


Abstract
Alternative splicing of pre-mRNA generates protein diversity. Dysfunction of splicing machinery and expression of specific transcripts has been linked to cancer progression and drug response. Exon microarray technology enables genome-wide quantification of expression levels of the majority of exons and facilitates the discovery of alternative splicing events. Analysis of exon array data is more challenging than the analysis of gene expression data and there is a need for reliable quantification of exons and alternatively spliced variants. We introduce a novel, computationally efficient methodology, Multiple Exon Array Preprocessing (MEAP), for exon array data pre-processing, analysis and visualization. We compared MEAP with existing pre-processing methods, and validation of six exons and two alternatively spliced variants with qPCR corroborated MEAP expression estimates. Analysis of exon array data from head and neck squamous cell carcinoma (HNSCC) cell lines revealed several transcripts associated with 11q13 amplification, which is related with decreased survival and metastasis in HNSCC patients. Our results demonstrate that MEAP produces reliable expression values at exon, alternatively spliced variant and gene levels, which allows generating novel experimentally testable predictions.
INTRODUCTION
Alternative splicing is a well-established post-transcriptional mechanism that has an essential role in regulating gene expression. Transcripts from ~95% of multiexon genes undergo alternative splicing and there are more than 100 000 intermediate- to high-abundance alternative splicing events in major human tissues (1). Transcript variation can be caused by multiple processes, such as alternative promoters or polyadenylation by utilizing different 5′ or 3′ exons or introns (2–4). Some transcript variants are associated with diseases such as spinal muscular atrophy, premature-aging disorder and familial dysautonomia (5). Furthermore, several reports have recently shown that alternatively spliced patterns significantly affect a number of cellular events critical for cancer development and progression, including cell proliferation, motility and drug response (6,7).
The abundance of alternatively spliced variants can be quantified with specific exon microarrays, such as Affymetrix Human Exon 1.0 ST Array. This microarray platform contains probes for ~80% of human exons and thus provides an option to quantify expression levels for exons, alternatively spliced variants and genes. Most of the studies using exon array data aim at detecting alternatively spliced events. The most popular method is the splicing index (SI), which measures the difference in the exon-gene expression ratio in two groups (8–13). Other published methods are based on outlier detection (14), correlation-based metrics (8) or weighted fold changes (15). While these methods list putative alternatively spliced events, they do not produce quantitative expression values at exon, transcript and gene levels for downstream analyses. Thus, there is a need for computationally efficient exon array data analysis methodologies that are able to produce reliable exon, alternative splice variant and gene expression levels enabling splicing event identification and other interesting biological studies.
We introduce here a Multiple Exon Array Preprocessing (MEAP) framework for Affymetrix Human Exon 1.0 ST microarray platform. MEAP is designed for large-scale exon array data analysis and is computationally efficient. A key feature in MEAP is a novel approach to estimate probe background using genomic and antigenomic background probes. This allows more reliable expression estimation than the existing background correction methods as shown in our case studies. Another novel feature in MEAP is its ability to calculate robust expression estimates especially for alternative splice variants. Here, we demonstrate the utility of MEAP by quantifying alternative splice variant expression levels from 15 head and neck squamous cell carcinoma (HNSCC) cell lines and verifying experimentally randomly selected findings.
MATERIALS AND METHODS
MEAP consists of background correction, normalization, data summarization, differential analysis and visualization as illustrated in Figure 1.
Figure 1.

MEAP workflow. The workflow contains modules for data pre-processing, differential expression analysis, and visualization. Data pre-processing uses a novel background estimation model (PM-BayesBG) and is followed by collective quantile normalization and multi-dimensional expression summarization based on user defined data type (probeset, exon, spliced variant or gene). Differential analysis enables finding biologically interesting targets. MEAP also includes web-based visualization for alternatively spliced events. See http://csbi.ltdk.helsinki.fi/meap/example/MEAPvisual/MEAP_visual_homepage.html for more information.
MEAP algorithm
MEAP is distributed as an R-package and tested on Linux. It is also implemented as a pipeline in the Anduril computational framework (16). The Anduril compliance enables the use of a wide variety of multivariate statistical tools, pathway and Gene Ontology methods for the MEAP processed exon microarray data. MEAP contains also Linear Models for Microarray Data R-package (limma) (17) in addition to t-test. The P-values from t-test are not corrected for multiple hypotheses testing by default. The MEAP package with a comprehensive user guide and precomputed probe annotations for human, mouse and rat are available at http://csbi.ltdk.helsinki.fi/meap/index.html.
MEAP annotations
Annotations for exon array probes should be updated when the reference genomes are updated (18). In MEAP, we construct annotation database to support summarization of either probeset or exon expression. Annotation was done by using BLAST alignment with 24 bp perfect match (identity 100%) for all main probes on Affymetrix Exon 1.0 array against Ensembl core database (version 58). We collected probes mapping to exonic regions and discarded probes mapping to different loci in the genome. MEAP annotation for human, rat and mouse can be downloaded at http://csbi.ltdk.helsinki.fi/meap/download.html. The probe annotations used in MEAP are updated regularly when updates to the reference genome are published.
MEAP background estimation model
Affymetrix GeneChip Exon 1.0 ST Array contains a pool of genomic and antigenomic background probes (BGP) that can be used to estimate background expression levels. So far published background correction models for exon array are global, PM-GCBG (19) and MAT (20,21). Briefly, the global background model estimates universal background from the median intensity of probes with different GC content. PM-GCBG predicts the background of a perfect match probe by the median intensity of BGP with the same GC content. The MAT model estimates probe background intensities with an 80-parameters linear model that considers the composition of nucleotides at each position in a probe sequence.
We introduce a novel sequence-specific background model (PM-BayesBG) that estimates the probe background signals from the nucleotide composition of a probe sequence. PM-BayesBG uses a naive Bayes approach for background signal estimation as follows. Let the random variable θ denote probe background intensity. The background estimation model estimates the probability of background intensity given a probe sequence s, i.e. P(θ|s). This can be formulated with Bayes' equation:
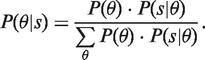 |
(1) |
The variables in Equation 1 are estimated from the background probe intensities. The continuous antigenomic and genomic background probe intensities are discretized into separated classes with user defined interval value. Thus, the prior probability of each background class is calculated by the number of probes in a class divided by the total number of background probes; 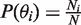 .
.
In each background class, the label is taken to be the median of the intensities belonging to that background class. For each background class we compute a base occurrence rate matrix (BORM) that contains probabilities  for the occurrence of the four possible nucleotides {A,T,G,C} at each locus j in s. An example of a BORM for a typical 25nt probe is given in Table 1.
for the occurrence of the four possible nucleotides {A,T,G,C} at each locus j in s. An example of a BORM for a typical 25nt probe is given in Table 1.
