

Abstract
3-Fluoro-1-((thiazol-4-yl)ethynyl)benzenes constitute an important class of high-affinity metabotropic glutamate subtype 5 receptor (mGluR5) ligands, some of which have been labeled with fluorine-18 (t1/2 = 109.7 min), to provide radioligands for molecular imaging of brain mGluR5 in living animal and human subjects with positron emission tomography (PET). Labeling in the 3-fluoro position of such ligands can be achieved through aromatic nucleophilic substitution of a halide leaving group with [18F]fluoride ion when a weakly activating m-nitrile group is present, but is generally very low yielding (< 8%). Here we used a microfluidic reaction platform to show that greatly enhanced (up to 6-fold) radiochemical yields can be achieved from suitably synthesized diaryliodonium tosylate precursors. The presence of a m-nitrile or other activating group is not required. Similar conditions were adopted in a more conventional automated radiochemistry platform having a single-pot reactor, to produce mGluR5 radioligands in useful radioactivities for PET imaging.
Introduction
The short-lived positron-emitter, fluorine-18 (t1/2 = 109.7 min), attracts intense interest because of its utility as a radiolabel in probes for molecular imaging in living animal and human subjects with positron emission tomography (PET).[1] Many of these probes, commonly called radioligands, are targeted at low concentration protein targets, such as brain neurotransmitter receptors, neurotransmitter transporters, channels and enzymes.[1] In order to avoid saturation of these generally low concentration binding sites with accompanying non-radioactive probe (carrier) and to avoid possible pharmacological effects, the radioligands must be prepared and administered with a high specific radioactivity i.e., with a high amount of radioactivity per unit mass of carrier.[1] Useful specific radioactivities typically exceed 1 Ci of radioligand per μmol of carrier. The source of fluorine-18 used to produce the probes must therefore have a specific radioactivity of at least this order of magnitude.
The cyclotron-promoted 18O(p,n)18F reaction on 18O-enriched water is the most attractive and popular route for producing high activities of fluorine-18 in high specific activities.[2] This process provides the fluorine-18 as [18F]fluoride ion. Hence, the first step in producing an 18F-labeled PET radioligand has generally been either an aliphatic or aromatic nucleophilic substitution reaction. Ideally, radioligand syntheses should require only one such step.
We have developed 3-fluoro-5-((2-([18F]fluoromethyl)thiazol-4-yl)ethynyl)benzonitrile ([18F]1, [18F]SP203, Chart 1) as an effective PET radioligand for quantifying metabotropic glutamate subtype 5 receptors (mGluR5) in human brain.[3,4] This radioligand is potentially useful for studying the involvement of mGluR5 in various neuropsychiatric disorders, such as autism (especially Fragile X syndrome) and drug addiction, and also for the development of drugs targeting mGluR5. The study of mGluR5 in living animal brain is also of related biomedical interest. However, [18F]SP203 has the radiolabel in an aliphatic position. Whereas this radiolabel adequately resists radiodefluorination in human subjects [4], this is not the case in rat and monkey [3]. In rat, this radiodefluorination occurs through a mercapturic acid pathway [5] and results in avid uptake of [18F]fluoride ion by bone, including skull. The proximity of radioactivity in skull to that in brain can compromise the PET measurement of animal brain mGluR5 densities with [18F]1. Hence, we sought an equivalent radioligand that does not suffer such radiodefluorination. Compound 1 with the radiolabel switched to the aryl fluoro position, which we call [18F]SP203B ([18F]2c, Chart 1), would be expected to meet this need, since most aryl C—F bonds are stable in vivo.
Chart 1.
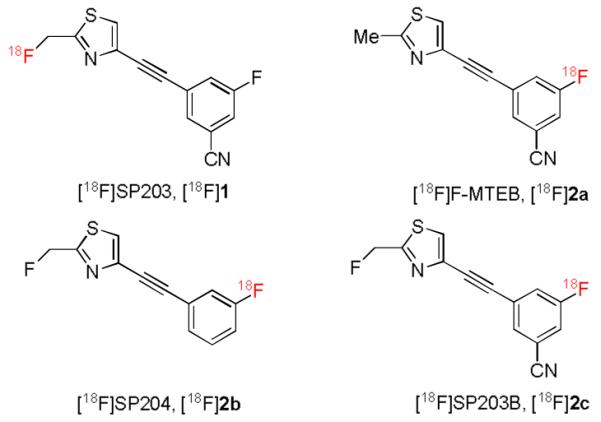
Structures of [18F]SP203 and target radioligands.
