

Abstract
Developing molecular markers that define high-risk lesions is clinically critical for improving the prognosis determination of the tumors and their treatment. We decided to focus on the two pathways involving FGFR3 and allelic losses at 9p22 to identify a potential combined role in predicting tumor recurrence, progression and/or muscle. Microsatellite and mutational FGFR3 status analyses was performed in tumor tissue of 58 patients in a prospective unicentre study. The results of microsatellite and FGFR3 analyses were dichotomized as follows: loss of heterozygos-ity (LOH) versus retention of heterozygosity (ROH) on the one hand; mutant FGFR3 (mtFGFR3) versus wild-type FGFR3 (wtFGFR3) on the other hand. The combined 9p22/FGFR3 status was strongly correlated with stage (p=0.001) and grade (p<0.001) whereas the single FGFR3 mutational status was not able to predict recurrence, progression or muscle invasion. The survival curves corresponding to each combined status (mtFGFR3/ROH, wtFGFR3/ROH, mtFGFR3/LOH, wtFGFR3/LOH) were significantly different for recurrence (p=0.008), progression (p=0.046) and progression to muscle invasive disease (p=0.004). In case of 9p22 LOH, the FGFR3 mutational status was strongly associated with different clinical outcomes. In a multivariate model, the combined wtFGFR3/9p22 LOH status remained significant in predicting oncologic outcomes. FGFR3 mutations strongly characterize tumors with low malignant potential and favourable clinical outcome in case of allelic losses at 9p22, whereas its prognostic value becomes null or slightly inverts in case of allelic stability. Thus, our findings may also lead to further experiments in order to study interactions between FGFR3 and genes located at 9p22, as CDKN2A.
Keywords: Bladder cancer, FGFR3, loss of heterozygosity, prognosis, mutations
Introduction
More than 70% of transitional cell carcinomas of the bladder are low stage at initial presentation, namely non-muscle invasive bladder cancer (NMI-BC), and may be surgically removed leaving no detectable disease [1]. To date, establishment of the prognosis of NMI-BC is based on the patient history of NMI-BC and on histopa-thological evaluation of the tumor [2]. Accordingly, there is a clear need to better identify patients with low probability of recurrence and/or progression in order to avoid aggressive follow-up and over treatment. Investigating the early urothelial lesions and developing molecular markers that define high-risk lesions is clinically critical for improving the prognosis determination of the tumors and their treatment. By providing a better understanding of tumor biology, molecular markers may help stratifying these heterogeneous tumors to guide the decision in patient management. Observations of transitional cell carcinomas of the bladder indicated that these tumors develop along two separate, although sometimes overlapping, pathways. The first is characterised by the presence of mutations of the tumor suppressor gene TP53 which are thought to inactivate one allele, followed by the loss of the second, wild-type allele, resulting in a complete loss-of-function of p53 protein. The second pathway involves somatic activating mutations of the FGFR3 gene in papillary tumors [3-6]. FGFR3 has garnered attention as being useful in risk stratification of high grade transitional cell carcinoma (TCC) [7]. However, the bladder cancer development remains a complex multistep process that is not clearly understood, and the study of single determinants does not reflect accurately the cascade of molecular aberrations. Loss of heterozygosity (LOH) has also been described as a distinct and frequent type of molecular alterations in bladder cancer, especially at chromosome 9 loci [8-13]. Specific locus at 9p21-22, referred to as CDKN2A, encodes two proteins, p16INK4A and p14ARF, which are frequently inactivated in bladder cancers [14-17]. However, the prognostic value of p16 was studied with conflicting results [18-20]. Our previous findings have determined the prognostic value of LOH at 9p22 in the prediction for tumor progression in NMI-BC [21].
We decided to focus on the two pathways involving FGFR3 and allelic loss at 9p22 in order to identify a potential combined role in predicting tumor recurrence, progression and/or muscle invasion in NMI-BC.
Materials and methods
Patients
We conducted a prospective monocentric study from January 2000 to November 2006 on patients treated at our institution for a NMI-BC. Microsatellite and mutational FGFR3 status analyses were performed in tumor tissue of 58 patients. The local ethical committee approved the study. Patients received a complete endoscopic resection of the bladder tumor. Tumor grading and staging were performed according to the 1973 World Health Organization grading system and the 1997 TNM classification respectively. At the time of endoscopic resection, venous blood samples were withdrawn from individual patients for constitutional DNA extraction. Analyses for each patient were established from tumor tissue. Surveillance by cystoscopy and cytology was performed according to the European guidelines [1]. Recurrence was defined as the first reappearance of a biopsy-proven urothelial cell carcinoma. Global progression was defined as the diagnosis of an urothelial cell carcinoma of a higher stage and/or grade than the previous occurrence. Progression to muscle-invasion was defined as the diagnosis of pT2 to pT4 urothelial cell carcinoma. Inclusion criteria were complete endoscopic resection of pTa, pT1 or CIS urothelial cell carcinoma of the bladder (NMI-BC) and signature of an informed consent. Exclusion criteria were incomplete resection and/or pT2 or higher urothelial cell carcinoma and/or non-urothelial cell carcinoma and/or urothelial cell carcinoma of the upper urinary tract and/or refusal to participate.
