


Abstract
Phenotypes are investigated in model organisms to understand and reveal the molecular mechanisms underlying disease. Phenotype ontologies were developed to capture and compare phenotypes within the context of a single species. Recently, these ontologies were augmented with formal class definitions that may be utilized to integrate phenotypic data and enable the direct comparison of phenotypes between different species. We have developed a method to transform phenotype ontologies into a formal representation, combine phenotype ontologies with anatomy ontologies, and apply a measure of semantic similarity to construct the PhenomeNET cross-species phenotype network. We demonstrate that PhenomeNET can identify orthologous genes, genes involved in the same pathway and gene–disease associations through the comparison of mutant phenotypes. We provide evidence that the Adam19 and Fgf15 genes in mice are involved in the tetralogy of Fallot, and, using zebrafish phenotypes, propose the hypothesis that the mammalian homologs of Cx36.7 and Nkx2.5 lie in a pathway controlling cardiac morphogenesis and electrical conductivity which, when defective, cause the tetralogy of Fallot phenotype. Our method implements a whole-phenome approach toward disease gene discovery and can be applied to prioritize genes for rare and orphan diseases for which the molecular basis is unknown.
INTRODUCTION
Animal models are used to investigate and understand the mechanisms underlying human disease (1,2). To facilitate the study and discovery of disease mechanisms, model organism databases include descriptions of the phenotypes that are associated with specific genotypes (e.g. an allelic composition in a background strain) in a defined environment. The ultimate aim of having a phenotypic description of null mutations for every gene in an organism is currently being pursued by the mouse community through the International Mouse Phenotyping Consortium (IMPC) (3) using systematic knockouts of all the protein coding genes in the mouse genome generated by the International Knockout Mouse Consortium (IKMC) (4). With the increasing availability of large volumes of phenotype data, automated comparative analyses that systematically relate and compare phenotypes within and across species become critical if we are to maximally exploit this rich data.
Several approaches to computational cross-species phenotype comparison have been explored to date, some making use of the semantic information contained in the ontologies (5) and others, such as PhenomicDB (6), using only lexical matching. More recently, in a predominantly lexical approach, the National Library of Medicine's UMLS thesaurus (7) has been used to map the Mammalian Phenotype ontology (MP) to human disease concepts (8). This approach suffers from the different conceptualizations of phenotype and disease in humans and mice and does not exploit the full semantic information contained in the relevant ontologies (2). To the best of our knowledge, our approach is the only one which utilizes the complete phenotypic repertoire of the organisms included in the framework, and uses automated reasoning over all of the phenotype ontologies to generate a representation that can be explored through measures of phenotypic similarity.
Biomedical ontologies are a means to capture and integrate research data across domains, species and levels of granularity. They formally specify the meaning of terms in a vocabulary so that the nature of the data can be understood and processed both by humans and machines. To express this meaning, ontologies utilize formal languages, i.e. languages that provide an explicit formal semantics. An example of such a language is the Web Ontology Language (OWL) (9) which is based on description logic and benefits from a plethora of software tools and libraries within the Semantic Web. In particular, OWL facilitates automated reasoning to exploit non-explicit knowledge in ontologies.
Species-specific phenotype ontologies are now well developed and are available from the OBO Foundry (10) for human, mouse, fly, worm and yeast. Mapping between these ontologies has recently been facilitated by the development of logical definitions for each class within them, which use species-independent ontologies such as the Gene Ontology (GO) to provide a common semantic level at which they can be integrated into a single framework (11,12).
The formal class definitions used are based on the Entity–Quality (EQ) method (12). In the EQ method, a phenotype is characterized by an affected Entity (from an anatomy or process ontology) and a Quality [from the Phenotype And Trait Ontology (PATO)] that specifies how the entity is affected (11). The affected entity can either be a biological function or process as specified in the GO (13) or an anatomical entity. Anatomical entities are commonly specified in terms of a species-specific anatomy ontology. To systematically relate classes in species-specific anatomy ontologies, the metazoan, species-independent UBERON ontology (12) is used to obtain mappings between classes in species-specific anatomy ontologies.
We describe a method that enables a whole-phenome approach to comparative phenomics and its application to disease gene discovery. Our method requires the formalization of anatomy and phenotype ontologies so that they can be integrated using the parthood relation. We have then generated a single, unified and logically consistent representation of phenotype data for multiple species annotated to the species-specific phenotype ontologies within our framework, which is amenable to automated reasoning. We make the resulting ontology and the software used to generate it freely available.
We apply this approach to generate PhenomeNET, a cross-species network of phenotypic similarity between genotypes and diseases. Based on the semantically and logically consistent cross-species ontology created through our method, we incorporate the phenotype annotations which are available in the mouse, zebrafish, fly, yeast and worm model organism databases. We further include the human phenotypes associated with inherited diseases in the Online Mendelian Inheritance in Man (OMIM) database (14) in the ontology. As a result, we obtain an ontology of more than 275 000 classes and more than a million axioms. This ontology includes classes for 86 203 complex phenotype annotations from the model organism databases and OMIM. Our approach is extensible in that further phenotype ontologies can be added, and updated phenotype or disease annotations can easily be incorporated within the same model.
The comparison of phenotypes has been shown to predict orthologous genes, genes involved in the same pathway and genes involved in a common disease (5). As orthologous genes tend to be associated with related phenotypes and share common patterns of gene expression across species (15,16), the former provides a useful validation of the PhenomeNET approach. We use the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (17) to quantify the performance of the network for predicting orthology and participation of disease gene products in a common pathway, and we use gene–disease associations in OMIM as well as disease model annotations in the MGI database (18) to quantify the network's performance for predicting disease genes. In contrast to the pairwise comparison of single genotypes and diseases, our approach scales to whole phenomes and can enable systematic analyses of phenotypic information. In our evaluation of PhenomeNET, we show that mutations in orthologous genes and genes in the same pathway have a significantly higher phenotypic similarity than genes that are not orthologous or participate in the same pathway, as previously predicted by Oti and Brunner (19) in their concept of the modular phenotype. We further demonstrate that PhenomeNET associates genes to diseases in which they are known to be involved significantly higher than to diseases in which they are not known to be involved. We use PhenomeNET to identify pathways that significantly correlate with diseases and make this list available on our website. Furthermore, PhenomeNET can identify novel gene–disease associations. Through manual analysis, we find evidence that Adam19 and Fgf15 are associated with the congenital cardiac malformation tetralogy of Fallot in mice, and from Zebrafish phenotypes that homologs of Cx36.7 and Nkx2.5 may be part of a pathway in mammals controlling cardiac morphogenesis and electrical conduction whose mutation is again associated with the tetralogy of Fallot syndrome.