Table 1.
An example of base occurrence rate matrix (BORM)
| Nucleotide/probe sequence | A | G | G | T | A | … |
|---|---|---|---|---|---|---|
| A | 0.6 | 0.3 | 0.1 | 0.1 | 0.7 | … |
| G | 0.1 | 0.4 | 0.3 | 0.1 | 0.1 | … |
| C | 0.2 | 0.3 | 0.5 | 0.6 | 0.2 | … |
| T | 0.1 | 0 | 0.1 | 0.2 | 0 | … |
The BORMs are used to calculate the likelihood for i-th background category (L(s|θi)), where we use the occurrences of each nucleotide in a probe sequence. From pj we choose a value for the nucleotide corresponding to the j-th nucleotide in s and denote it as pi,sj. For example, in Table 1, if j = 2 then 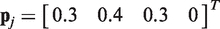 , sj = {G} and pi,sj = 0.4. We assume that each locus j in the probe sequence s is independent from its neighbor nucleotides. Therefore, for i-th BORM we can use pi,sj to calculate the likelihood function:
, sj = {G} and pi,sj = 0.4. We assume that each locus j in the probe sequence s is independent from its neighbor nucleotides. Therefore, for i-th BORM we can use pi,sj to calculate the likelihood function:
| (2) |
The background intensity for a probe, which is used in the MEAP background correction step, is estimated from the maximum posterior probability 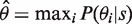 , where
, where
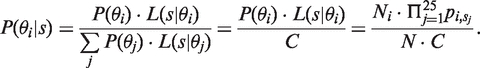 |
(3) |
Expression summarization
MEAP annotation gives mappings for ‘probe-probeset’ and ‘probe-exon’. Mapping probes to their corresponding exons, i.e. skipping the probeset level, allows the use of a larger number of probe intensity values to summarize exon expression values than probesets having four probes per set. Median polishing (22) is used in MEAP exon summarization where probes are mapped uniquely to the exon regions.
In MEAP, the alternatively spliced variant level expression is quantified by considering the problem of transforming the exon-level data to transcripts as a least squares problem. The idea is similar to non-negative matrix factorization approach introduced by Wang et al. (23) and further developed in (24) and (25) for exon or junction probes. For i-th gene having m exons and n transcripts in Ensembl by transcript quality control, we define  and
and  . Transcript expression values
. Transcript expression values 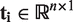 are solved from the equation Ati = e using the QR decomposition to ensure numerical stability (26). If the equation cannot be solved or negative solutions appear in some samples, transcripts' expression values are denoted as missing values. If there are negative solutions or no solution for all samples, the transcripts of the query gene will be removed from the final expression matrix.
are solved from the equation Ati = e using the QR decomposition to ensure numerical stability (26). If the equation cannot be solved or negative solutions appear in some samples, transcripts' expression values are denoted as missing values. If there are negative solutions or no solution for all samples, the transcripts of the query gene will be removed from the final expression matrix.
As an example, suppose that a gene g contains three transcripts {t1,t2,t3} and three exons {e1,e2,e3} with summarized intensity values {1024,724.1,512}. Assume further that the transcripts t1, t2 and t3 are composed of {e1,e2,e3}, {e1,e3} and {e1,e2}, respectively, giving:
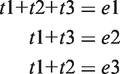 |
(4) |
Thus, the transcript expression vector tg can be solved by
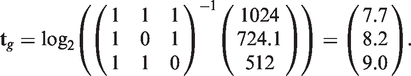 |
(5) |
MEAP also allows the use of transcript quality in the analysis. Ensembl transcript annotation data, which are used in the MEAP quality-control step, are fetched from several databases including UniProt-SwissProt, UniProt-TrEMBL and NCBI Refseq. In the quality-control step only transcripts that are curated and thus match to any of CCDS, UCSC, NCBI RefSeq or Havana/Ensembl merges are retained in the downstream analysis.
A schematic of steps used to produce gene level data is illustrated in Supplementary Figure S1. Briefly, probes that map to the user specified number of the gene's alternatively spliced variants (default 60%) are used to summarize the expression at the gene level.
In qPCR, Ct (cycle threshold) value represents the cycle number at which a reaction reaches a fluorescent intensity that supersedes fluorescence background. Thus, we use here −Ct values to indicate gene expression levels. Correlations between MEAP estimated gene expression values and qPCR normalized −Ct values for eight reference genes from 15 HNSCC cell lines are shown in Supplementary Figure S2.
Parallel computing in MEAP
The calculations in MEAP are executed in parallel to achieve efficient memory and CPU consumption. The parallelization is implemented with the OpenMPI (27) and Rmpi (28) R-packages.
Head and neck cancer cell culture
Head and neck squamous cell carcinoma (HNSCC) cell lines from tongue UT-SCC-21, UT-SCC-24B, UT-SCC-30, UT-SCC-67, UT-SCC-73, UT-SCC-76A, UT-SCC-81, UT-SCC-87, UT-SCC-95 and larynx UT-SCC-8, UT-SCC-11, UT-SCC-75 were provided by the Department of Otorhinolaryngology-Head and Neck Surgery at the Turku University Central Hospital (Turku, Finland). Cells were cultured in DMEM supplemented with l-glutamine and 10% fetal bovine serum at 37°C in an atmosphere of 5% CO2. HNSCC cell lines SCC-4, SCC-9, SCC-25 were ordered from American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured according to the ATCC recommendations.
Exon expression array experiments
HNSCC samples were preprocessed for Affymetrix Human Exon 1.0 ST microarrays using Affymetrix GeneChip Whole Transcript (WT) Sense Target Labeling Assay according to the manufacturer's instructions (manual version 4). Total RNA from HNSCC cell lines was isolated using RNeasy mini kit from Qiagen. The quality of total RNA was assessed with Agilent Bioanalyzer. The starting amount of total RNA was 1 μg. Single-stranded cDNA was first synthesized from total RNA using engineered random primers with T7 promoter sequences. Single-stranded cDNA was then converted to double-stranded cDNA which was further used as a template for IVT reaction to synthesize and amplify antisense cRNA with T7 RNA polymerase. Sense-stranded cDNA was produced by the reverse transcription of cRNA using random primers. cRNA template was hydrolyzed with RNase H leaving single-stranded cDNA. 5.5 μg of single-stranded cDNA was enzymatically fragmented and end-labeled with Affymetrix DNA Labeling Reagent. Labeled cDNA was hybridized on Human Exon 1.0 ST microarray for 17 h, washed and stained using Affymetrix Fluidics Station and scanned with Affymetrix GeneChip Scanner. The exon array data are deposited to GEO with accession number GSE27501.