[18F]3-Fluoro-5-((2-methylthiazol-4-yl)ethynyl)benzonitrile ([18F]F-MTEB, [18F]2a) is a known mGluR5 radioligand [6] that has very similar structure to [18F]2c (Chart 1). [18F]2a was prepared by Hamill et al. through nucleophilic substitution of a chloro leaving group with cyclotron-produced [18F]fluoride ion, but only in low decay-corrected radiochemical yield (RCY; 4 ± 0.9%).[6] The meta-nitrile group is expected to impart only weak activation for aromatic nucleophilic substitution with [18F]fluoride ion [1] and this likely accounts for the reported low radiochemical yield. Thus, we predicted that a similar labeling approach would also give a low radiochemical yield for [18F]2c.
Simple diaryliodonium salts usefully allow efficient introduction of high specific radioactivity [18F]fluoride ion into aryl rings, whether electron-rich or poorly activated for conventional aromatic nucleophilic substitution reactions.[7-9] It is increasingly evident [8] that these reactions occur by a mechanism that is distinct from classical aromatic nucleophilic substitution. Secondary bonding of the incoming [18F]fluoride ion to the hypervalent iodine atom before transfer to either of the aryl carbon atoms bonded to the same iodine atom likely occurs.[8] Recently we have also shown that diaryliodonium salts may be used to prepare simple meta-substituted [18F]fluoroarenes.[9] We considered that the use of diaryliodonium salt precursors might deliver higher radiochemical yields of [18F]2c and related radioligands, and thereby allow adequate amounts to be prepared regularly for mGluR5 imaging in animal or human subjects with PET. Therefore we set out to prepare and test diaryliodonium tosylates as precursors to [18F]2c and to the two closely related radioligands [18F]2a [6] and [18F]2b [3].
Here we describe the successful syntheses of appropriate diaryliodonium tosylates and their use to synthesize [18F]2a–c in usefully high radiochemical yields, whether in a microfluidic apparatus [8-10] or in a more conventional radiotracer production platform having a single-pot reactor (Synthia) [11].
Results and Discussion
The main objective of this study was to develop an adequately high yielding radiosynthesis of 18F-labeled mGluR5 radioligands, based on a 3-fluoro-1-((thiazol-4-yl)ethynyl)benzene scaffold, in which the fluorine-18 label is incorporated rapidly into the aryl ring. The target radioligands were [18F]2a–c (Chart 1). Each of these ligands is known to have high affinity for mGluR5 in the nanomolar or sub-nanomolar range [3,6], and one ([18F]2a) is already known to be an effective imaging agent [6]. Our aim was accomplished through the reaction of novel diaryliodonium salt precursors with high specific radioactivity [18F]fluoride ion.
Chemistry
Six diaryliodonium salts were required for this study (Scheme 1). Treatment of tri(alkyl)stannylarenes with Koser’s reagent [PhI(OH)OTs] is a useful and established approach for the preparation of diaryliodonium tosylates.[12] To make use of this approach, three iodo analogs (5a–c) of the fluoro compounds 2a–c were needed as precursors for the tri(n-butyl)stannylarenes, 6a–c. These iodo compounds were synthesized through Sonogashira cross-coupling reactions between the trimethylsilyl synthons 3a or 3b and the iodoarenes, 4a or 4b in 32–42% yields. The tri(n-butyl)stannylarenes 6a–c were obtained in 39–57% yield by treating the iodo compounds 5a–c, respectively, with hexa-n-butylditin.[13]
Scheme 1.

Syntheses of bromo-precursors 5d and 5e, and the diaryliodonium tosylates 7a–c and 8a–c.