Previous approaches to disease gene prioritization often rely on additional sources of information other than the phenotype involved. In particular, several systems make use of functional, pathway and literature annotations of known diseases and disease genes to prioritize novel disease gene candidates (20–23). However, analyses that rely on the availability of gene annotations (such as GO-based functional annotations) may not always provide results when genes of unknown function are implicated in the analysis and the pathobiology of the disease is uncharacterized. Since PhenomeNET's predictions are based on information about phenotypes alone, it can be applied to identify candidate genes for diseases with an unknown molecular basis.
To allow researchers to explore PhenomeNET and use its predictions for the prioritization of genes that may be involved in diseases, we have made a web server available that enables access to our results. The web server as well as the raw data and source code we produced are freely available from http://phenomeblast.googlecode.com.
MATERIALS AND METHODS
Ontologies, software tools and libraries
We used the ontology files available for download from the OBO Foundry website (http://obofoundry.org). The ontology files on which we base our results were downloaded on 3 February, 2011, except for the definitions for the MP, HPO, FPO and WPO ontologies which were obtained on 9 February 2011. The definition file for the MP was obtained from the source code repository at http://code.google.com/p/phenotype-ontologies/. The definition file for the HPO was obtained from http://www.human-phenotype-ontology.org.
Implementation
The software we produce uses the OWL API (24) and is written in Groovy. We use the EL Vira software (25) to convert the generated OWL files to the OWL EL subset and enable tractable automated reasoning over the combined ontologies. To query the ontology, we combine the CB reasoner (26) and the CEL reasoner (27). A part of the software is available as the PhenomeBLAST software tool for aligning phenotypes across species.
We implemented a novel conversion software for phenotype ontology definitions, following the patterns described below. The software is implemented as a script in Groovy and available in the source code repository at our project website. Similarly, we implement a conversion for phenotype annotations in various model organism databases according to our method and make the software available in the source code repository. The specific versions of the ontologies we used as well as th software we generated is available on our project website (http://phenomeblast.googlecode.com).
The ontology we created includes representations of phenotypes associated with genotypes in the yeast, worm, fly, fish and mouse model organism databases, disease phenotypes based on OMIM as well as the core set of phenotype and anatomy ontologies. The resulting ontology contains more than 275 000 classes, 372 000 subclass axioms, 152 000 equivalent class axioms and 133 object properties, and combines the information of five species' phenomes as well as phenotypes of human disease.
Phenotype annotations
We converted the phenotype annotations available in various model organism databases. The following annotation files were used:
SGD annotations (phenotype_data.tab at http://downloads.yeastgenome.org),
WormBase annotations available from WormMart (http://www.wormbase.org/biomart/martview),
FlyBase annotations (allele_phenotypic_data_fb_2011_01.tsv.gz at http://flybase.org)
MGI annotations (MGI_PhenoGenoMP.rpt at ftp://ftp.informatics.jax.org/pub/reports/),
HPO annotations of OMIM (phenotype_annotation.omim at http://www.human-phenotype-ontology.org/), and
ZFIN annotation (phenotype.txt at http://zfin.org/data_transfer/Downloads/).
Phenotype representation
The basic components of the definition of a phenotype class are a quality (Q) and an entity (E), such that the quality inheres in the entity: Q and inheres-in some E (12).
We observe at least four basic kinds of subclass relation in the asserted taxonomic structure of the phenotype ontologies. A phenotype class P1 (based on Q1 and E1) is a subclass of the phenotype class P2 (based on Q2 and E2), if any of the following conditions or a combination thereof is true:
Q1 is a subclass of Q2 and E1 is equivalent to E2; e.g. Abdominal distention (HP:0003270) is a subclass of Abnormality of the abdomen (HP:0001438) and both affect the Abdomen (E) in different ways (Q);
E1 is a subclass of E2, while Q1 and Q2 are equivalent; e.g. Abnormality of the 3rd finger (HP:0004150) is a subclass of Abnormality of the fingers (HP:0001167), and Middle finger (E1) is a subclass of Finger (E2);
E1 is a part of E2 (i.e. every instance of E1 is a part of some instance of E2) and Q1 and Q2 are equivalent; e.g. Abnormality of the diaphragm (HP:0000775) is a subclass of Abnormality of the abdomen (HP:0001438), and Diaphragm is a part of the Abdomen;
E1 is either a function of E2 or a process that realizes a function of E2 and Q1 and Q2 are equivalent; e.g. Hearing abnormality (HP:0000364) is a subclass of Abnormality of the ears (HP:0000598) (28).
To reflect the asserted taxonomic structure of the phenotype ontologies in their definitions and utilize the assertion of part-hood in the anatomy ontologies for inferences, we restructured the phenotype ontologies' definitions to reflect these assumptions. When a definition is based on an entity E and the quality class PATO:0000001 (labelled Quality), we represent the phenotype as
has-part some (part-of some E and has-quality some PATO:0000001)
Furthermore, we represent phenotypes based on the entity E and a quality Q as descriptions of the class
has-part some (E and has-quality some Q)
The use of ‘part-of’ and ‘has-part’ establishes a basic overlap in relations between phenotype and anatomy ontologies and permits the use of inferences made in anatomy ontologies to infer information about phenotypes.
Statistical testing
For each disease in OMIM, we generate a distribution Δ of semantic similarity values for each gene node in the phenotype network. We filter all disease–gene pairs in which the gene is not present in KEGG. As a result, we obtain a distribution Δ of the phenotypic similarities of disease–gene pairs. For each KEGG pathway P, we identify all participating genes and generate the distribution ΔP⊆Δ of genes that participate in P. We then perform a Wilcoxon signed-rank test on Δ and ΔP. The result is a list of P-values for each disease-pathway pair. On this list, we applied the Holm method and the Benjamini–Hochberg corrections for multiple testing.
We find that PhenomeNET contains only a single, under-annotated genotype for genes in the pathways ‘Sulfur relay system’ (ko04122), ‘Lipoic acid metabolism’ (ko00785) and ‘Indole alkaloid biosynthesis’ (ko00901). As a result, we obtain only a single P-value for all tests involving these pathways, and we therefore eliminate these pathways from further analyses.