Sample preparation for qRT-PCR
RNase-Free DNase Set (Qiagen) was used to remove contaminating genomic DNA in RNA samples. Concentration and purity of RNA was determined using the Qubit Quantification Platform (Invitrogen) and Quant-iT RNA assay kit (Invitrogen). Quant-iT dsDNA HS Assay Kit was used to evaluate DNA contamination of RNA samples. Reverse transcription was carried out using QuatiTect Reverse Transcription Kit with additional elimination of genomic DNA (Qiagen). One microgram of RNA was incubated in gDNA Wipeout Buffer for 2 min at 42°C and placed on ice. RNA templates were added to reverse transcription master mix containing optimized blend of oligo-dT and random primers. The reactions were incubated for 15 min at 42°C and heat-inactivated for 3 min at 95°C.
Primer design and qRT-PCR
qRT-PCR was carried out using the LightCycler480 and SYBR Green dye system (Roche) for exon expression analysis or Probe Master TaqMan (Roche) for transcript expression analysis. RT-PCR primers for reference genes and exons were designed by SIGMA (oligo.design@sial.com). Sufficient qRT-PCR assay efficiencies were determined by standard curves of serial dilutions of cell line cDNA. The TaqMan assays for the quantification of the differentially expressed transcripts were designed by TIB MOLBIOL (www.tibmolbiol.de). All primer/probe information is given in Table 2, Table 3 and Supplementary Table S1. Ensembl IDs and corresponding annotations are from Ensembl v.58. TaqMan assay for human β-actin has been published by Kreuzer et al. (29): b-ACTIN forward primer AGCCTCGCCTTTGCCGA, b-ACTIN reverse primer CTGGTGCCTGGGGCG, ACTIN TM 6FAM-CCgCCgCCCgTCCACACCCgCC–BBQ. Each sample was measured in duplicate and the data were analyzed by the ΔCt method for comparing relative expression results.
Table 2.
Primer sequences designed for exon qRT-PCR validation
| Exon | Gene | Forward | Reverse |
|---|---|---|---|
| ENSE00000833461 | TTC26 | AGTATATTCTCAAAGGAGTG | ACCCATTTCCTGGCCAA |
| ENSE00000855493 | PMP22 | AGAACTTGCCGCCAGAAT | GTGGAGGACGATGATACTCAG |
| ENSE00001382781 | PDE4A | TTGTCAGGAGTCGAGGAA | CTGTGCCATAACTTCCAA |
| ENSE00001131505 | EMP3 | CCTCATTCTCTGCTGTCT | GCATAGAAGAGACCTCCT |
| ENSE00001628012 | DBNL | CGGCTTGGCAGACTCA | GGGATGCAGGAAGGATGT |
| ENSE000001442185 | KCNK1 | CTGAGGGTTTTATCTCCTGATTTG | GCTCTCTCCTTTAGGCACTT |
| ENSE00001597418 | ACTB | CTTCACCACCACGGC | CCATCTCTTGCTCGAAG |
| ENSE00001904310 | HPRT1 | GCCTAAGATGAGAGTTCAAGTT | AACAACAATCCGCCCAAA |
Table 3.
Primers and probes for TaqMan assays
| Transcript | EnsemblID | Primer/probe name | Targeted exon index |
|---|---|---|---|
| ORAOV1-201 | ENST00000279147 | ORAOV1_F forward | Exon 3 |
| ORAOV1_147R reverse | Exon 5 | ||
| ORAOV1_V1_TM | Exon 4 | ||
| ORAOV1-202 | ENST00000376587 | ORAOV1_F forward | Exon 3 |
| ORAOV1_587R reverse | Exon 6 | ||
| ORAOV1_V1_TM | Exon 4 | ||
| NEO1-201 | ENST00000339362 | NEO_V2_S forward | Exon 25–26 |
| NEO_V2_A reverse | Exon 26 | ||
| NEO_V2_TM | Exon 26 | ||
| NEO1-202 | ENST00000379842 | NEO_V3_F forward | Exon 18–19 |
| NEO_V3_A reverse | Exon 20–21 | ||
| NEO_V3_TM | Exon 20 |
The transcript names in the 1st column are used in the text. The targeted exons and primer/probe sequences are given in Supplementary Table S1.
Determination of endogenous reference genes
A panel of eight reference genes was used, comprising commonly used reference genes and genes identified as being consistently expressed across the exon array data set at a level similar to the expression level of exons selected for validation. Expression of reference genes was evaluated from 15 HNSCC cell lines. Samples were tested in duplicate and median values were calculated. Using geNorm (30) and BestKeeper (31) software, β-actin and HPRT1 were selected as best housekeeping genes for the cell lines used in this study.
Statistical analysis of the qRT-PCR data
qRT-PCR data were analyzed according to the ΔCt approach. In short, the mean of duplicate measures of the exon target in each sample was normalized using the mean of the two selected endogenous reference genes. β-actin served as an endogenous control to normalize the expression levels of NEO1 and ORAOV1 splice variants. Exons and splice variants expression fold changes were calculated by dividing the mean expression of the 11q13+ samples by the mean expression of the 11q13− samples. Two separated one-sided Mann–Whitney tests were applied to evaluate the statistical significance of differences in the relative mRNA expression levels of exons and splice variants between 11q13+ and 11q13− samples.
RESULTS
Background correction comparison using colon cancer data
Background correction is one of the most important steps in quantifying the probe intensities as the errors in correcting for background can propagate to downstream analyses, which may severely bias the results.
To illustrate the impact of different background correction approaches to probe intensities, we tested four approaches [global, PM-GCBG (19), MAT (20,21) and here introduced PM-BayesBG] using a colon cancer exon microarray data set consisting of 14 samples from Affymetrix public resources. Each exon array contains 37 687 antigenomic and genomic background probes. As these sequences should not be present in the samples, the ratio between a probe intensity and the estimated background signal should equal to one. With this experimental setting we can compare the performance of different background models.
The results for background correction comparison are illustrated in Figure 2. While all methods resulted in a number of outliers, global, PM-GCBG and MAT models produced several gross outliers (>10× fold change) that are absent with PM-BayesBG. We further used ANOVA to test whether the means of ratios are significantly different. With the significance level of P < 0.05, the means for the PM-GCBG and global background models were higher than the ones obtained with PM-BayesBG and MAT. The mean of ratios based on the MAT method is also slightly higher than the one resulted by PM-BayesBG but the difference is not statistically significant.
Figure 2.