DNA extraction
At the time of endoscopic resection, a biopsy was taken from the main tumor for immediate DNA extraction (Figure 1). The remaining tumor was sent separately for pathological analysis in order to assess the inclusion criteria and the existence of more than 80% cancer cells in the biopsy. DNA was extracted from the tumor biopsy and from blood samples using Qiagen Tissue and Blood Kits (Qiagen, Valencia, CA) according to the manufacturer's instructions. Aliquots were removed to measure DNA concentrations using the DNA-binding fluorochrome Hoechst 33258 (DyNA Quant 200, Hoefer Pharmacia Biotech, San Francisco, CA) before freezing at -20°C until use. Genomic control DNA of Family 134702 was the gift of Dr. H. Cann (Centre d'Etude de Polymorphismes Humains, CEPH, Paris, France).
Figure 1.

Protocol of DNA and RNA extraction from tumor tissue.
Microsatellite analysis
Microsatellite analysis was performed blind from clinical and pathological data in 2 independent experiments to control for reproducibility. Two microsatellite markers at 9p22 (p16 locus) listed in the UCSC database, were used: IFNA.1, and IFNA. Microsatellite analysis was performed as previousy described [21]. Informative cases were scored as LOH when the intensity of the signal for one allele was decreased by at least 50% relative to the allele control i.e. when tumor to normal allelic ratio decreased by 50% or more. In the case of similar tumor and blood allelic ratios (< 10% decrease) the locus was scored as retention of heterozygosity (ROH) in this tumor. A homozygous locus exhibit 2 alleles of identical size so that both alleles appear as a single allele-peak upon electrophoresis of PCR product of normal DNA. In such a case a locus is scored non-informative because tumor allele profiling of this locus cannot allow evaluating the eventual loss of one of the 2 alleles being of identical size despite the presence of an allele-peak of identical size as in the corresponding normal DNA.
FGFR3 mutational status
The mutational status of FGFR3 was assessed by Allele-Specific-PCR (AS-PCR) as described elsewhere with minor modifications [22]. Confirmation of individual mutations was achieved subsequently by direct sequence analysis. The rationale of the AS-PCR is the preferential and specific amplification of a mutant allele achieved by the design of a mutation-specific primer. In TCC of the bladder four activating FGFR3 missense mutations account for >95% of all mutations documented. Two mutations (R248C & S249C) occur at exon 7 and the two others (G372C & Y375C) occur at exon 10. The mutations at codon 248 and 372 were simultaneously detected by multiplex AS-PCR containing the following final concentrations at 20μl volume: 20ng DNA, 1X Buffer II, 5% DMSO, 0.2 mM dNTP, 2 units of TaqGold DNA polymerase (Applied Biosystems) and 0.2 μM of each of the following primers (Each forward primer was end-labelled with a specific fluorophore for subsequent PCR product detection; Applied Biosystems): F248-9: 6FAM 5'CAGTGG CGGTGGTGGTGAGG; R248,5'ATGGGCCGGTGCG GGGACCA; F-372,5'-HEX-5'ATGTCTTTGCAGCC GAGGAGGAG; R372,5'AGCTGAGGATGCCGG CATACACACTGCA; FGLO,NED-5'CCTTTGGGGATC TGTCCACTCCTGA; RGLO,5'GTTGTCCAGGT GAGCCAGGCCAT. The latter couple of primers allowed the amplification of b-globin gene as an internal control. The reaction tubes were incubated in The GeneAmp PCR System 9700 (Applied Biosystems). After one cycle of denaturation and TaqGold activation, 35 cycles were done each consisted of 15 sec at 95 °C, 15 sec at 61°C and 15 sec at 72°C, then a final elongation step of 7 min at 72°C. The mutations at codon 249 and 375 were similarly detected by multiplex AS-PCR containing the following final concentrations at 20μl volume: 20ng DNA, 1X Buffer II, 10% DMSO, 0.2 mM dNTP, 2 units of TaqGold DNA polymerase (Applied Biosystems) and 0.2 μM of each of the following primers: F248-9: 6FAM-5'-CAGTGGCGGTGGTGGTGAGG; R249: 5'-CAGGATGGGCCGGTGCGAGC; F-372: 5'-HEX-5'-ATGTCTTTGCAGCCGAGGAGGAG; R375: 5'-ACCCCGTAGCTGAGGATGCCTTGAC; FGLO: NED-5'-CCTTTGGGGATCTGTCCACTCCTGA; RGLO: 5'-GTTGTCCAGGTGAGCCAGGCCAT. In each experiment wild-type and mutant FGFR3 DNA (A gift of Dr. François Radvanyi, Institut Curie, Paris, France) were included as control. Each individual DNA was analyzed in 2 independent experiments. The resulting PCR products were analyzed on an Applied Biosystems 3130xl genetic analyzer using the GeneMapper software.