RESULTS AND DISCUSSION
Formalizing anatomy
As the first step in our method, illustrated in Table 1 and Figure 1, we utilize the species-independent ontology UBERON (5,12) to construct a cross-species bridging anatomy ontology. This cross-species ontology integrates classes and axioms for species-independent anatomical entities with those for species-specific anatomy ontologies. UBERON contains species-independent anatomical classes as well as mappings to species-specific anatomical entities, including classes from the Foundational Model of Anatomy (FMA) (29), Mouse Anatomy Ontology (MA) (30), Worm Anatomy (WAO) (31), Fly Anatomy (32) and Zebrafish Anatomy (33). The mappings between species-specific anatomy ontologies could then be used to derive mappings between species-specific phenotype ontologies, e.g. we use the mappings between ‘Liver’ in MA and ‘Liver’ in FMA to derive a mapping between the phenotypes ‘Abnormality of liver morphology’ (MP:0000598) and ‘Liver abnormality’ (HP:0001392). To achieve this goal, we treat the mappings between species-specific anatomical entities as statements of equivalence between classes.
Table 1.
General overview of the method
| Step | Method | Materials used | Example |
|---|---|---|---|
| Formalization of cross-species anatomy | Assign equivalent class statements based on UBERON cross-species mappings; integrate all ontologies and equivalence statements in single ontology. | Gene Ontology, Mouse Anatomy, Worm Anatomy and Development, Zebrafish Anatomy and Development, Fly Anatomy, Foundational Model of Anatomy, UBERON | Both the classes ‘Tail’ from Mouse Anatomy and ‘Caudal fin’ from Zebrafish Anatomy are declared equivalent to the class ‘Tail’ in UBERON. |
| Consistency verification and removal of contradictions | Remove disjointness statements from UBERON. | UBERON ontology, processing of OBO Flatfile Format | The disjointness between ‘Material anatomical entity’ (UBERON:0000465) and ‘Immaterial anatomical entity’ (UBERON:0000466) is removed. |
| Formalization of cross-species phenotypes | Convert phenotype ontologies' definitions to enable interoperability with anatomy (using has-part and part-of relations); combine all phenotype ontologies, their class definitions and the cross-species anatomy ontology in a single ontology. | Yeast Phenotype, FlyBase Controlled Vocabulary, Worm Phenotype, Mammalian Phenotype, Human Phenotype; related ontologies: PATO, ChEBI, Gene Ontology, Mouse Pathology, Celltype, Protein Ontology | Define the mouse phenotype ‘Matted coat’ as shown in Figure 2. |
| Represent phenotype annotations | Phenotype annotations in model organisms databases or of diseases are represented as class C; the class C is then asserted as equivalent to the intersection of the annotated phenotypes. | HPO-based phenotype annotations of OMIM; phenotype annotations in model organism databases | The class ‘Alport syndrome’ is defined as equivalent to the intersection of the disease's phenotypic characteristics: ‘Renal failure, Nephritis, Hearing loss’ and ‘Hematuria’. |
| Inference of cross-species phenotype representation | Using automated reasoning, each class that represents a genotype annotations or disease is examined and all its super-classes in each species-specific phenotype ontology are inferred. The result is a representation of the annotated phenotypes based on five species-specific phenotype ontologies. | OWL reasoners (CB and CEL) | The worm phenotype ‘Abnormal apoptosis’ is inferred as a super-class of the human phenotype ‘Defective lymphocyte apoptosis’. |
| Application of semantic similarity | A semantic similarity measure is applied to compensate for missing information and noisy data. | Jaccard metric weighted by information content; implemented parallel algorithm for computation | The phenotype of an allele of the Adam19 gene (MGI:3028702) is similar to tetralogy of Fallot phenotype. |
| Quantitative evaluation | KEGG and known disease models provide gene–gene and gene–disease associations which are compared within the network. Orthologous genes and genes in the same pathway are phenotypically similar; gene–disease pairs of known gene–disease associations are similar. | KEGG, OMIM and Morbidmap, mouse model annotations in MGI | The area under the receiver operator characteristic curve (a plot of the true positive rate as function of the false positive rate) for pathways is 0.59, for orthology 0.62 and for disease 0.68. |
Figure 1.

Overview over ontology-based data analysis. First, the ontologies have to be formalized before their consistency can be verified. If contradictory axioms are identified, they must be removed. Using the ontology, biological data is represented within the same model so that the biological questions across the data can be asked in flexible ways. If necessary, statistical approaches are applied to complete missing information and results can then be inferred over the combined representation.
For example, UBERON provides the class ‘Islet of Langerhans’ (UBERON:0000006) and mappings to ‘Pancreatic islet’ in human anatomy (FMA:16016) and mouse anatomy (MA:0000127). Based on these mappings, we declare these three classes as equivalent. As a consequence of these classes' becoming equivalent, the axioms involving ‘Pancreatic islet’ in species-specific anatomy ontologies are combined: according to the FMA, ‘Pancreatic islet’ is part of the ‘Endocrine pancreas’ (a kind of ‘Set of organ components’) and the ‘Endocrine system’, according to the MA, is part of the ‘Endocrine pancreas’ (a kind of ‘Endocrine gland’) and part of the ‘Pancreas’. According to UBERON ‘Pancreatic islet’ is a kind of ‘Organ part’ and part of the ‘Endocrine pancreas’. Due to further mappings in the UBERON ontology we create cross-species equivalences for ‘Endocrine pancreas, Pancreas, Endocrine system’ and ‘Organ part’. Consequently, each of these axioms restricts the classes in all three ontologies.
We demonstrate this integration in our framework using the FMA, MA, WAO, ZFA and Fly Anatomy ontologies. The result is an ontology including more than 86 000 classes in which the subsumption hierarchies and the axioms that obtain in the species-dependent anatomy ontologies are formally translated across all species. Therefore, such an integrated ontology can serve as an ‘interlingua’ across anatomy.
Removing inconsistencies
Using an OWL reasoner on this ontology allows us to identify several thousand unsatisfiable classes, i.e. classes that cannot have any instance because their definition contains a contradiction. For example, the class ‘Anus’ (FMA:15711) is a subclass of ‘Anatomical orifice’ (FMA:3724) which we declare equivalent to the class ‘Orifice’ (UBERON:0000161), and all these classes are unsatisfiable. In UBERON, ‘Orifice’ is a subclass of ‘Material anatomical entity’ while ‘Anatomical orifice’ is a subclass of ‘Immaterial anatomical entity’ in the FMA. ‘Immaterial anatomical entity’ (FMA:67112) is mapped to ‘Immaterial anatomical entity’ (UBERON:0000466) which is declared disjoint from ‘Material anatomical entity’ (UBERON:0000465). According to UBERON, an immaterial anatomical entity is an anatomical entity that has no mass while material anatomical entities always have a mass.