Boxplot for the ratios of raw intensity of each probe versus estimated background signal. We used human colon cancer exon array data from Affymetrix public resource and calculated signals of antigenomic and genomic background probes on global, PM-GCBG, MAT and PM-BayesBG background correction models. Ideally, the background intensities of these probes from a background model should be the same as their detected signals. MEAP has the lowest mean ratio according to the mean ratios labeled in the figure.
These results indicate that the PM-BayesBG and MAT background models are better in estimating probe background signals than PM-GCBG and global background models. In particular, the global background correction model results in a very large number of outlier values, which are likely to affect the downstream analyses negatively. PM-BayesBG resulted in fewer outliers than the other methods and it requires less parameters to be estimated than in MAT, which uses an 80-parameters linear model (21).
Identification of differentially expressed exons in HNSCC
Identification of differentially expressed genes or transcripts between two or more conditions is one of the most important objectives in studies using transcriptome data. As exon array gives an opportunity to quantify also exon expression levels, we compared the performance of four pre-processing methods (RMA, FIRMA, PLIER, MEAP) in identification of differentially expressed exons (DEEs) in HNSCC cell lines.
One of the most common genomic alterations in HNSCC is amplification of the chromosome region 11q13. This alteration is present in 30–50% of HNSCC patients (32) and patients with this amplification are shown to have higher propensity for metastasis and decreased survival (33–35). Here, we use exon array data from seven HNSCC cell lines having 11q13 amplification (11q13+) and eight cell lines without this amplification (11q13−) to compare exon array pre-processing algorithms.
We quantified exon level expression with RMA, PLIER (Affymetrix Power Tool with PM-GCBG background correction) and FIRMA by translating the probeset values to exon level by taking a median of probesets belonging to the exon region according to the Ensembl database (v.58) (36). For MEAP we generated exon level expression directly from probes as explained in ‘Materials and Methods’ section. The number of DEEs varied greatly between the pre-processing methods (RMA: 4420; PLIER: 7620; FIRMA: 4303; MEAP: 7284). The methods agreed in 2383 DEEs (nominal P-value <0.05 and absolute fold change >2) out of 12 650 unique DEEs identified by at least one method. The number of common DEEs is unexpectedly small and indicates that the choice of the exon array pre-processing method has a large impact to the results.
We then validated randomly chosen four exons that were identified as DEEs by MEAP alone, as well as two exons that were not identified as DEEs by most methods (Table 4). The expressions of these six exons were quantitated with qRT-PCR in all 15 HNSCC cell lines and the results are shown in Figure 3. The box plots show three outliers in three exons and these were excluded from the statistical analyses. Four exons that were identified as DEEs by MEAP alone were statistically significantly different also in the qRT-PCR experiments. Likewise, the two exons that were not identified as DEEs by most of the methods were not significantly different in the qRT-PCR experiments. These results demonstrate that MEAP is able to produce exon expression estimates that allow reliable identification of differentially expressed exons.
Table 4.
Fold changes and P-values of six randomly selected exons with MEAP, RMA, PLIER and FIRMA
| ExonID | MEAP | PLIER | RMA | FIRMA |
|---|---|---|---|---|
| P-value, FC | P-value, FC | P-value, FC | P-value, FC | |
| ENSE00001442185 | 0.0272, 1.7658 | 0.0278, 1.4750 | 0.0179, 2.1503 | 0.0472, 1.8533 |
| ENSE00001628012 | 0.0422, 0.6100 | 0.0387, 0.6588 | 0.0065, 0.5370 | 0.0139, 0.4833 |
| ENSE00000833461 | 0.0328, 0.2246 | 0.1218, 0.6572 | 0.0075, 0.7272 | 0.0111, 0.7761 |
| ENSE00000855493 | 0.0130, 0.2531 | 0.2772, 0.5612 | 0.0191, 0.6641 | 0.0256, 0.6345 |
| ENSE00001382781 | 0.0043, 2.2765 | 0.0443, 1.4478 | 0.0230, 1.5832 | 0.0132, 1.7690 |
| ENSE00001131505 | 0.0376, 0.3468 | 0.0392, 0.6565 | 0.0324, 0.7334 | 0.0332, 0.7119 |
Quantitative exon expression values were generated from the median intensity of corresponding probeset expression values for PLIER, RMA, FIRMA and directly from probe intensities for MEAP. Four differentially expressed exons found by MEAP alone are ENSE00000833461, ENSE00000855493, ENSE00001382781 and ENSE00001131505. The other two exons (ENSE00001442185 and ENSE00001628012) without significant differential expression are found by most of the methods. The threshold defining a significant differentially expressed exon was P < 0.05 and linear-scale fold change more than two. The first column of the bolded values denotes significances (P-values). The bolding refers to exons that were detected by MEAP alone.
Figure 3.

Boxplot of the normalized −Ct values for randomly selected exons in HNSCC samples. Exons ENSE00000833461, ENSE00000855493, ENSE00001131505 and ENSE00001382781 are statistically significantly differentially expressed between 11q13+ and 11q13− groups (**P < 0.01; *P < 0.05). ○ represents an outlier.
Identification of differentially expressed alternative splice variants in HNSCC
Quantification of alternative splice variants is a non-trivial task because an exon may belong to several variants. As an exon expression value may have been influenced by several alternatively spliced variants, simply averaging exons associated with an alternatively spliced variant may severely bias the transcript expression values. In order to address this issue, we use a linear algebra based approach to quantify alternatively spliced variants (see ‘Materials and Methods’ section for details).
To demonstrate the benefits of the MEAP in quantifying the expression values for alternatively spliced variants, we analyzed alternatively spliced variants of three genes (ORAOV1, ANO1 and PPFIA1) located in the 11q13 amplified region and associated with HNSCC. ORAOV1 is involved in the regulation of cell growth, apoptosis and tumor angiogenesis (37, 38). Furthermore, the amplification of ORAOV1 is associated with several clinicopathological features (39, 40). ANO1 has been reported to function as a calcium-dependent chloride channel (CaCC) (41). Overexpression of ANO1 in cancer cells with low endogenous ANO1 levels stimulates cell migration and channel activity is required for this effect (42). Alternative splicing of ANO1 is an important mechanism to regulate channel properties, such as sensitivity to calcium (43). PPFIA1 is a member of the LAR protein-tyrosine phosphatase-interacting protein (liprin) family and it is involved in the regulation of cell spreading and migration (44–46). Alternatively spliced variants of PPFIA1 are regulated in a developmental- or region-specific manner (47).