Statistical analysis
The results of microsatellite and FGFR3 analyses were dichotomized as follows: loss of heterozygosity (LOH) versus retention of heterozygosity (ROH) on the one hand; mutant FGFR3 (mtFGFR3) versus wild-type FGFR3 (wtFGFR3) on the other hand. The associations between 9p22/FGFR3 statuses and clinico-pathological parameters were tested with two-sided Fisher's exact test or chi-square test for qualitative data and Mann-Whitney and Krus kall-Wallis tests for quantitative data. Kaplan-Meier survival curves were computed by 9p22/FGFR3 statuses with the date of first transurethral resection as the starting point for analysis. End points were recurrence, progression in stage or grade, and progression to muscle invasive disease. Groups were compared using the log-rank test according to recurrence, progression and muscle-invasion free survivals. The limit of statistical significance was defined as p<0.05. Cox regression model was applied with a backward step-wise procedure, in order to assess whether the combined 9p22/FGFR3 statuses were independent predictor of recurrence, progression or muscle invasion. In order to perform multivariate analysis, we decided to adjust the results to the score that each tumor obtained with the EORTC scoring system [2]. Hazard ratios (HR) and their 95% confidence interval (CI) were estimated to obtain risks of each outcome for cases. Tumors displaying non-informative microsatellite markers were considered as tumors with conservation of heterozygosity. All statistical calculations were performed using SPSS 13.0 (Chicago, Illinois) software.
Results
Clinical and pathological characteristics
Characteristics of the patients are summarized in Table 1. Fifty-five percent of patients were smokers. The distribution of initial tumors was: 14 TaG1, 22 TaG2, 3 T1G2, 19 T1G3. CIS was associated with papillary tumors in 7 cases (12%). Intravesical therapy after resection was used as follows: BCG therapy in 26 cases (44.8%), mitomycine C in 17 cases (29.3%). Recurrent disease was observed in 18 cases (31.0%). Late recurrence (≥12 months) occurred in 8 patients, and early recurrence (<12 months) in 10 cases. Progression to higher stage or grade was noted in the course of 6 cases (10.3%). Progression to muscle invasive disease was diagnosed in 4 cases (6.9%).
Table 1.
Patient's cohort characteristics
| Age, years: | |
| Mean | 62.5 |
| Median | 62.0 |
| Range | 39.0-81.4 |
| Smokers, n (%) | 32 (55.2) |
| Multifocality of NMI-BC, n (%) | 28 (48.3) |
| Previous history of NMI-BC, n (%) | 25 (43.1) |
| Stage, n (%) | |
| pTa | 36 (62.1) |
| pT1 | 22 (37.9) |
| Grade, n (%) | |
| G1 | 14 (24.1) |
| G2 | 25 (43.1) |
| G3 | 19 (32.8) |
| Presence of CIS, n (%) | 7 (12.1%) |
| Follow-up, months: | |
| Mean | 28.3 |
| Median | 28.0 |
| Range | 2-98 |
Microsatellite and FGFR3 mutation analyses
LOH on chromosome 9p22 was detected in 14 of the 58 (24.1%) patients.
FGFR3 mutations were detected in 50% of tumor specimens (n=29). Mutations in codon 249 (S249C) occurred most frequently (20 cases). Codons 372 (G372C), 248 (R248C), and 375 (Y375C) mutations were found in 4, 3, and 2 cases, respectively.
Overall, FGFR3 mutation was detected in 50% of tumors with LOH and in 50% of tumor with ROH. Thus, no significant correlation between 9p22 LOH and FGFR3 mutations was reported (p=1.00).
Clinico-pathologic parameters in relation to LOH at 9p22, FGFR3 mutational status and combined 9p22/FGFR3 status
To gain additional information, associations between LOH at 9p22, FGFR3 mutational status combined 9p22/FGFR3 status, and clinico-pathological criteria were analyzed (Table 2). There was no strong significant difference in 9p22 LOH frequency or in FGFR3 mutations according to age, smoking habits or multifocality of NMI-BC. FGFR3 mutations were significantly more detected in tumors with previous history of NMI-BC than in initial NMI-BC (68.0% versus 36.4%, p=0.017). No significant association between CIS and analyses findings was found.
Table 2.