Unsatisfiable classes such as ‘Anus’ are the sub-class of all classes in the ontology (i.e. every class in the ontology is a super-class of or equivalent to every unsatisfiable class). Because all asserted sub-classes of an unsatisfiable class are also unsatisfiable, a contradictory definition of a very general class (with respect to the ontology's taxonomy) can result in a large number of classes becoming unsatisfiable. For example, because the class ‘Anatomical orifice’ is unsatisfiable, it has all classes in our ontology as super-classes (or equivalent classes), including ‘Tail, Caudal fin’ and ‘Anus’. All asserted sub-classes of ‘Anatomical orifice’, including ‘Oral cavity’ and ‘Anus’, will also be unsatisfiable. Unsatisfiable classes can not be used to establish meaningful relations between species-specific anatomical entities within our ontology.
Due to these problems, we removed all disjointness statements from the anatomy ontologies in order to derive a consistent representation. As a direct consequence of the monotonicity of first-order logic (i.e. if a set of axioms S is a subset of a set of axioms T, then the inferences that can be drawn from S are a subset of the inferences that can be drawn from T) (34), reducing an ontology's axioms will reduce the number of inferences that can be drawn from it but not their nature. Consequently, we obtain fewer inferences from the disjointness-free module of the anatomy ontology than could be made when the disjointness axioms are included, but gain the advantage of being able to accurately use these ontologies for reasoning. Removing the asserted disjointness statements does not remove the underlying conflicts in the conceptualization of the anatomy ontologies and the mappings between them. Therefore, the accurate alignment of the anatomy ontologies is an important subject for future work. Since the ontologies and their alignments are collaboratively developed within the OBO Foundry (10), we are working with the ontology developers on resolving these issues.
Formalizing phenotype definitions
Based on the integrated cross-species anatomy ontologies, we add species-dependent phenotype ontologies to our framework in order to derive a ‘cross-species’ ontology of phenotypes. We include the Human Phenotype Ontology (HPO) (35), the Mammalian Phenotype Ontology (MP) (36), the Worm Phenotype Ontology (WPO) (37), Yeast Phenotype Ontology (APO) (38) and the Fly Phenotype Ontology (FPO) (32) since formal definitions have been created for these ontologies' classes (11,12).
In the phenotype ontologies' definitions, the affected entity of a phenotype can either be an anatomical structure, a process or a function. To accommodate classes that refer to processes or functions, we further add GO to our combined ontology, which provides processes and functions as well as additional anatomical structures (in GO's Cellular Component branch). Furthermore, to specify phenotypes such as ‘Abnormal triglyceride level’ (MP:0000187) or ‘Abnormal hair cell morphology’ (MP:0000045), we include the ChEBI ontology of chemical entities (39) and the Celltype Ontology (40). We further modified the phenotype definitions to accurately reflect the assumptions behind the phenotype ontologies' asserted taxonomic structure and interoperate with anatomy ontologies (28, 41).
The precise formulation of phenotype classes based on their definitions is described in the ‘Materials and Methods’ section. The link between the phenotype ontologies is established through species-independent ontologies such as UBERON, GO, MPATH, ChEBI or the Celltype Ontology, combined with the PATO ontology of qualities. The resulting ontology, however, is more than a combination of phenotype and anatomy ontologies: due to axioms that relate classes in all these ontologies, subclass relations and domain-specific axioms are propagated across all ontologies and therefore restrict phenotypes and anatomical entities in all species for which we include an ontology.
Since this integrated ontology contains species-specific classes, we can use the resulting ontology as an ‘interlingua’ between human, mouse, worm, fish and yeast phenotypes. For example, the MP class ‘Matted coat’ (MP:0003846) is mapped to the most specific superclasses ‘Abnormality of the skin’ (HP:0000951) and ‘Hair abnormality’ (HP:0001595) in the HPO. Figure 2 illustrates parts of the inferences that lead to this mapping.
Figure 2.

Illustration of assertions and inferences about the class Matted coat. Blue-colored shapes represent qualities, gray-colored shapes represent anatomical entities and green-colored shapes represent phenotypes. Dashed lines represent inferred associations.
Representing annotated data
The Human Phenotype Ontology is used to annotate Mendelian diseases represented in the OMIM database (14). We use these annotations to define classes corresponding to the intersection of the HPO-based disease phenotypes. For example, for the OMIM disease ‘Alport Syndrome’ (OMIM:203780), we add a class to our ontology that is defined as equivalent to the intersection of the disease's phenotypic characteristics: ‘Renal failure’ (HP:0000083), ‘Nephritis’ (HP:0000123), ‘Hearing loss’ (HP:0000365) and ‘Hematuria’ (HP:0000790). The HPO classes for OMIM diseases are based on the HPO annotations of OMIM (see ‘Materials and Methods’ section).
Using automated reasoning in our cross-species phenotype framework, we obtain a rich characterization of diseases based on classes from all the included phenotype ontologies. For example, from the definition of the OMIM disease ‘Alport Syndrome’ (OMIM:104200), we can infer through automated reasoning that 57 classes from MP characterize this disease in addition to 55 classes from HPO, including ‘abnormal kidney physiology’ (MP:0002136) and ‘abnormal sensory capabilities/reflexes/nociception’ (MP:0002067).
The use of automated reasoning for translating representations of disease phenotypes into a species-specific representation is more powerful than a translation based on mappings between the ontologies alone (41,42). OWL reasoning on complex disease phenotype classes can utilize inferences of additional phenotypes from complex class definitions. For example, the class tetralogy of Fallot (HP:0001636) is defined (according to the HPO definitions) as a phenotype of things having ‘Overriding aorta’ (HP:0002623), ‘Ventricular septal defect’ (HP:0001629), ‘Pulmonic stenosis’ (HP:0001642) and ‘Right ventricular hypertrophy’ (HP:0001667). The phenotype tetralogy of Fallot would automatically be inferred to be a phenotype of an entity that is characterized by all four individual phenotypes, i.e. if all phenotypes that are sufficient for having the disease are available, the additional information about having the disease is inferred.
Several model organism databases use species-specific phenotype ontologies to formally represent phenotypes. We can use our ontology framework to automatically translate model organism phenotypes coded by their cognate phenotype ontology into a human-specific representation based on HPO. To demonstrate this application, we include within our framework the MP-based phenotypic characterization of mouse models, the WPO-based representation of worm phenotypes, the FPO-based representation of fly phenotypes and the APO-based representation of yeast phenotypes. Each genotype is represented as a class in the ontology that is defined to be equivalent to the intersection of its phenotype annotations.