In the radiofluorination of diaryliodonium salts that do not carry ortho substituents, the [18F]fluoride ion usually prefers to enter the least electron-rich ring.[7] We therefore aimed to prepare diaryliodonium salt precursors to the target radioligands in which one of the aryl rings would be either a phenyl or 4-methoxyphenyl group, since these are electron-rich either to a similar or to a greater extent relative to the aryl ring progenitors of the radioligands. Treatment of compound 6a with Koser’s reagent in dichloromethane at room temperature for two days gave the phenyl(aryl)iodonium tosylate 7a in high purity after double recrystalization in 14% yield. Similar, treatment of 6b and 6c gave the phenyl(aryl)iodonium salts 7b and 7c in 36% and 18% isolated yields, respectively.
For the syntheses of the required 4-methoxyphenyl(aryl)iodonium salts, the required iodine(III) reagent [4-MeO-C6H4I+(OH)TsO−] was prepared by adding a solution of p-TsOH·H2O in MeCN to a solution of 4-MeO-C6H4I(OAc)2 in MeCN at room temperature. Formation of the iodine (III) reagent was indicated by the immediate appearance of a bright yellow color, sometimes quickly accompanied by a bright yellow precipitate. This reagent is unstable under solvent-free conditions [14] and was therefore used in situ to treat the tri(n-butyl)stannylarenes 6a–c in MeCN-dichloromethane. This method gave the target 4-methoxyphenyl(aryl)iodonium tosylates 8a–c in high purity after double recrystalization in 22, 50 and 23% isolated yields, respectively. All prepared diaryliodonium salts were stable for at least 12 months when stored at 4–5 °C, in the dark and under argon.
Additionally, we wished to compare the radiofluorination of diaryliodonium salt precursors with the radiofluorination of halo-precursors for conventional aromatic nucleophilic substitution (SNAr). Therefore, two bromo compounds, 5d and 5e, were prepared as potential precursors for the radiosyntheses of [18F]2a and [18F]2c, respectively. These compounds were obtained through cross-coupling of the trimethylsilyl synthons 3a or 3b with 3,5-dibromobenzonitrile (4c) (Scheme 1).
Radiochemistry
The radiosynthesis of [18F]2a has been achieved previously by classical aromatic nucleophilic substitution in a chloro-precursor with [18F]fluoride ion under microwave heating. However, the isolated RCY was very low (RCY; 4 ± 0.9%).[6] We prepared the corresponding bromo precursor, 5d, and also obtained a low isolated RCY from its use to prepare [18F]2a (7–8%). Similarly, the microwave-assisted radiofluorination of the bromo compound 5e in DMSO gave [18F]2c, but the isolated RCY was again very low (4.0 ± 0.7%, n = 3) (Scheme 2). The RCY did not improve when DMF was used as solvent. We considered that further optimization of such radiosyntheses with conventional reaction platforms would be challenging because of the consumption of appreciable amounts of precursor and the need to manipulate radioactive solutions safely.
Scheme 2.
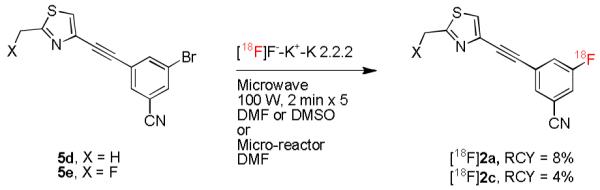
Syntheses of [18F]2a and [18F]2c via nucleophilic substitution in bromo precursors 5d and 5e, respectively.
We have recently shown that a NanoTek apparatus equipped with a microfluidic reactor (internal volume, ~ 31.4 μL) is an excellent platform for optimizing radiofluorination reactions.[8,9] This apparatus allows sequential radiofluorination reactions to be carried out remotely with small amounts of non-radioactive precursor and with very controlled reagent concentrations, reaction times and reaction temperature. Therefore, we used this apparatus to test whether the RCY of [18F]2c from 5e could be improved. Only reactions conducted above 180 °C produced [18F]2c and the highest RCY (just 6%), was obtained when the reaction was performed at 220 °C in DMF. Thus, overall our results confirmed that halo-precursors are poorly reactive in SNAr reactions to produce either [18F]2a or [18F]2c, and that they would be unsuitable for producing these radioligands in adequate activities for PET studies in human subjects. We therefore proceeded to explore diaryliodonium salts as potentially more promising precursors for the target radioligands, [18F]2a–c (Scheme 3).
Scheme 3.