Expression values of alternatively spliced variants and fold changes for ORAOV1, ANO1 and PPFIA1 genes are given in Table 5. ORAOV1-201 and ORAOV1-203 are clearly overexpressed in 11q13+ samples, whereas ORAOV1-202 expression is almost identical in both the 11q13+ and 11q13− samples. This is evident also from Figure 4 that contains MEAP visualization for the ORAOV1 gene. For ANO1, all variants were 1.9–2.8 times more expressed in 11q13+ than in 11q13− samples. Our results further suggest that alternatively spliced variant PPFIA1-201 is very strongly expressed whereas PPFIA1-202 is weakly expressed. Furthermore, PPFIA1-201 is three times more expressed in 11q13+ samples as in 11q13− samples.
Table 5.
MEAP quantification of transcripts for genes ANO1, PPFIA1, ORAOV1 and NEO1
| Transcript | EnsemblID | GeneName | 11q13+ | 11q13− | Fold change |
|---|---|---|---|---|---|
| ANO1-201 | ENST00000316296 | ANO1 | 92.347 | 48.840 | 1.891 |
| ANO1-202 | ENST00000355303 | ANO1 | 188.707 | 67.696 | 2.788 |
| ANO1-203 | ENST00000398543 | ANO1 | 321.795 | 115.520 | 2.786 |
| PPFIA1-201 | ENST00000253925 | PPFIA1 | 629.473 | 209.528 | 3.004 |
| PPFIA1-202 | ENST00000389547 | PPFIA1 | 19.522 | 12.159 | 1.606 |
| ORAOV1-201 | ENST00000279147 | ORAOV1 | 91.076 | 23.687 | 3.845 |
| ORAOV1-202 | ENST00000376587 | ORAOV1 | 16.268 | 15.649 | 1.040 |
| ORAOV1-203 | ENST00000441922 | ORAOV1 | 62.813 | 23.884 | 2.630 |
| NEO1-001 | ENST00000261908 | NEO1 | 38.639 | 16.000 | 2.415 |
| NEO1-201 | ENST00000339362 | NEO1 | 4.780 | 1.456 | 3.283 |
| NEO1-202 | ENST00000379842 | NEO1 | 46.656 | 18.975 | 2.459 |
In our HNSCC dataset, there are 7 samples in 11q13+ and 8 samples in 11q13− group. Median is applied to represent the expression of each transcript in 11q13+ or 11q13− group. Expression and fold changes in this table are on linear-scale.
Figure 4.

MEAP splice visualization plot for gene ORAOV1. Top ‘P-value’, ‘Fold Change’ and ‘Expression Curve’ sections display the t-test P-values (−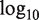 ), fold change (
), fold change (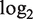 ), expression intensities of each exon in a gene between two sample groups (Group1: 11q13+; Group2: 11q13−). Gaps may occur when there are no probes designed against a specific exon region. Sections `Expression' and `Transcript' give the exon expression profile and the expression of each splice variant of ORAOV1 in 11q13+/11q13−. G1/G2 in the exon expression profile corresponds to the fold change between 11q13+ and 11q13− samples, in which green represents downregulation and red represents upregulation. Expression values are on
), expression intensities of each exon in a gene between two sample groups (Group1: 11q13+; Group2: 11q13−). Gaps may occur when there are no probes designed against a specific exon region. Sections `Expression' and `Transcript' give the exon expression profile and the expression of each splice variant of ORAOV1 in 11q13+/11q13−. G1/G2 in the exon expression profile corresponds to the fold change between 11q13+ and 11q13− samples, in which green represents downregulation and red represents upregulation. Expression values are on 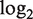 -scale.
-scale.
In addition to analyzing genes inside the 11q13 region, we identified all differentially expressed transcripts (DETs) between 11q13+ and 11q13− samples. This resulted in 615 DETs with P < 0.05 and absolute fold change of at least two. In this experimental setting amplifications and deletions independent of the 11q13 amplification may confound the results as these 11q13-independent genomic aberrations, which are abundant in these 15 HNSCC cell lines (48), may introduce high or low expression values. In order to overcome this confounding factor, we used copy number data from the same 15 cell lines (Lepikhova et al. in preparation) and excluded 285 DETs that mapped to amplified/deleted regions. The list of the remaining 330 11q13-specific DETs and their expression levels is given in Supplementary Table S2.
To verify the results produced by MEAP, we performed qRT-PCR for alternatively spliced variants of ORAOV1 and NEO1 in all 15 HNSCC cell lines. NEO1 (neogenin homolog 1), whose alternative splice variants' fold changes were 2.4–3.3, was randomly chosen from the list of 330 11q13-specific DETs. Two NEO1 alternative splice variants (NEO1-202 and NEO1-001) were 8–13 fold up-regulated as compared to NEO1-201 in 11q13+ and 11q13− samples. For NEO1 we experimented the expression levels of the two transcripts NEO1-201 and NEO1-202, since there is no specific assay for the third alternative splice variant NEO1-001. The hypothesis here is that NEO1-202 is expressed at a higher level than NEO1-201 in both 11q13+ and 11q13− samples, and both NEO1-201 and NEO1-202 are overexpressed in 11q13+ samples. For ORAOV1, we experimented whether ORAOV1-201, but not ORAOV1-202, is significantly overexpressed in 11q13+ samples as predicted by MEAP.
The qRT-PCR results for NEO1 and ORAOV1 are shown in Figure 5. We performed two independent one-sided Mann–Whitney U tests using the normalized Ct values (ΔCt) between the 11q13+ and 11q13− HNSCC samples. There is a significant increase in the expression of ORAOV1-201 (P < 0.0006) in the 11q13+ group, whereas no significant changes were observed for ORAOV1-202 (P < 0.0837). Both NEO1-201 (P < 0.0320) and NEO1-202 (P < 0.0246) were significantly upregulated in the 11q13+ samples. Furthermore, NEO1-201 was expressed at a lower level as compared to NEO1-202 in both sample sets. These results corroborate the predictions made with MEAP and demonstrate that MEAP is able to produce reliable expression values for alternatively spliced variants.
Figure 5.

Relative expression level of spliced variants from qRT-PCR. NEO1-201 and NEO1-202 both have high relative expression values in the 11q13+ group compared to the 11q13− group. Meanwhile, the expression level of NEO1-201 is lower than NEO1-202 in both the 11q13+ and 11q13− groups. For ORAOV1, transcript ORAOV1-201 is overexpressed in the 11q13+ group. The SD of ΔCt values in the 11q13+ group for transcript ORAOV1-202 is high and such a variant does not have significant expression changes between two groups. (**P < 0.01; *P < 0.05).