Some model organism databases do not utilize their own species-specific phenotype ontology but rather employ post-composed phenotype terms based on the EQ method (11). For example, the Zebrafish Model Organism Database (ZFIN) (33) contains phenotypic descriptions of zebrafish with a particular genotype. For this purpose, only the entity (either from GO or ZFA) and the quality (from PATO) are recorded. Utilizing our cross-species anatomy and phenotype ontology, we can automatically incorporate such phenotype descriptions, thereby enabling automatic translation of research results across species even when no species-specific phenotype ontology is available. The EQ phenotypes from ZFIN are formalized following the same approach as the EQ-based class definitions (see ‘Materials and Methods’ section), and we represent each genotype annotated with a set of EQ statements as a class that is equivalent to the intersection of the EQ-based phenotype classes.
Cross-species phenotype representations
Based on OWL reasoning, we automatically generate a phenotype representation based on MP, HPO, WPO, APO and FPO for each genotype and disease annotated using a set of phenotype classes. For example, the zebrafish genotype ZDB-GENO-091204-5 is annotated with ‘Caudal fin’ (ZFA:0000017) and the quality ‘Decreased length’ (PATO:0000574). Through inference within our cross-species ontology framework, we obtain, among others, the phenotype ‘Short tail’ (MP:0000592) as a mapping of this phenotype to the MP. All cross-species phenotype representations are available on our website.
Constructing a cross-species phenotype network
After including classes that represent complex phenotypes associated with either diseases or specific genotypes [i.e. their phenoset (2)], we can investigate the relation between these classes in the classified ontology. In particular, a class representing a disease could be a sub- or super-class of a class that represents a phenotype annotation of a genotype within a model organism database. If the phenotypes that we associate with diseases would be sufficient for having the disease (i.e. having the phenotypes would necessarily imply having the disease), we could infer (i.e. provide a formal proof for the fact) that, if a class representing a disease has sub-classes that represent genotypes, then these genotypes are necessarily associated with the disease. However, at least two obstacles impair the automatic inference of this information: the incompleteness of phenotypic characterization of diseases and animal models as well as incomplete mappings between species-specific anatomy and phenotype ontologies. To account for incomplete information and reduce the impact that potentially incorrect assertions have in our framework, we explore the relation between phenotypes using a measure of phenotypic similarity.
Using the cross-species representation of genotypes and diseases, we construct a network in which nodes represent phenotypes, and edges the similarity between phenotypes. As nodes, we select all phenotypes that are associated with a particular genotype in the mouse, worm, fish, fly and yeast model organism databases. Additionally, we add disease nodes that are based on the phenotype associated with a disease in OMIM. Our network contains 86 203 nodes.
Since our method relies on inference over several ontologies and complex phenotype descriptions may refer to many nodes in these ontologies, the ontologies' graph structure is not readily available to us for measuring similarity. Consequently, we use the Jaccard metric for comparing sets and assign weights to set members based on the information content of an ontology term (43).
We define the information content I(t) of an ontology class t based on the probability P(X = t) that a genotype or disease is characterized with t:
| (1) |
The probability P(X = t) is empirically derived within the corpus of 86 203 complex phenotypes used for the construction of the phenotype network.
Given two complex phenotypes P and R, where P is characterized by the ontology classes Cl(P) = P1,...,Pn and R is characterized by the classes Cl(R) = R1,...,Rm, we define the similarity between P and R as:
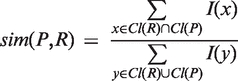 |
(2) |
sim(P,R) is the weighted Jaccard index between Cl(P) and Cl(R). Using this metric, we perform a pairwise comparison of phenotype nodes in the network. We then add the similarity measure as the weight of the edge between two phenotype nodes. The result of applying this method is an adjacency matrix of a cross-species phenotype and disease network.
Phenome Browser
The Phenome Browser is a web server that allows researchers to explore the PhenomeNET and its predictions. It allows the retrieval of nodes in the PhenomeNET based either on their name or their identifier within a model organism database. For example, the node representing the disease ‘tetralogy of Fallot’ (OMIM:187500) can be retrieved either through its OMIM identifier (OMIM:187500), its name or any part of its name. Similarly, the search string ‘SSH’ can be used to retrieve nodes representing the sonic hedgehog (SHH) gene (and associated genotypes) in all species included in PhenomeNET, and the search string ‘FBal0055336’ (a FlyBase accession number) can be used to retrieve the node representing SHH in fly.
For each phenotype node, all nodes that are related with a similarity score >0.1 can be explored. The related nodes are presented by species and ranked according to their similarity score. For example, exploring the node representing ‘Sd’ (MGI:1857746) will show 76 OMIM entries with a similarity score >0.1 (VACTERL association with hydrocephalus (OMIM:276950) on Rank 1 with similarity score 0.23), 1422 mouse genes and genotypes, 19 nodes representing worm genes and genotypes as well as 40 zebrafish genotype nodes. No nodes representing yeast or fly phenotypes are identified with a similarity >0.1. Each of the phenotype nodes can further be explored in a similar way, thereby allowing to navigate through PhenomeNET based on phenotypic relatedness between phenotype nodes.
Evaluation
Comparison of phenotypes has been shown to predict orthologous genes, genes involved in the same pathway and genes involved in a common disease (5). We use the KEGG database (17) to evaluate PhenomeNET's performance for predicting orthology and participation in a common pathway, and we use gene–disease associations in OMIM as well as disease model annotations in the MGI database (18) to quantify the network's performance for predicting disease genes. To quantify the performance, we create the receiver operator characteristic (ROC) curve for the three tasks and report the area under the ROC curve (AUC). A ROC curve is a plot of the true positive rate as a function of the false positive rate. To generate the ROC curve for our evaluation, we first filter the list of associations in PhenomeNET for gene–gene and gene–genotype associations in the case of predicting pathway and orthology, and for gene–disease associations when predicting gene–disease associations. We then identify the positive samples in the ranked list of the phenotype nodes associated with each node in PhenomeNET and treat all remaining pairs as negative samples. For example, to evaluate PhenomeNET's capability for predicting participation in the same pathway, we identify all pairs of nodes that represent genes (or associated genotypes) participating in the same pathway as positive samples, all remaining pairs of genes as negative samples. Based on these positive and negative examples, we calculate the true and false positive rates based on the rank of phenotypic similarity with which nodes are associated in PhenomeNET. The diagonal line in a ROC curve represents a classifier that guesses randomly, and the AUC for a random classifier is expected to be 0.5. The AUC is equivalent to the probability that a randomly chosen positive instance is ranked higher than a randomly chosen negative instance (44). For example, when evaluating PhenomeNET by predicting associations between genes in the same pathway, the AUC will be the probability that a randomly chosen pair of genes that participate in the same pathway have a higher score than a randomly chosen pair of genes that do not participate in the same pathway.