Correlations between clinico-pathologic features and 9p22, FGFR3 and combined FGFR3/9p22 statuses
| Combined FGFR3/9p22 status | FGFR3 status | 9p22 status | ||||||
|---|---|---|---|---|---|---|---|---|
| mtFGFR3/ROH | wtFGFR3/ROH | mtFGFR3/LOH | wtFGFR3/LOH | wt | mt | ROH | LOH | |
| Age: | ||||||||
| <65y | 11 | 14 | 3 | 5 | 19 | 14 | 25 | 8 |
| >65y | 11 | 8 | 4 | 2 | 10 | 15 | 19 | 6 |
| p value | 0.573 | 0.185 | 0.983 | |||||
| Multifocality: | ||||||||
| No | 9 | 11 | 4 | 6 | 17 | 13 | 20 | 10 |
| Yes | 13 | 11 | 3 | 1 | 12 | 16 | 24 | 4 |
| p value | 0.223 | 0.293 | 0.090 | |||||
| Previous BC | ||||||||
| No | 9 | 16 | 3 | 5 | 21 | 12 | 25 | 8 |
| Yes | 13 | 6 | 4 | 2 | 8 | 17 | 19 | 6 |
| p value | 0.127 | 0.017 | 0.983 | |||||
| Smokers : | ||||||||
| No | 9 | 11 | 3 | 3 | 14 | 12 | 20 | 6 |
| Yes | 13 | 11 | 4 | 4 | 15 | 17 | 24 | 8 |
| p value | 0.941 | 0.597 | 0 | |||||
| Adjuvant treatment: | ||||||||
| No | 9 | 3 | 2 | 1 | 4 | 11 | 12 | 3 |
| Yes | 13 | 19 | 5 | 6 | 25 | 18 | 32 | 11 |
| p value | 0.001 | 0.036 | 0.664 | |||||
| Stade : | ||||||||
| pTa | 20 | 11 | 4 | 1 | 12 | 24 | 31 | 5 |
| pT1 | 2 | 11 | 3 | 6 | 17 | 5 | 13 | 9 |
| p value | 0.001 | 0.001 | 0.020 | |||||
| Grade: | ||||||||
| G1 | 10 | 4 | 0 | 0 | 4 | 10 | 14 | 0 |
| G2 | 12 | 5 | 5 | 3 | 8 | 17 | 17 | 8 |
| G3 | 0 | 13 | 2 | 4 | 17 | 2 | 13 | 6 |
| p value | <0.001 | <0.001 | 0.053 | |||||
| CIS: | ||||||||
| No | 21 | 18 | 6 | 6 | 24 | 27 | 39 | 12 |
| Yes | 1 | 4 | 1 | 1 | 5 | 2 | 5 | 2 |
| p value | 0.570 | 0.423 | 0.710 | |||||
A strong correlation between FGFR3 mutations and stage was found. Mutations occurred in 66.7% of pTa tumors, versus 22.7% of pT1 tumors (p=0.001). Mutations were also significantly more detected in G1 (71.5%) and G2 (68.0%) tumors than in G3 tumors (10.5%, p<0.001). No 9p22 LOH was reported in pTaG1 tumors, compared with 37.5% of pT1G3 tumors (p=0.019). LOH at 9p22 was more frequently reported in pT1 tumors compared with pTa tumors (p=0.020). Difference did not reach significance concerning the grade (p=0.053).
The combined 9p22/FGFR3 status was significantly correlated with stage (p=0.001) and grade (p<0.001). Thus, a combination of ROH and FGFR3 mutation was seen in 71.4% of pTaG1 tumors, compared with none of pT1G3 tumors.
Oncologic outcome in relation to LOH at 9p22, FGFR3 mutational status and combined 9p22/FGFR3 status
The FGFR3 mutational status was not able to predict recurrence, progression or muscle invasion in univariate or in a Cox regression model (p=0.277, 0.319, and 0.280, respectively).
The 9p22 LOH was significantly correlated with progression to muscle invasive disease (p=0.040), as previously described (Ploussard et al). Nevertheless, 9p22 LOH was not a significant predictor of recurrence (p=0.819) or overall progression (p=0.145).
When studying the value of combined FGFR3/9p22 status in survival curves, the survival curves corresponding to each status (mtFGFR3/ROH, wtFGFR3/ROH, mtFGFR3/LOH, wtFGFR3/LOH) were significantly different by using the log-rank test, for recurrence (p=0.008, Figure 2), progression (p=0.046, Figure 3), and progression to muscle invasive disease (p=0.004, Figure 4). The most interesting profile was seen in tumors expressing 9p22 LOH. In these 9p22 LOH cases, the FGFR3 mutational status was strongly associated with different clinical outcomes. No recurrence was reported in case of FGFR3 mutation, whereas the recurrence-free survival rates at 1 and 2 years were 47.6% and 23.8% when no FGFR3 mutation was detected (p=0.015). The progression-free survival rate at 2 years was 50% wtFGFR3 cases, compared with 100% in mtFGFR3 cases (p=0.032).
Figure 2.
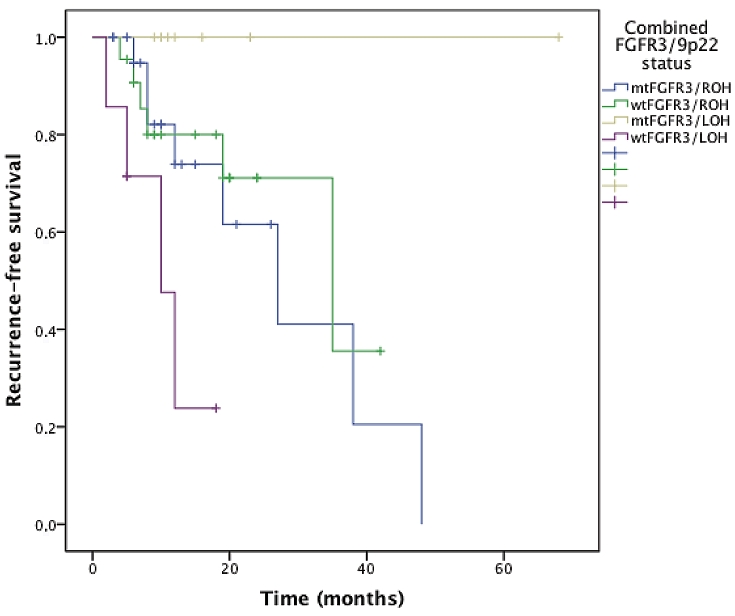
Recurrence-free survival curves stratified on the combined FGFR3/9p22 status.