DISCUSSION
Alternative splicing plays an important role in a wide spectrum of biological processes. Exon microarray technology enables quantitative measurement of exon-specific expressions, which allows advanced analysis of alternatively spliced transcripts in complex diseases. We have designed and implemented a comprehensive exon-array data processing approach that is able to produce trustworthy data at probeset, exon, alternatively spliced variant and gene levels using a novel background estimation model. MEAP is implemented to take advantage of distributed computing and thus it is able to process large sets of exon arrays rapidly. For instance, analysis of 500 glioblastoma multiforme primary tumors subjected to exon arrays (49) with MEAP took 4 h with a small cluster (data not shown). MEAP also has a visualization interface that facilitates identification of novel alternatively spliced variants.
In order to assess performance of MEAP and three other exon array processing methods, we assessed their performance in background correction, identification of differentially expressed exons and alternatively spliced variants. The key observations were further validated with qRT-PCR. The results demonstrate that the MEAP generated expression values for exons and alternatively spliced variants are trustworthy, which facilitates advanced downstream analyses. The major reason for MEAP to outperform other exon array pre-processing methods is its background correction model together with linear algebra-based method to calculate expressions of alternatively spliced variants using exon expression values. A drawback of the linear algebra approach is that sometimes expression value for an alternatively spliced variant may become negative. This is due to small values within some exons in the transcript as well as missing probes for an exon (Affymetrix exon array contains probes for ~80% of the exons). However, MEAP is able to result in an expression value at the gene level even in cases where alternatively spliced variant expressions cannot be produced as shown in Supplementary Figure S2 in which the MEAP gene expression values correlate well (R = 0.76, … , 0.95) with qPCR quantifications.
We identified 330 differentially expressed transcripts between 11q13+ and 11q13− samples. These variants belong to several genes involved in the regulation of proliferation, survival, adhesion and invasion, including NEO1, NME1, FAP, CYP26A1. After identifying the differentially expressed alternatively spliced variants between 11q13+ and 11q13− samples we further corroborated the expression pattern of NEO1 alternatively spliced variants with qRT-PCR.
NEO1 encodes a protein with 50% amino acid homology to the human tumor suppressor molecule deleted in colon cancer (DCC) though DCC and neogenin mediate different responses (50–52). Neogenin is present in tissues where active growth takes place, and ubiquitous expression of neogenin was observed in a wide variety of human cancers (50,51). The role of NEO1 in cancer progression, however, is controversial (53,54). Our results show that all alternatively spliced variants of NEO1 were co-expressed in 15 HNSCC cell lines and overexpressed in 11q13+ compared to 11q13− sample groups. However, NEO1-201 was expressed 8–10 times lower as compared to NEO1-001 and more than 10 times lower as compared to NEO1-202 in 11q13+ and 11q13− cell lines. In contrast, the difference in expression level between NEO1-201 and NEO1-001 or NEO1-202 variants in two normal keratinocyte cell lines was only 7-fold (data not shown). As neogenin is a multi-functional receptor regulating many diverse processes (55,56), both the nature of the ligand and alternatively spliced variants could be responsible for the different functional outcomes of neogenin activation. This could explain different expression levels of alternatively spliced variants not only in cancer such as HNSCC but also during development [57]. NEO1-201 differs from other variants by exclusion of exon 26 which contains several potential phosphorylation sites. Thus, our results suggest that the phosphorylation pattern of neogenin could determine the downstream intracellular signaling through the binding of different molecules, and deletion of exon 26 could lead to abolished interactions and consequently lower expression of NEO1-201.
As alternatively spliced variants of several cancer-related genes located at 11q13 amplicon have been reported to differ significantly in their ability to promote tumor progression (58,59), we studied further the alternatively spliced variants located in the 11q13 region. For example, two alternative splice variants of human cortactin, which lack exon 11 or exons 10 and 11, show reduced ability to induce cell migration when compared with the form of cortactin that includes exon 11 (58). While upregulation of a cancer associated splice variant does not necessarily correlate with the gene amplification, there is evidence for amplified and overexpressed genes to show distinct pattern of splice variants in comparison to tumors without amplification (60). Accordingly, we tested whether expression levels of alternative splice variants belonging to the 11q13 region are associated with the amplification status using exon array data from 15 HNSCC cell lines.
Our results suggest that ANO1-203 variant with deletion of exons 13 and 15 is expressed at a higher level compared to ANO1-202 with inclusion of both exon 13 and 15 in all tested HNSCC cell lines. Interestingly, skipping of exon 13 and exon 15 in ANO1 has a strong effect on the properties of CaCC (43). Both ANO1-202 and ANO1-203 were 2.8 times more expressed in the 11q13+ group while the difference in the expression level of ANO1-201 was only 1.9-fold. Thus, our results point to the direction that different ANO1 transcripts are expressed in 11q13+ cases, which could result in alterations in the properties of CaCC.
In summary, these findings indicate that MEAP is able to quantify expression of alternatively spliced variants in complex diseases, such as cancer. When splice variants are translated, this results in a set of closely related, but different proteins that could have different functional properties in normal and cancer cells. Our results highlight a number of alternatively spliced variants that may have an impact in development of head and neck squamous cell carcinoma. While more work is needed to firmly establish the role of these alternatively spliced variants in HNSCC progression, our results demonstrate that MEAP can quantify exon and alternatively spliced transcripts establishing a solid basis for generating experimentally testable predictions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Academy of Finland (grant number 125826), Biocentrum Helsinki; Finnish Graduate School in Computational Sciences (PC); the Finnish Cancer Society; Sigrid Jusélius Foundation. Funding for open access charge: Academy of Finland (project 125826).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Riku Louhimo for the integration study on CGH array and exon array HNSCC data, Javier Núñez-Fontarnau, Ville Rantanen for helping with parallel implementation and memory recording, Kimmo Mattila at CSC—Scientific Computing Ltd for help with installing the genome database and parallel computing with blast. Taru Perttula and Teija Inkinen are thanked for their contribution in HNSCC sample processing and exon array hybridizations. We also thank Ensembl helpdesk for the discussion relating the transcript quality control.