Within the phenotype network, orthologous genes have a significantly higher phenotypic similarity than non-orthologous genes (AUC 0.62), genes in the same pathway have a significantly higher similarity than genes that do not participate in the same pathways (AUC 0.59), and genes have a significantly higher phenotypic similarity to the diseases in which they are known to be involved than to diseases for which the involvement is not known (AUC 0.68). Figure 3 shows the ROC curve for all three evaluations.
Figure 3.

ROC curves for predicting disease, participation in a common pathway and orthology using PhenomeNET. The ROC curves for pathway and orthology predictions are obtained by comparison with KEGG, while the gene-disease predictions are derived from OMIM and the annotated disease models in the MGI. AUC for pathways is 0.59, for orthology 0.62 and for disease 0.68.
In this evaluation, we assumed that gene–disease associations that are not known to be true are false. Therefore, the true performance of our phenotype network for predicting disease genes will likely be higher than shown in Figure 3. For example, while the four phenotypically most similar mouse models for tetralogy of Fallot (TOF, OMIM:187500) are already associated with the syndrome in the MGI, the genotype node representing an allele of the Adam19 gene (MGI:3028702) is phenotypically very similar to TOF. However, while Adam19 is known to play an essential role in cardiovascular morphogenesis (45), neither the gene nor the allele are currently associated with the syndrome. Similarly, the phenotype of an allele of Fgf15 (MGI:3044957) is highly similar to TOF, and although Fgf15 is known to be required for the development of the cardiac outflow tract (46), it has not yet been associated with the syndrome.
We further identified pathways in KEGG that significantly overlap with diseases and make these lists available on our project website. For example, for the TOF, we identify the cytokine–cytokine receptor interaction pathway (ko04060) as significant (P = 5 × 10−7, Wilcoxon signed-rank test, Benjamini–Hochberg correction). The cytokine–cytokine receptor interaction pathway is known to be involved in embryonic heart development (47). Another significantly overlapping pathway includes the TGF signalling pathway (P = 0.006, Wilcoxon signed-rank test, Benjamini–Hochberg correction) which may result in TOF when disrupted (48).
It is possible to query the network for specific phenotypes common to one or more model organisms in order to explore, for example, common morphogenetic pathways. We have analyzed the set of phenotypes for the mouse Hey2 gene (MGI:1341884), and, by querying across the network, a mutant in the Zebrafish Cx36.7 gene (a member of the super-family of connexin genes) was identified as the phenotypically most similar genotype in fish (Rank 1, similarity score 0.22). While the published description of this zebrafish ENU mutant (ftk) does not represent a classical manifestation of TOF (which is not possible in a fish), it does include morphogenetic defects in the atrial and ventricular walls. The authors noted that expression of the transcription factor Nkx2.5 was dramatically reduced in ftk mutants and provide evidence that, in these fish, the mutant phenotype can be explained entirely by the action of Cx36.7 mediated by the transcription factor Nkx2.5 (49). These candidates for the morphological defects seen in TOF were then examined in MGI and OMIM. While other connexins are associated with TOF in mouse mutants, notably connexins 40, 43 and 45 (50), there is no asserted implication in OMIM although the literature records, as with mice, an association with connexin 43 (51). Intriguingly, Sultana et al. (49) show that expression of the transcription factor Nkx2.5 is dramatically decreased with loss of the Cx36.7 gene. OMIM associates Nkx2.5 with TOF, strongly suggesting that the two might act in a pathway which affects heart morphogenesis (52). The linkage between the phenotypes generated by mutations in a connexin and Nkx2.5 has never been made for either the human or the mouse. Further, the association between cardiac electrical conductance and morphogenesis has only been previously associated with the connexins (50). This example demonstrates the power of exploring the network of model organism phenotypes in generating new hypotheses for gene function.
While PhenomeNET is based exclusively on information about phenotypes, previous approaches combine several data sources to make predictions for gene–disease associations (20–23). In particular, many of these systems make use of known disease genes' ontology annotations from GO or phenotype ontologies to identify genes with a similar functional or phenotypic profile. As a result, these systems can predict gene–disease associations with a significantly higher sensitivity (i.e. true positive rate) than PhenomeNET and these systems achieve an AUC for predicting gene–disease associations of >0.9 (20,23) (in contrast to 0.68 for PhenomeNET). However, systems that are based on known annotations assume that the disease genes will be consistent with the known pathobiology of a disease and its genetic basis, and therefore rely on the availability of information about the molecular mechanisms underlying a disease (23). They can not be applied when the information about the molecular origins of a disease is not available. Therefore, while other gene prioritization methods can provide more accurate predictions for candidate genes when incorporating the molecular and functional information of known disease genes, our method can complement these approaches as it relies on information about phenotypes alone and can therefore be applied when the description of the disease phenotype is the only information available. For example, PhenomeNET suggests the Slc34a1 gene (MGI:1345284) as a candidate for Fanconi renotubular syndrome 1 (OMIM:134600) (Rank 2, similarity score 0.44). Slc34a1 has recently been identified as a cause of an autosomal recessive form of Fanconi renotubular syndrome 1 (53), and since this information is not yet available in OMIM's gene–disease association map, no GO-based gene prioritization system was able to provide predictions for this syndrome. We make our data and source code freely available so that the developers and maintainers of other gene prioritization systems can incorporate our method and extend their systems.
CONCLUSION
We have developed a method to compare phenotypes across species. We applied this method to the discovery of disease gene candidates based on information about the phenotype alone and could identify several novel disease gene candidates. The prime application of the PhenomeNET framework is to suggest candidate genes for rare and orphan diseases without a known molecular basis, and we provide a web server to give access to our results. This web server can be used by researchers to explore candidate genes for heritable diseases within the phenotype data provided by five model organism databases. Thereby, PhenomeNET can provide a means for the analysis of phenotype data and improve the speed by which primary research data from phenotype studies can be translated into a better understanding of human disease.