Figure 3.
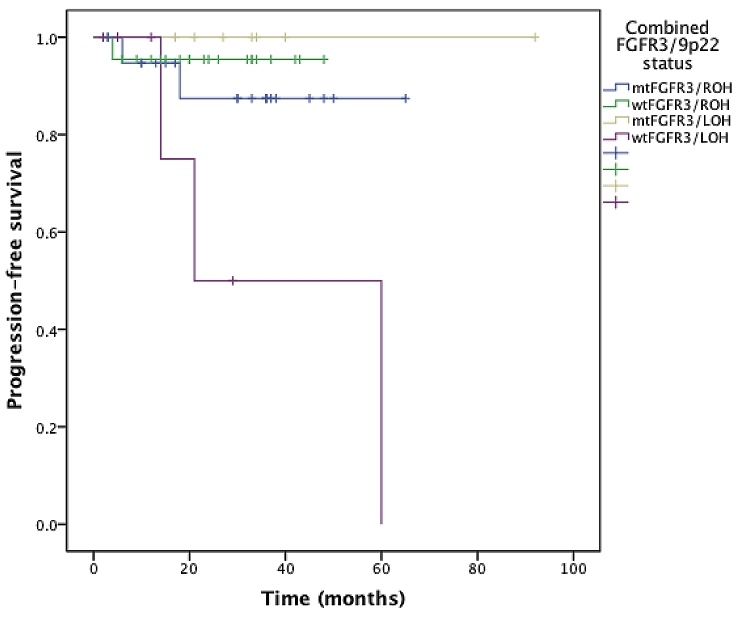
Progression-free survival curves stratified on the combined FGFR3/9p22 status.
Figure 4.
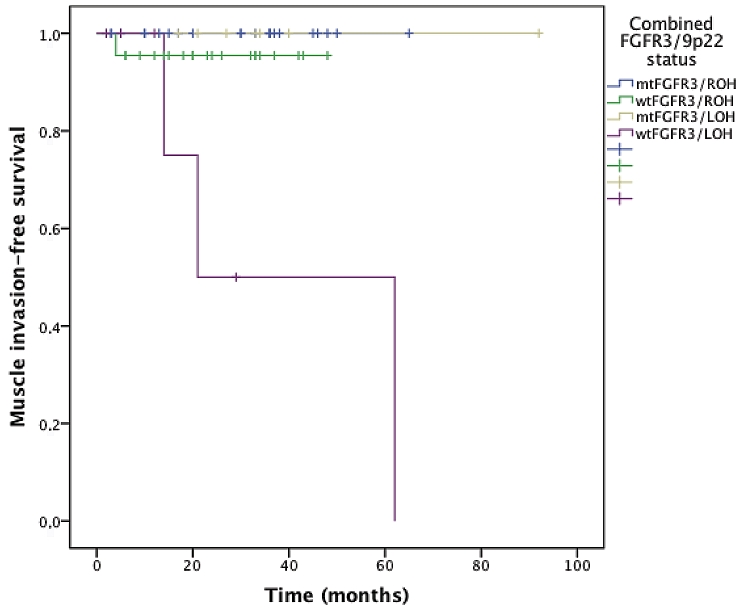
Muscle invasive disease free survival curves stratified on the combined FGFR3/9p22 status.
By contrast, the FGFR3 mutational status was not predictive for recurrence (p=0.746), progression (p=0.499), or progression to muscle invasive disease (p=0.340), in tumors with 9p22 ROH. Survival cures did not differ significantly when 9p22 ROH was observed. The recurrence-free survival rate at 2 years was 71.1% and 61.6% in case of wtFGFR3 and mtFGFR3, respectively. The progression-free survival rate at 2 years was 95.5% and 87.4% in case of wtFGFR3 and mtFGFR3, respectively.
In the Cox regression model after adjustment for age and Bladder Calculator, the combined wtFGFR3/9p22 LOH status remained significant in predicting recurrence, global progression and progression to invasive disease (p=0.007, 0.023 and 0.015, respectively). The HR of the combined wtFGFR3/9p22 LOH for recurrence was 5.5 (95%CI: 1.6-18.9). The HR of the combined wtFGFR3/9p22 LOH for progression was 8.0 (95% CI: 1.3-47.3).