REFERENCES
- 1.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 2.Modrek B, Lee C. A genomic view of alternative splicing. Nat. Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 3.Tian B, Pan Z, Lee JY. Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 2007;17:156–165. doi: 10.1101/gr.5532707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trinklein ND, Aldred SJ, Saldanha AJ, Myers RM. Identification and functional analysis of human transcriptional promoters. Genome Res. 2003;13:308–312. doi: 10.1101/gr.794803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochim. Biophys. Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? Int. J. Biochem. Cell Biol. 2007;39:1432–1449. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Fackenthal JD, Godley LA. Aberrant RNA splicing and its functional consequences in cancer cells. Dis. Model Mech. 2008;1:37–42. doi: 10.1242/dmm.000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Affymetrix Alternative transcript analysis methods for exon arrays. Affymetrix Whitepaper. 2005 http://media.affymetrix.com/support/technical/whitepapers/exon_alt_transcript_analysis_whitepaper.pdf (11 September 2005, date last accessed) [Google Scholar]
- 9.Clark TA, Schweitzer AC, Chen TX, Staples MK, Lu G, Wang H, Williams A, Blume JE. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007;8:R64. doi: 10.1186/gb-2007-8-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gellert P, Uchida S, Braun T. Exon Array Analyzer: a web interface for Affymetrix exon array analysis. Bioinformatics. 2009;25:3323–3324. doi: 10.1093/bioinformatics/btp577. [DOI] [PubMed] [Google Scholar]
- 11.Laajala E, Aittokallio T, Lahesmaa R, Elo LL. Probe-level estimation improves the detection of differential splicing in Affymetrix exon array studies. Genome Biol. 2009;10:R77. doi: 10.1186/gb-2009-10-7-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xing Y, Stoilov P, Kapur K, Han A, Jiang H, Shen S, Black DL, Wong WH. MADS: a new and improved method for analysis of differential alternative splicing by exon-tiling microarrays. RNA. 2008;14:1470–1479. doi: 10.1261/rna.1070208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen S, Warzecha CC, Carstens RP, Xing Y. MADS+: discovery of differential splicing events from Affymetrix exon junction array data. Bioinformatics. 2010;26:268–269. doi: 10.1093/bioinformatics/btp643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irizarry R, Bolstad B, Collin F, Cope L, Hobbs B, Speed T. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moller-Levet CS, Betts GN, Harris AL, Homer JJ, West CM, Miller CJ. Exon array analysis of head and neck cancers identifies a hypoxia related splice variant of LAMA3 associated with a poor prognosis. PLoS Comput. Biol. 2009;5:e1000571. doi: 10.1371/journal.pcbi.1000571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ovaska K, Laakso M, Haapa-Paananen S, Louhimo R, Chen P, Aittomäki V, Valo E, Nún̈ez-Fontarnau J, Rantanen V, Karinen S, et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010;2:65. doi: 10.1186/gm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 18.Yates T, Okoniewski MJ, Miller CJ. X:Map: annotation and visualization of genome structure for Affymetrix exon array analysis. Nucleic Acids Res. 2008;36:D780–D786. doi: 10.1093/nar/gkm779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Affymetrix. Exon array background correction. Affymetrix Whitepaper. 2005 http://media.affymetrix.com/support/technical/whitepapers/exon_background_correction_whitepaper.pdf (27 September 2005, date last accessed) [Google Scholar]
- 20.Johnson WE, Li W, Meyer CA, Gottardo R, Carroll JS, Brown M, Liu XS. Model-based analysis of tiling-arrays for ChIP-chip. Proc. Natl Acad. Sci. USA. 2006;103:12457–12462. doi: 10.1073/pnas.0601180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapur K, Xing Y, Ouyang Z, Wong W. Exon arrays provide accurate assessments of gene expression. Genome Biol. 2007;8:R82. doi: 10.1186/gb-2007-8-5-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okoniewski M, Miller C. Comprehensive analysis of Affymetrix exon arrays using BioConductor. PLoS Comput. Biol. 2008;4:e6. doi: 10.1371/journal.pcbi.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Hubbell E, Hu JS, Mei G, Cline M, Lu G, Clark T, Siani-Rose MA, Ares M, Kulp DC, et al. Gene structure-based splice variant deconvolution using a microarray platform. Bioinformatics. 2003;19:i315–i322. doi: 10.1093/bioinformatics/btg1044. [DOI] [PubMed] [Google Scholar]
- 24.Anton MA, Gorostiaga D, Guruceaga E, Segura V, Carmona-Saez P, Pascual-Montano A, Pio R, Montuenga LM, Rubio A. SPACE: an algorithm to predict and quantify alternatively spliced isoforms using microarrays. Genome Biol. 2008;9:R46. doi: 10.1186/gb-2008-9-2-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anton MA, Aramburu A, Rubio A. Improvements to previous algorithms to predict gene structure and isoform concentrations using Affymetrix Exon arrays. BMC Bioinformatics. 2010;11:578. doi: 10.1186/1471-2105-11-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golub GH, Van Loan CF. Matrix Computations. 1996. 3rd edn. The Johns Hopkins University Press, Baltimore, Maryland, 21218–4319. [Google Scholar]
- 27.Edgar G, Graham EF, George B, Thara A, Jack JD, Jeffrey MS, Vishal S, Prabhanjan K, Brian B, Andrew L, et al. Open MPI: goals, concept, and design of a next generation MPI implementation. 2004 In Proceedings, 11th European PVM/MPI Users’ Group Meeting, Budapest, Hungary, pp. 97–104. [Google Scholar]
- 28.Hao Y. Rmpi: interface (Wrapper) to MPI (Message-Passing Interface) 2010. [Google Scholar]
- 29.Kreuzer KA, Lass U, Landt O, Nitsche A, Laser J, Ellerbrok H, Pauli G, Huhn D, Schmidt CA. Highly sensitive and specific fluorescence reverse transcription-PCR assay for the pseudogene-free detection of beta-actin transcripts as quantitative reference. Clin. Chem. 1999;45:297–300. [PubMed] [Google Scholar]
- 30.Vandesompele J, De PK, Pattyn F, Poppe B, Van RN, De PA, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:34. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004;26:509–515. doi: 10.1023/b:bile.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 32.Muller D, Millon R, Velten M, Bronner G, Jung G, Engelmann A, Flesch H, Eber M, Methlin G, Abecassis J. Amplification of 11q13 DNA markers in head and neck squamous cell carcinomas: correlation with clinical outcome. Eur. J. Cancer. 1997;33:2203–2210. doi: 10.1016/s0959-8049(97)00198-6. [DOI] [PubMed] [Google Scholar]
- 33.Bockmühl U, Schlüns K, Schmidt S, Matthias S, Petersen I. Chromosomal alterations during metastasis formation of head and neck squamous cell carcinoma. Genes Chromosome. Canc. 2002;33:29–35. [PubMed] [Google Scholar]