We intend to update the data underlying PhenomeNET on a regular basis and make new versions available to the scientific community when new phenotype data becomes available or the structure, definitions or content of the phenotype ontologies changes. Furthermore, we intend to integrate new phenotype ontologies of different species when they become available to extend PhenomeNET's coverage to further model organisms. We plan to extend the PhenomeNET method and web server into an analysis platform for phenotype data that is based on real-time analyses. In such a system, scientists would be able to describe phenotypes using a supported phenotype ontology and explore similar phenotypes across multiple model organism databases. The integration of other ontologies and data characterized by them, information about genotypes, genes and gene products, pathways and disease can enable the development of an analysis platform for high-throughput phenotype data that is based on the combination of ontology-based data integration, automated reasoning and statistical and similarity-based approaches to complete missing information.
Our method constitutes a general approach toward knowledge discovery with biomedical ontologies and can be applied in all domains that apply ontologies for the annotation of data. It further provides a means for the qualitative evaluation and validation of both ontologies and biological data through automated reasoning based on the axioms and constraints provided by ontology developers.
FUNDING
Funding for open access charge: NIH (grant number R01 HG004838-02). R.H. was funded by the European Commission's 7th Framework Programme, RICORDO project (grant number 248502), P.S. by the NIH (grant number R01 HG004838-02) and G.V.G. by the BBSRC (grant BBG0043581).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Michael Dannemann for help with the statistic analyses and Michael Ashburner for his continued enthusiastic support.
REFERENCES
- 1.Rosenthal N, Brown S. The mouse ascending: perspectives for human-disease models. Nat. Cell. Biol. 2007;9:993–999. doi: 10.1038/ncb437. [DOI] [PubMed] [Google Scholar]
- 2.Schofield PN, Gkoutos GV, Gruenberger M, Sundberg JP, Hancock JM. Phenotype ontologies for mouse and man: bridging the semantic gap. Dis. Model. Mech. 2010;3:281–289. doi: 10.1242/dmm.002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbott A. Mouse megascience. Nature. 2010;465:526. doi: 10.1038/465526a. [DOI] [PubMed] [Google Scholar]
- 4.Collins FS, Finnell RH, Rossant J, Wurst W. A new partner for the international knockout mouse consortium. Cell. 2007;129:235. doi: 10.1016/j.cell.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Washington NL, Haendel MA, Mungall CJ, Ashburner M, Westerfield M, Lewis SE. Linking human diseases to animal models using ontology-based phenotype annotation. PLoS Biol. 2009;7:e1000247. doi: 10.1371/journal.pbio.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groth P, Pavlova N, Kalev I, Tonov S, Georgiev G, Pohlenz H-D, Weiss B. PhenomicDB: a new cross-species genotype/phenotype resource. Nucleic Acids Res. 2006;35(Suppl. 1):D696–D699. doi: 10.1093/nar/gkl662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004;32(Suppl. 1):D267–D270. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sardana D, Vasa S, Vepachedu N, Chen J, Gudivada RC, Aronow BJ, Jegga AG. PhenoHM: human-mouse comparative phenome-genome server. Nucleic Acids Res. 2010;38(Suppl. 2):W165–W174. doi: 10.1093/nar/gkq472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grau B, Horrocks I, Motik B, Parsia B, Patelschneider P, Sattler U. OWL 2: The next step for OWL. Web Semant. Sci., Serv. Agents World Wide Web. 2008;6:309–322. [Google Scholar]
- 10.Smith B, Ashburner M, Rosse C, Bard J, Bug W, Ceusters W, Goldberg LJ, Eilbeck K, Ireland A, Mungall CJ, et al. The OBO foundry: coordinated evolution of ontologies to support biomedical data integration. Nat. Biotech. 2007;25:1251–1255. doi: 10.1038/nbt1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gkoutos GV, Green EC, Mallon A-MM, Hancock JM, Davidson D. Using ontologies to describe mouse phenotypes. Genome Biol. 2004;6:R8. doi: 10.1186/gb-2004-6-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mungall C, Gkoutos G, Smith C, Haendel M, Lewis S, Ashburner M. Integrating phenotype ontologies across multiple species. Genome Biol. 2010;11:R2. doi: 10.1186/gb-2010-11-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry MJ, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amberger J, Bocchini C, Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM) Hum. Mutat. 2011;32:564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 15.McGary KL, Park TJ, Woods JO, Cha HJ, Wallingford JB, Marcotte EM. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc. Natl Acad. Sci. 2011;107:6544–6549. doi: 10.1073/pnas.0910200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng-Bradley X, Rung J, Parkinson H, Brazma A. Large scale comparison of global gene expression patterns in human and mouse. Genome Biol. 2010;11:R124. doi: 10.1186/gb-2010-11-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanehisa M. The KEGG database. Novartis Found Symp. 2002;247:91–101. [PubMed] [Google Scholar]
- 18.Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT, the Mouse Genome Database Group The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res. 2011;39(Suppl. 1):D842–D848. doi: 10.1093/nar/gkq1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oti M, Brunner HG. The modular nature of genetic diseases. Clinical Genet. 2007;71:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- 20.Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS. SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics. 2006;22:773–774. doi: 10.1093/bioinformatics/btk031. [DOI] [PubMed] [Google Scholar]
- 21.Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent L-C, De Moor B, Marynen P, Hassan B, et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Xu H, Aronow B, Jegga A. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics. 2007;8:392. doi: 10.1186/1471-2105-8-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlicker A, Lengauer T, Albrecht M. Improving disease gene prioritization using the semantic similarity of Gene Ontology terms. Bioinformatics. 2010;26:i561–i567. doi: 10.1093/bioinformatics/btq384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horridge M, Bechhofer S, Noppens O. Igniting the OWL 1.1 Touch Paper: the OWL API. Proceedings of OWLED 2007: Third International Workshop on OWL Experiences and Directions; 2007. (CEURS-WS.org, volume 258), RWTH Aachen, Aachen, Germany. [Google Scholar]