Discussion
The recent reports have led to the suggestion that urothelial carcinoma develop through at least two molecular pathways, one related to FGFR3 and one related to TP53 [23]. The aim of such a pathway-driven approach was to identify strong and comprehensible gene signatures associated with carcinogenesis of bladder cancer [24]. In previous findings, FGFR3 mutations in nonmuscle invasive bladder carcinoma were reported to be associated with low recurrence rate [22,26]. Further studies revealed contradictory findings. In a large prospective study [25], FGFR3 mutations were associated with increased recurrence rate only in TaG1 tumors. Bladder cancer development is a complex multistep process and the pathway from normal to malignant urothelium is not clearly understood. Molecular alterations in tumors are not isolated and the study of single determinants does not reflect accurately the cascade of molecular aberrations.
We previously found that 9p22 LOH was able to discriminate potentially progressing tumors from non-progressing ones, in patients presenting with primary or recurrent NMI-BC, and that LOH at 9p22 had an independent prognostic value for progression to muscle invasive disease. Observation of LOH at a specific chromosomal marker in cells from the tumor suggests the presence of a closely linked tumor suppressor gene, the loss of which is involved in pathogenesis of the tumor. Specific locus at 9p21-22, referred to as CDKN2A, encodes two proteins, p16INK4A and p14ARF, which are frequently inactivated in bladder cancers [14-17]. The protein p16 plays an important role in cell cycle, and blocks the cell division at G1/S checkpoint [27]. The prognostic value of loss of p16 was studied in the literature [18-20]. Moreover, association between 9p22 LOH and loss of p16 function have been previously demonstrated by immunohistochemical analysis in NMI-BC [14]. These findings have led us to address the question whether the combined FGFR3/9p22 status might be predictive for clinical outcome in NMI-BC.
As previously described, we found FGFR3 mutations in 50% of all tumors [28]. We also noted in line with published findings that the mutation frequency of FGFR3 was significantly associated with tumor stage and grade [25-26,28]. FGFR3 mutations characterize low grade and low stage NMI-BC with favourable histological features. The 9p22 LOH was more frequently detected in high grade and high stage tumors, but difference was not so markedly detected than FGFR3 mutational status.
Losses at 9p22 were similarly distributed in groups with or without FGFR3 mutations [28]. Thus, a correlation of FGFR3 mutations and alterations at 9p22 does not exist in NMI-BC. We could not conclude on the fact that losses on 9p22 occur earlier than FGFR3 mutations. However, the presence of allelic losses at 9p22 might modulate the mechanisms of FGFR3 mutations on the disease recurrence and progression.
Our observations suggest that allelic losses at 9p22 locus or FGFR3 mutations are not significantly associated with changes leading to tumor recurrence and global progression when these molecular alterations are taking into account separately, except for deletion of genes at 9p22 that may be more likely to lead to muscle invasion.
In spite of limitations due to the low number of cases, we think our findings are of great value and may participate in the understanding of the conflicting results reported in the literature concerning the prognostic value of the FGFR3 mutational status. In the present series, we did not found significant association with isolated FGFR3 status and propensity for recurrence or progression. Nevertheless, our findings emphasized that the clinical relevance of the FGFR3 mutational status depended on the allelic status at 9p22. In tumors showing allelic losses at 9p22 locus, the presence of FGFR3 mutation (in 50% of these cases) characterized low malignant tumors with favourable clinical outcome that are at low risk of recurrence and progression. By contrast, combination of 9p22 LOH and absence of FGFR3 mutation was associated with tumors at high malignant potential with early recurrence, progression and muscle invasion. Interestingly, the risk of recurrence was slightly higher in tumors with FGFR3 mutation when no allelic loss was found at 9p22 (recurrence-free survival rate at 2 years: 61.6% versus 71.1%). Nevertheless, difference did not reach significance. As tumors without 9p22 LOH represents 75% of the NMI-BC of our study, it is not surprising that large prospective studies report a higher recurrence rate in NMI-BC with FGFR3 mutations [25].
Recently, Sylvester and co-workers retrospectively analyzed 2596 EORTC bladder tumor patients and proposed a scoring system predicting the risk of recurrence and of progression of NMI-BC [2]. We decided to integrate this tool in the multivariate analysis in order to reduce the number of integrated variables and, thus, to keep the statistical relevance for the Cox model. In our multivariate analysis, the combined FGFR3/9p22 status was associated with an increased risk of recurrence, progression and muscle-invasion independent of other prognostic parameters such as age and Bladder Calculator. The tumors displaying wtFGFR3 and 9p22 LOH had a risk of recurrence and progression increased by 5.5 and 8-fold, respectively. In spite of the weak rate of events, strong significant differences were detected that bore witness of strong value of such a marker combination. Another interest of our methodology was to evaluate any patient with NMI-BC at any time of the follow-up, reflecting every day clinical practice.
However, the short follow-up of our cohort constituted an important limitation to end in such observations. Our follow-up data were not able to evaluate the correlation between the combined FGFR3/9p22 status and progression, in subgroups of patients stratified on stage and grade, due to the small number of progression observed. Given the mid-term follow-up of our cohort, we cannot conclude whether the combined mutational status impact the patient survival. Longer follow-up and larger cohort remain warranted.