- 34.Xu C, Liu Y, Wang P, Fan W, Rue TC, Upton MP, Houck JR, Lohavanichbutr P, Doody DR, Futran ND, et al. Integrative analysis of DNA copy number and gene expression in metastatic oral squamous cell carcinoma identifies genes associated with poor survival. Mol. Canc. 2010;9:143. doi: 10.1186/1476-4598-9-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibcus JH, Mastik MF, Menkema L, de Bock GH, Kluin PM, Schuuring E, van der Wal JE. Cortactin expression predicts poor survival in laryngeal carcinoma. Br. J. Cancer. 2008;98:950–955. doi: 10.1038/sj.bjc.6604246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flicek P, Aken BL, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Coates G, Fairley S, et al. Ensembl's 10th year. Nucleic Acids Res. 2010:D557–D562. doi: 10.1093/nar/gkp972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang L, Zeng X, Yang H, Wang Z, Shen J, Bai J, Zhang Y, Gao F, Zhou M, Chen Q. Oral cancer overexpressed 1 (ORAOV1): a regulator for the cell growth and tumor angiogenesis in oral squamous cell carcinoma. Int. J. Cancer. 2008;123:1779–1786. doi: 10.1002/ijc.23734. [DOI] [PubMed] [Google Scholar]
- 38.Jiang L, Zeng X, Wang Z, Ji N, Zhou Y, Liu X, Chen Q. Oral cancer overexpressed 1 (ORAOV1) regulates cell cycle and apoptosis in cervical cancer HeLa cells. Mol. Canc. 2010;9:20. doi: 10.1186/1476-4598-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia J, Chen Q, Li B, Zeng X. Amplifications of TAOS1 and EMS1 genes in oral carcinogenesis: association with clinicopathological features. Oral Oncol. 2006;43:508–514. doi: 10.1016/j.oraloncology.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 40.Komatsu Y, Hibi K, Kodera Y, Akiyama S, Ito K, Nakao A. TAOS1, a novel marker for advanced esophageal squamous cell carcinoma. Anticancer Res. 2006;26:2029–2032. [PubMed] [Google Scholar]
- 41.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 42.Ayoub C, Wasylyk C, Li Y, Thomas E, Marisa L, Robé A, Roux M, Abecassis J, de Reynias A, Wasylyk B. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br. J. Cancer. 2010;103:715–726. doi: 10.1038/sj.bjc.6605823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrera L, Caputo A, Ubby I, Bussani E, Zegarra-Moran O, Ravazzolo R, Pagani F, Galietta LJ. Regulation of TMEM16A chloride channel properties by alternative splicing. J. Biol. Chem. 2009;284:33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen JC, Unoki M, Ythier D, Duperray A, Varticovski L, Kumamoto K, Pedeux R, Harris CC. Inhibitor of growth 4 suppresses cell spreading and cell migration by interacting with a novel binding partner, liprin alpha1. Cancer Res. 2007;67:2552–2558. doi: 10.1158/0008-5472.CAN-06-3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Curtis I. Function of liprins in cell motility. Exp. Cell Res. 2011;317:1–8. doi: 10.1016/j.yexcr.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 46.Astro V, Asperti C, Cangi G, Doglioni C, de Curtis I. Liprin-alpha1 regulates breast cancer cell invasion by affecting cell motility, invadopodia and extracellular matrix degradation. Oncogene. 2010;30:1841–1849. doi: 10.1038/onc.2010.562. [DOI] [PubMed] [Google Scholar]
- 47.Zürner M, Schoch S. The mouse and human Liprin-alpha family of scaffolding proteins: genomic organization, expression profiling and regulation by alternative splicing. Genomics. 2009;93:243–253. doi: 10.1016/j.ygeno.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 48.Järvinen AK, Autio R, Kilpinen S, Saarela M, Leivo I, Gronman R, Makitie AA, Monni O. High-resolution copy number and gene expression microarray analyses of head and neck squamous cell carcinoma cell lines of tongue and larynx. Genes Chromosome. Canc. 2008;47:500–509. doi: 10.1002/gcc.20551. [DOI] [PubMed] [Google Scholar]
- 49.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vielmetter J, Kayyem JF, Roman JM, Dreyer WJ. Neogenin, an avian cell surface protein expressed during terminal neuronal differentiation, is closely related to the human tumor suppressor molecule deleted in colorectal cancer. J. Cell Biol. 1994;127(6 Pt 2):2009–2020. doi: 10.1083/jcb.127.6.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyerhardt JA, Look AT, Bigner SH, Fearon ER. Identification and characterization of neogenin, a DCC-related gene. Oncogene. 1997;14:1129–1136. doi: 10.1038/sj.onc.1200935. [DOI] [PubMed] [Google Scholar]
- 52.Ren XR, Hong Y, Feng Z, Yang HM, Mei L, Xiong WC. Tyrosine phosphorylation of netrin receptors in netrin-1 signaling. Neurosignals. 2008;16:235–245. doi: 10.1159/000111566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho SM, Lau KM, Mok SC, Syed V. Profiling follicle stimulating hormone-induced gene expression changes in normal and malignant human ovarian surface epithelial cells. Oncogene. 2003;22:4243–4256. doi: 10.1038/sj.onc.1206437. [DOI] [PubMed] [Google Scholar]
- 54.Lee JE, Kim HJ, Bae JY, Kim SW, Park JS, Shin HJ, Han W, Kim SW, Kang KS, Noh DY. Neogenin expression may be inversely correlated to the tumorigenicity of human breast cancer. BMC Can. 2005;5:154. doi: 10.1186/1471-2407-5-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cole SJ, Bradford D, Cooper HM. Neogenin: a multi-functional receptor regulating diverse developmental processes. Int. J. Biochem. Cell Biol. 2007;39:1569–1575. doi: 10.1016/j.biocel.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 56.Wilson NH, Key B. Neogenin: one receptor, many functions. Int. J. Biochem. Cell Biol. 2007;39:874–878. doi: 10.1016/j.biocel.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 57.Keeling SL, Gad JM, Cooper HM. Mouse Neogenin, a DCC-like molecule, has four splice variants and is expressed widely in the adult mouse and during embryogenesis. Oncogene. 1997;15:691–700. doi: 10.1038/sj.onc.1201225. [DOI] [PubMed] [Google Scholar]
- 58.van Rossum AG, de Graaf JH, Schuuring-Scholtes E, Kluin PM, Fan YX, Zhan X, Moolenaar WH, Schuuring E. Alternative splicing of the actin binding domain of human cortactin affects cell migration. J. Biol. Chem. 2003;278:45672–45679. doi: 10.1074/jbc.M306688200. [DOI] [PubMed] [Google Scholar]
- 59.Comstock CE, Augello MA, Benito RP, Karch J, Tran TH, Utama FE, Tindall EA, Wang Y, Burd CJ, Groh EM, et al. Clin. Cancer Res. 2009;15:5338–5349. doi: 10.1158/1078-0432.CCR-08-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maas RM, Reus K, Diesel B, Steudel WI, Feiden W, Fischer U, Meese E. Amplification and expression of splice variants of the gene encoding the P450 cytochrome 25-hydroxyvitamin D(3) 1,alpha-hydroxylase (CYP 27B1) in human malignant glioma. Clin. Cancer Res. 2001;7:868–875. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.