- 25.Hoehndorf R, Dumontier M, Oellrich A, Wimalaratne S, Rebholz-Schuhmann D, Schofield P, Gkoutos GV. A common layer of interoperability for biomedical ontologies based on OWL EL. Bioinformatics. 2011;27:1001–1008. doi: 10.1093/bioinformatics/btr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazakov Y. Consequence-driven reasoning for horn SHIQ ontologies. Proceedings of the 21st International Conference on Artificial Intelligence (IJCAI 2009); San Francisco, CA, USA, Morgan Kaufmann Publishers Inc. 2009. pp. 2040–2045. [Google Scholar]
- 27.Baader F, Lutz C, Suntisrivaraporn B. CEL – a polynomial-time reasoner for life science ontologies. In: Furbach U, Shankar N, editors. Proceedings of the 3rd International Joint Conference on Automated Reasoning (IJCAR'06); 2006. Berlin, Germany, Springer Verlag, Vol. 4130, of Lecture Notes in Artificial Intelligence, pp. 287–291. [Google Scholar]
- 28.Hoehndorf R, Ngomo A-CN, Kelso J. Applying the functional abnormality ontology pattern to anatomical functions. J. Biomed. Semant. 2010;1:4. doi: 10.1186/2041-1480-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosse C, Mejino JLV. A reference ontology for biomedical informatics: the foundational model of anatomy. J. Biomed. Inform. 2003;36:478–500. doi: 10.1016/j.jbi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Hayamizu TF, Mangan M, Corradi JP, Kadin JA, Ringwald M. The adult mouse anatomical dictionary: a tool for annotating and integrating data. Genome Biol. 2005;6:R29. doi: 10.1186/gb-2005-6-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee RYN, Sternberg PW. Building a cell and anatomy ontology of Caenorhabditis elegans. Comp. Funct. Genomics. 2003;4:121–126. doi: 10.1002/cfg.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tweedie S, Ashburner M, Falls K, Leyland P, Mcquilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, et al. FlyBase: enhancing Drosophila gene ontology annotations. Nucleic Acids Res. 2009;37(Suppl. 1):D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sprague J, Bayraktaroglu L, Bradford Y, Conlin T, Dunn N, Fashena D, Frazer K, Haendel M, Howe DG, Knight J, et al. The zebrafish information network: the zebrafish model organism database provides expanded support for genotypes and phenotypes. Nucleic Acids Res. 2008;36(Suppl. 1):D768–D772. doi: 10.1093/nar/gkm956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barwise J, Etchemendy J. Stanford, CA, USA: CSLI Publications, Stanford University; 2002. Language, Proof and Logic. [Google Scholar]
- 35.Robinson PN, Koehler S, Bauer S, Seelow D, Horn D, Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CL, Goldsmith C-AW, Eppig JT. The mammalian phenotype ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2004;6:R7. doi: 10.1186/gb-2004-6-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schindelman G, Fernandes J, Bastiani C, Yook K, Sternberg P. Worm phenotype ontology: integrating phenotype data within and beyond the C. elegans community. BMC Bioinformatics. 2011;12:32. doi: 10.1186/1471-2105-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engel SR, Balakrishnan R, Binkley G, Christie KR, Costanzo MC, Dwight SS, Fisk DG, Hirschman JE, Hitz BC, Hong EL, et al. Saccharomyces genome database provides mutant phenotype data. Nucleic Acids Res. 2010;38(Suppl. 1):D433–D436. doi: 10.1093/nar/gkp917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Degtyarenko K, de Matos P, Ennis M, Hastings J, Zbinden M, McNaught A, Alcantara R, Darsow M, Guedj M, Ashburner M. ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008;36(Suppl. 1):D344–D350. doi: 10.1093/nar/gkm791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bard J, Rhee SY, Ashburner M. An ontology for cell types. Genome Biol. 2005;6:R21. doi: 10.1186/gb-2005-6-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoehndorf R, Oellrich A, Rebholz-Schuhmann D. Interoperability between phenotype and anatomy ontologies. Bioinformatics. 2010;26:3112–3118. doi: 10.1093/bioinformatics/btq578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jimenez-Ruiz E, Grau BC, Berlanga R, Rebholz-Schuhmann D. First steps in the logic-based assessment of post-composed phenotypic descriptions. Proceedings of Semantic Web Applications and Tools for Life Sciences (SWAT4LS); 2010. http://arxiv.org/abs/1012.1659 (23 June 2011, date last accessed) [Google Scholar]
- 43.Xu T, Du L, Zhou Y. Evaluation of GO-based functional similarity measures using S. cerevisiae protein interaction and expression profile data. BMC Bioinformatics. 2008;9:472. doi: 10.1186/1471-2105-9-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fawcett T. An introduction to ROC analysis. Pattern Recogn. Lett. 2006;27:861–874. ROC Analysis in Pattern Recognition. [Google Scholar]
- 45.Zhou H-M, Weskamp G, Chesneau V, Sahin U, Vortkamp A, Horiuchi K, Chiusaroli R, Hahn R, Wilkes D, Fisher P, et al. Essential role for ADAM19 in cardiovascular morphogenesis. Mol. Cell. Biol. 2004;24:96–104. doi: 10.1128/MCB.24.1.96-104.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vincentz JW, McWhirter JR, Murre C, Baldini A, Furuta Y. Fgf15 is required for proper morphogenesis of the mouse cardiac outflow tract. Genesis. 2005;41:192–201. doi: 10.1002/gene.20114. [DOI] [PubMed] [Google Scholar]
- 47.Chowdhury S, Erickson SW, MacLeod SL, Cleves MA, Hu P, Karim MA, Hobbs CA. Maternal genome-wide DNA methylation patterns and congenital heart defects. PLoS ONE. 2011;6:e16506. doi: 10.1371/journal.pone.0016506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Runge MS, Patterson C, editors. Principles of Molecular Cardiology (Contemporary Cardiology) 1st edn. New York, NY, USA: Humana Press; 2005. [Google Scholar]
- 49.Sultana N, Nag K, Hoshijima K, Laird DW, Kawakami A, Hirose S. Zebrafish early cardiac connexin, Cx36.7/Ecx, regulates myofibril orientation and heart morphogenesis by establishing Nkx2.5 expression. Proc. Natl Acad. Sci. USA. 2008;105:4763–4768. doi: 10.1073/pnas.0708451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu H, Smith FC, Taffet SM, Delmar M. High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 2003;93:201–206. doi: 10.1161/01.RES.0000084852.65396.70. [DOI] [PubMed] [Google Scholar]
- 51.Kolcz J, Drukala J, Bzowska M, Rajwa B, Korohoda W, Malec E. The expression of connexin 43 in children with Tetralogy of Fallot. Cell. Mol. Biol. Lett. 2005;10:287–303. [PubMed] [Google Scholar]
- 52.Goldmuntz E, Geiger E, Benson DW. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565–2568. doi: 10.1161/hc4601.098427. [DOI] [PubMed] [Google Scholar]
- 53.Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, Selig S, Lapointe JY, Zelikovic I, Skorecki K. A loss-of-function mutation in NaPi-IIa and renal Fanconi's syndrome. N. Engl. J. Med. 2010;362:1102–1109. doi: 10.1056/NEJMoa0905647. [DOI] [PubMed] [Google Scholar]