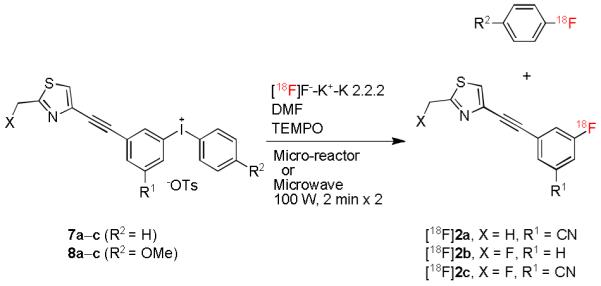
Radiosyntheses of target radioligands [18F]2a–c from diaryliodonium tosylates.
In the micro-reactor, brief (251 s) treatment of the phenyl(aryl)iodonium salt precursor 7c with [18F]fluoride ion in DMF in the presence of potassium-K 2.2.2 complex produced [18F]2c with RCY increasing up to 19% over the temperature range 60–160 °C (Figure 1). The selectivity for producing [18F]2c over [18F]fluorobenzene was very high (~ 35-fold) across the whole temperature range (60–200 °C). Changing the aryl partner in the iodonium salt from phenyl to the more electron-rich 4-methoxyphenyl, as in 8c, gave [18F]2c in an improved RCY of 25 to 28% between 140 and 200 °C, with complete product specificity. The RCY of [18F]2c from 8c increased two-fold between 120 and 140 °C but did not improve much more at yet higher temperatures (Figure 1).
Figure 1.
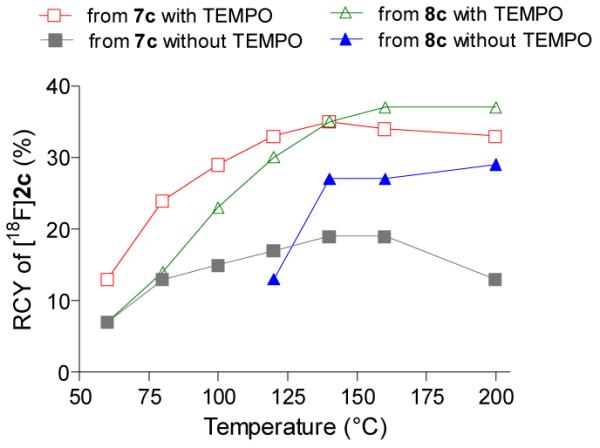
Effects of aryl partner and TEMPO on RCY of [18F]2c in the microfluidic apparatus from precursors 7c (aryl partner, Ph) and 8c (aryl partner, 4-MeOC6H4).
High concentrations of radical scavengers, and especially 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), have been reported to improve the yields and reproducibility of the fluorinations of diaryliodonium salts.[15] The radiofluorination of 7c in the presence of 2 equivalents of TEMPO gave appreciable RCYs of [18F]2c, even at moderate temperature (Figure 1). Between 60 and 200 °C the RCYs of [18F]2c were between 1.9- and 2.5-fold greater for reactions conducted in the presence of TEMPO over reactions without TEMPO, and high product selectivity was retained.
The radiofluorination of 4-methoxyphenyl(aryl)iodonium salt 8c in the presence of TEMPO also gave an appreciable RCY (14%) of [18F]2c at moderate temperature (80 °C) and this yield increased to 37% at 160 °C. Overall, the presence of TEMPO led to a modest 1.4-fold increase in RCYs between 140 and 200 °C.
In our previous studies on the radiofluorination of relatively simple diaryliodonium salts, in the same micro-reactor, we did not observe any great effect of added TEMPO on RCY.[8,9] Thus, the benefit of added TEMPO may depend on the structure of the diaryliodonium salt and perhaps its consequent susceptibility to free radical-induced decomposition. The photosensitivity of diaryliodonium salts is well-known [16] and in some cases they have been developed as photoinitiators of polymerization reactions.[17] Reactions in the micro-reactor occur in the dark and this may be a factor in suppressing any free radical-induced decomposition of diaryliodonium salt precursors.