The findings of this study support the notion that only a combination of molecular markers may characterize a subgroup of bladder cancer with poor prognosis. Our prognostic evaluation of the combined FGFR3/9p22 status seems to be interesting to better identify patients at high risk of poorer clinical outcome. The strengths of our study were the prospective design, the relative high number of cases, and the homogeneity of our cohort, consisting in NMI-BC. One concern was the possible presence of concomitant CIS in our tumor cohort. We systematically performed random quadrant biopsies in all cases and found 7 CIS cases. However, the possibility of undetected concomitant CIS cannot be entirely ruled out because resection was performed under white-light cystoscopy. Such a consideration is important as CIS plays an important role in tumor progression and may affect the genetic profile. Nevertheless, no significant association between CIS and combined FGFR3/9p22 status was found.
Conclusions
Correlations of the combined FGFR3/9p22 status with clinical outcome provides important dimension to such an analysis with biological implication in NMI-BC. This marker combination represents a valuable prognostic marker of recurrence and progression in NMI-BC. Our results emphasize that the clinical relevance of the FGFR3 mutational status depends on the presence of allelic losses at 9p22. FGFR3 mutations strongly characterize tumors with low malignant potential and favourable clinical outcome in case of allelic losses at 9p22, whereas its prognostic value becomes null or slightly inverts in case of allelic stability. Thus, our findings may also lead to further experiments in order to study interactions between FGFR3 and genes located at 9p22, as CDKN2A.
Acknowledgments
The authors have no conflict of interest and nothing to disclose.
References
- 1.Stenzl A, Cowan NC, De Santis M, Jakse G, Kuczyk MA, Merseburger AS, Ribal MJ, Sherif A, Witjes JA. The Updated EAU Guidelines on Muscle-Invasive and Metastatic Bladder Cancer. Eur Urol. 2009;55:815–25. doi: 10.1016/j.eururo.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Sylvester RJ, van der Meijden AP, Oosterlinck W, Witjes JA, Bouffioux C, Denis L, Newling DW, Kurth K. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: a combined analysis of 2596 patients from seven EORTC trials. Eur Urol. 2006;49:466–5. doi: 10.1016/j.eururo.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 3.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 4.Bakkar AA, Wallerand H, Radvanyi F, Lahaye JB, Pissard S, Lecerf L, Kouyoumdjian JC, Abbou CC, Pairon JC, Jaurand MC, Thiery JP, Chopin DK, de Medina SG. FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Res. 2003;63:8108–12. [PubMed] [Google Scholar]
- 5.van Rhijn BW, van der Kwast TH, Vis AN, Kirkels WJ, Boevé ER, Jöbsis AC, Zwarthoff EC. FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Res. 2004;64:1911–4. doi: 10.1158/0008-5472.can-03-2421. [DOI] [PubMed] [Google Scholar]
- 6.Lamy A, Gobet F, Laurent M, Blanchard F, Varin C, Moulin C, Andreou A, Burger M, van der Aa MN, van Oers JM, Brinkmann A, van der Kwast TH, Steyerberg EC, Stoehr R, Kirkels WJ, Denzinger S, Wild PJ, Wieland WF, Hofstaedter F, Hartmann A, Zwarthoff EC. Prediction of progression of non-muscle-invasive bladder cancer by WHO 1973 and 2004 grading and by FGFR3 mutation status: a prospective study. Eur Urol. 2008;54:835–43. doi: 10.1016/j.eururo.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 7.Burger M, Burger SJ, Denzinger S, Wild PJ, Wieland WF, Blaszyk H, Obermann EC, Stoehr R, Hartmann A. Elevated microsatellite instability at selected tetranucleotide repeats does not correlate with clinicopathologic features of bladder cancer. Eur Urol. 2006;50:770–5. doi: 10.1016/j.eururo.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 8.Catto JW, Xinarianos G, Burton JL, Meuth M, Hamdy FC. Differential expression of hMLH1 and hMSH2 is related to bladder cancer grade, stage and prognosis but not microsatellite instability. Int J Cancer. 2003;105:484–90. doi: 10.1002/ijc.11109. [DOI] [PubMed] [Google Scholar]
- 9.Berger AP, Parson W, Stenzl A, Steiner H, Bartsch G, Klocker H. Microsatellite alterations in human bladder cancer: detection of tumor cells in urine sediment and tumor tissue. Eur Urol. 2002;41:532–9. doi: 10.1016/s0302-2838(02)00073-8. [DOI] [PubMed] [Google Scholar]
- 10.Miyao N, Tsai YC, Lerner SP, Olumi AF, Spruck CH, 3rd, Gonzalez-Zulueta M, Nichols PW, Skinner DG, Jones PA. Role of chromosome 9 in human bladder cancer. Cancer Res. 1993;53:4066–70. [PubMed] [Google Scholar]
