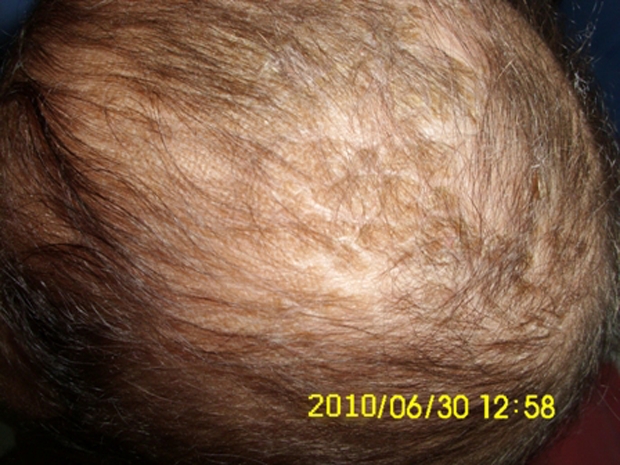
Seborrhoeic dermatitis of the scalp with alopecia.
Investigations
Laboratory investigations revealed a normal haemogram, urine routine, liver profile, serum electrolytes, serum zinc and ammonia. Serum lactate was raised (56.8 mg/dl). Sepsis screen was negative, arterial blood gas and cerebrospinal fluid analyses were normal. Hair microscopy was normal.
Pretreatment serum biotinidase levels showed a profound deficiency (0.10 nmol/min/ml serum, normal range 2.67–5.35 nmol/min/ml serum, profoundly deficient if the levels are less than 0.7 nmol/ml/min serum). A repeat assay done after 6 months showed persistently low levels of biotinidase (0.39 nmol/min/ml serum). Brainstem evoked response audiogram and visual evoked potentials were normal. MRI of the brain done on follow-up was found to be normal.
Differential diagnosis
Though the dermatological lesions were similar to those seen in acrodermatitis enteropathica, the constellation of seizures, abnormal muscle tone and typical skin lesions precluded diagnoses other than biotinidase deficiency.
Treatment
The infant was admitted to the intensive care unit and treated for seizures initially with phenytoin and subsequently with phenobarbitone despite which he continued to have seizures. Within 24 h of hospitalisation, the possibility of biotinidase deficiency was entertained and oral biotin was started empirically at a dose of 10 mg twice a day, after obtaining a blood sample for biotinidase assay.
Outcome and follow-up
There was total cessation of seizures and considerable improvement in the rash by 48 h. The hypertonia in the upper limbs resolved within 72 h though it persisted in the lower limbs, even at discharge, albeit much less than before. The balanitis and blepharitis also resolved by 72 h.
Serum biotinidase levels showed a profound deficiency. The child was then started on regular biotin supplementation and was discharged after 5 days. Anticonvulsants were successfully stopped without recurrence of seizures.
The child is currently 10 months old, healthy, with adequate weight gain and normal developmental milestones. At follow-up, there are no sequelae except for a mild persisting lower limb hypertonia.
Discussion
Biotinidase deficiency is an autosomal recessive disorder; the incidence of severe biotinidase deficiency (levels less than 10% normal) being 1:137401 in population. The disease is characterised by defective recycling of biotin leading to multiple carboxylase deficiency thereby resulting in defective gluconeogenesis, fatty acid catabolism, amino acid metabolism and defective immunity.3
The manifestations may be (a) neurological (seizures, hypotonia and developmental delay), (b) dermatological (eczematous skin rash, seborrhoeic dermatitis and alopecia), (c) pulmonological (hyperventilation, laryngeal stridor and apnoea), (d) ophthalmological (conjunctivitis) and (e) metabolic ketoacidosis and organic aciduria.
These manifestations of biotinidase deficiency, though, are highly variable.4 Although hypotonia is commonly seen in this condition, this infant had hypertonia with exaggerated deep tendon reflexes in all the four limbs. Three cases of biotinidase deficiency, with unexplained hypertonia as an unusual feature, have been reported from India.5
Ketoacidosis and organic aciduria (absent in this child) may not be seen in all cases. Thus, metabolic acidosis may not be a constant feature and urinary organic acid excretion may be unremarkable, particularly early in the disease.4 Also, phenotypic variations have been observed and the absence of ketoacidosis does not go against a diagnosis of biotinidase deficiency.6 Though, five among the six cases of biotinidase deficiency reported from India, had ketoacidosis and organic aciduria.1 3 7 8 biotinidase deficiency is also associated with developmental delay.1 3 Our patient had already attained social smile at the time of admission and continued to achieve normal milestones during follow-up, due to the early diagnosis and treatment. Older children and adolescents may exhibit limb weakness, sensorineural hearing loss and eye problems that is, optic atrophy and scotomata.3
Learning points.
-
▶
Consider biotinidase deficiency in an infant with refractory seizures along with characteristic neurological and dermatological manifestations.
-
▶
A high index of suspicion should be maintained.
-
▶
Biotinidase deficiency can be treated very effectively with biotin.
-
▶
Though rare, all newborns should be screened for biotinidase deficiency as the treatment is inexpensive and very gratifying.
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Ananth N, Praveen Kumar GS. Biotinidase deficiency-diagnosis by enzyme assay and a follow-up study. Indian J Clin Biochem 2003;18:23–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taitz LS, Leonard JV, Bartlett K. Long-term auditory and visual complications of biotinidase deficiency. Early Hum Dev 1985;11:325–31 [DOI] [PubMed] [Google Scholar]
- 3.Bhardwaj P, Kaushal RK, Chandel A. Biotinidase deficiency: a treatable cause of infantile seizures. J PediatrNeurosci 2010;5:82–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wastell HJ, Bartlett K, Dale G, et al. Biotinidase deficiency: a survey of 10 cases. Arch Dis Child 1988;63:1244–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rathi N, Rathi M. Biotinidase deficiency with hypertonia as unusual feature. Indian Pediatr 2009;46:65–7 [PubMed] [Google Scholar]
- 6.Wolf B, Grier RE, Allen RJ, et al. Phenotypic variation in biotinidase deficiency. J Pediatr 1983;103:233–7 [DOI] [PubMed] [Google Scholar]
- 7.Dahiphale R, Jain S, Agrawal M. Biotinidase deficiency. Indian Pediatr 2008;45:777–9 [PubMed] [Google Scholar]
- 8.Gulati S, Passi GR, Kumar A, et al. Biotinidase deficiency–a treatable entity. Indian J Pediatr 2000;67:464–6 [DOI] [PubMed] [Google Scholar]