We also examined the radiofluorination of precursors 8a and 8b to synthesize the two other target mGluR5 ligands, [18F]2a and [18F]2b (Figure 2). In the radiofluorination of 8a, the RCY of [18F]2a gradually increased from 10% to 25% between 80 and 140 °C and increased slightly at higher temperature. No unwanted 4-[18F]fluoroanisole was detected. In the radiofluorination of 8b, the highest RCY of [18F]2b (42%) was obtained at 180 °C. The selectivity for producing [18F]2b over 4-[18F]fluoroanisole was also very high (92 ± 48; n = 5).
Figure 2.
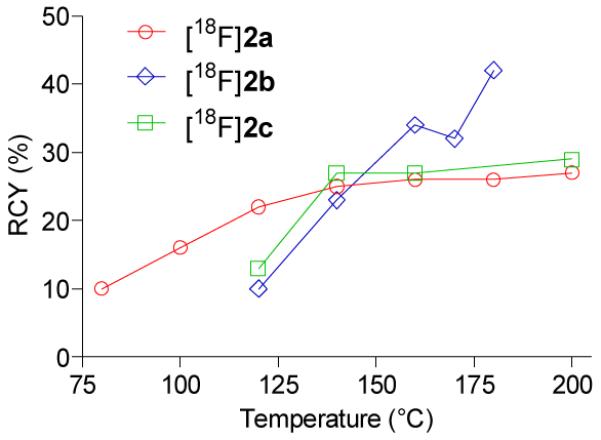
Temperature dependence of RCYs of [18F]2a–c from 8a–c, respectively, in the microfluidic apparatus. Residence times were 251 (○), 188 (◇) and 251 s (□).
To test whether [18F]2c could be produced from the microfluidic apparatus in useful activities for imaging, we also worked with higher amounts of radioactivity (120–200 mCi). Reactions were first performed at 180 °C with 7c as precursor in the absence of TEMPO and were conducted in two ways, either with separate continuous infusion of precursor and [18F]F−-K+-K 2.2.2 reagent solutions or continuous infusion of a premixed solution of precursor and [18F]F−-K+-K 2.2.2 reagent. The RCY of [18F]2c ranged from 10–15% (Table 1). From these sub-optimal reactions, 7–10 mCi of radiochemically and chemically pure [18F]2c were isolated after preparative HPLC. These amounts are adequate for clinical PET experiments. The product had a high specific radioactivity of > 3.4 Ci/μmol corrected to the end of radionuclide production. Inclusion of TEMPO in the reaction gave no significant increase in RCY.
Table 1.
Productions of [18F]2c from diaryliodonium tosylate 7c in a microfluidic reactor.a
| Methodb | Infused conc’n. (mM) |
Flow rate (μL/min) |
Residence time (s) |
RCY (%)c | Selectivity for [18F]2c over [18F]PhF |
Specific activity (Ci/μmol)d |
||
|---|---|---|---|---|---|---|---|---|
| 7c | TEMPO | 7c | [18F]F−–K+–K 2.2.2 | |||||
| A (n = 3) | 6 | 0 | 15 | 10 | 75 | 10 ± 4 | 22 ± 5 | 3.4 ± 0.5 |
| B (n = 1) | 5.7 | 0 | 13 | 10 | 82 | 10 | 18 | 4.2 |
| C (n = 2) | 7 | 0 | 10 | 10 | 94 | 15 ± 1.4 | 15 ± 4 | 4.8 ± 0.3 |
| D (n = 2) | 7 | 14 | 10 | 10 | 94 | 14 ± 0.7 | 20 ± 2 | 8.7 ± 2.4 |
| E (n = 2) | 10 | 0 | 10 | 188 | 10 ± 0.7 | 23 ± 2 | 6.4 ± 1.4 | |
All the reactions were carried out in DMF at 180 °C.
Methods A–D infused precursor and radioactive solutions separately. Method E used infusion of a premixed solution of precursor and [18F]F−–K+–K 2.2.2.
Based on radioactivity infused into the reactor.
Decay-corrected to end of radionuclide production.