- 11.Stadler WM, Sherman J, Bohlander SK, Roulston D, Dreyling M, Rukstalis D, Olopade OI. Homozygous deletions within chromosomal bands 9p21-22 in bladder cancer. Cancer Res. 1994;54:2060–3. [PubMed] [Google Scholar]
- 12.Mhawech-Fauceglia P, Cheney RT, Schwaller J. Genetic alterations in urothelial bladder carcinoma: an updated review. Cancer. 2006;106:1205–16. doi: 10.1002/cncr.21743. [DOI] [PubMed] [Google Scholar]
- 13.Bartoletti R, Cai T, Nesi G, Roberta Girardi L, Baronti G, Dal Canto M. Loss of p16 Expression and Chromosome 9p21 LOH in Predicting Outcome of Patients Affected by Superficial Bladder Cancer. J Surg Res. 2007;143:422–7. doi: 10.1016/j.jss.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Czerniak B, Chaturvedi V, Li L, Hodges S, Johnston D, Roy JY, Luthra R, Logothetis C, Von Eschenbach AC, Grossman HB, Benedict WF, Batsakis JG. Superimposed histologic and genetic mapping of chromosome 9 in progression of human urinary bladder neoplasia: implications for a genetic model of multistep urothelial carcinogenesis and early detection of urinary bladder cancer. Oncogene. 1999;18:1185–96. doi: 10.1038/sj.onc.1202385. [DOI] [PubMed] [Google Scholar]
- 15.Cairns P, Tokino K, Eby Y, Sidransky D. Homozygous deletions of 9p21 in primary human bladder tumors detected by comparative multiplex polymerase chain reaction. Cancer Res. 1994;54:1422–4. [PubMed] [Google Scholar]
- 16.Devlin J, Keen AJ, Knowles MA. Homozygous deletion mapping at 9p21 in bladder carcinoma defines a critical region within 2cM of IFNA. Oncogene. 1994;9:2757–60. [PubMed] [Google Scholar]
- 17.Hitchings AW, Kumar M, Jordan S, Nargund V, Martin J, Berney DM. Prediction of progression in pTa and pT1 bladder carcinomas with p53, p16 and pRb. Br J Cancer. 2004;91:552–7. doi: 10.1038/sj.bjc.6601954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedrich MG, Blind C, Milde-Langosch K, Erbersdobler A, Conrad S, Loning T, Hammerer P, Huland H. Frequent p16/MTS1 inactivation in early stages of urothelial carcinoma of the bladder is not associated with tumor recurrence. Eur Urol. 2001;40:518–24. doi: 10.1159/000049829. [DOI] [PubMed] [Google Scholar]
- 20.Korkolopoulou P, Christodoulou P, Lazaris A, Thomas-Tsagli E, Kapralos P, Papanikolaou A, Kalliteraki I, Davaris P. Prognostic implications of aberrations in p16/pRb pathway in urothelial bladder carcinomas: a multivariate analysis including p53 expression and proliferation markers. Eur Urol. 2001;39:167–77. doi: 10.1159/000052432. [DOI] [PubMed] [Google Scholar]
- 21.Ploussard G, Dubosq F, Soliman H, Verine J, Desgrandchamps F, De Thé H, Mongiat-Artus P. Prognostic value of loss of heterozygosity at chromosome 9p in non-muscle-invasive bladder cancer. Urology. 2010;76:513–518. doi: 10.1016/j.urology.2010.03.037. [DOI] [PubMed] [Google Scholar]
- 22.Bakkar AA, Quach V, Le Borgne A, Toublanc M, Henin D, Wallerand H, Radvanyi F, Bittard H, Ravery V, Gibod LB, de Medina SG, Chopin DK, Grandchamp B. Sensitive allele specific PCR assay able to detect FGFR3 mutations in tumors and urine from patients with urothelial cell carcinoma of the bladder. Clin Chem. 2005;51:1555–7. doi: 10.1373/clinchem.2005.049619. [DOI] [PubMed] [Google Scholar]
- 23.Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nat Rev Cancer. 2005;5:713–25. doi: 10.1038/nrc1697. [DOI] [PubMed] [Google Scholar]
- 24.Mitra AP, Pagliarulo V, Yang D, Waldman FM, Datar RH, Skinner DG, Groshen S, Cote RJ. Generation of a concise gene panel for outcome prediction in urinary bladder cancer. J Clin Oncol. 2009;27:3929–37. doi: 10.1200/JCO.2008.18.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernandez S, Lopez-Knowles E, Lloreta J, Kogevinas M, Amoros A, Tardón A, Carrato A, Kishore S, Serra C, Malats N, Real FX. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J Clin Oncol. 2006;24:3664–71. doi: 10.1200/JCO.2005.05.1771. [DOI] [PubMed] [Google Scholar]
- 26.van Rhijn BW, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001;61:1265–8. [PubMed] [Google Scholar]
- 27.Wolff EM, Liang G, Jones PA. Mechanisms of Disease: genetic and epigenetic alterations that drive bladder cancer. Nat Clin Pract Urol. 2005;2:502–10. doi: 10.1038/ncpuro0318. [DOI] [PubMed] [Google Scholar]
- 28.Junker K, van Oers JM, Zwarthoff EC, Kania I, Schubert J, Hartmann A. Fibroblast growth factor receptor 3 mutations in bladder tumors correlate with low frequency of chromosome alterations. Neoplasia. 2008;10:1–7. doi: 10.1593/neo.07178. [DOI] [PMC free article] [PubMed] [Google Scholar]