In our laboratory, the production of 18F-labeled radioligands from [18F]fluoride ion for clinical use is usually carried out on a more conventional reaction platform. This consists of a semi-robotic apparatus (Synthia) configured with a capped glass vial (1 mL), housed within a microwave heating cavity.[11] We therefore tested whether the useful RCYs obtained for the targeted radioligands in the microfluidic apparatus could also be obtained in this more conventional apparatus (Table 2). We first studied the production of [18F]2c from 7c in which the reaction mixture was subjected to four successive 2-min microwave irradiations. An aliquot was taken from the reaction mixture after each irradiation and analyzed by HPLC. RCY did not improve after the second microwave irradiation. Based on this observation, we conducted two 2-min irradiations in all further radiofluorination reactions. The radiofluorination of 7c (5 μmol) in DMF gave [18F]2c in quite low isolated RCY (8%) but with high selectivity for [18F]2c over [18F]fluorobenzene byproduct (~ 30-fold). Under the same reaction conditions, [18F]2c was also obtained in a low RCY (4%) as the single radioactive product from 8c.
Table 2.
Productions of [18F]2a–c from diaryliodonium tosylates 7a–c and 8a–c in a Synthia apparatus under microwave conditions.a
| Precursor | Product | Without TEMPO |
With 2 eq. of TEMPO |
||
|---|---|---|---|---|---|
| RCY (%) | SA (Ci/μmol)b | RCY (%) | SA (Ci/μmol)b | ||
| 7c | [18F]2c | 8 ± 2 (n = 6) | 4.5 ± 1.2 | 30 ± 1.5 | 7.8 ± 1.9 |
| 8c | [18F]2c | 4 ± 1 (n = 4) | 1.6 ± 1.7 | 33 ± 8 | 6.8 ± 3.0 |
| 7b | [18F]2b | 20 ± 13 | NDc | ||
| 8b | [18F]2b | 40 ± 8 | 7.5 ± 6.6 | ||
| 7a | [18F]2a | 7 ± 2 (n = 2) | 6.5 ± 0.3 | ||
| 8a | [18F]2a | 20 ± 1.1 | 4.8 ± 2.3 | ||
All the experiments were performed in triplicate, except where specified.
Decay-corrected to end of radionuclide production.
HPLC conditions led to co-elution of [18F]2b along with a non-radioactive impurity with strong UV-absorbance, and therefore SA was not determined (ND).
TEMPO had a dramatic effect on the RCYs of these microwave-promoted reactions (Table 2). Thus, inclusion of 2 equivalents of TEMPO in the reaction mixtures increased the RCY of [18F]2c from 7c about 4-fold. Selectivity for [18F]2c over [18F]fluorobenzene was again above 30-fold. Up to 15 mCi of [18F]2c in a formulated dose ready for intravenous injection were prepared from 200 mCi of [18F]fluoride ion. In a separate experiment, where precursor amount was halved to 2.5 μmol of 7c, the RCY was not greatly diminished. The radiofluorination of 8c in the presence of TEMPO gave [18F]2c as the only radioactive product in 8-fold improved RCY (33%).
The radiofluorinations of 7a, 7b, 8a and 8b were also performed with added TEMPO under these conditions (Table 2). The phenyl(aryl)iodonium salt 7a gave [18F]2a in low RCY (7%) with 30-fold selectivity over [18F]fluorobenzene. Use of the corresponding 4-methoxyphenyl(aryl)iodonium salt 8a as precursor increased the RCY of [18F]2a 3-fold to 20% and gave no radioactive byproduct. The radiofluorination of the phenyl(aryl)iodonium salt 7b gave [18F]2b in 20% RCY, with 2.4-fold selectivity over [18F]fluorobenzene. Use of the corresponding 4-methoxyphenyl(aryl)iodonium salt 8b almost doubled the RCY of [18F]2b to 40% and also greatly increased selectivity for this product to ~ 50-fold. These results highlight the benefit of using the highly electron-rich 4-methoxyphenyl ring to direct radiofluorination to the opposite aryl ring, especially when the target ring is also quite electron-rich. In the case of 8b, where the target ring is not activated, it is extremely unlikely that useful activities of [18F]2b could have been obtained by classical SNAr reactions.
Finally, specific radioactivities of the radioligands [18F]2a-c were high at the end of radiosyntheses (Tables 1 & 2). A fluorine atom bonded to an aliphatic carbon could be a potential source of carrier fluoride in classic SNAr reactions. The presence of such a fluorine atom in the diaryliodonium salt precursors in this work did not adversely affect the specific radioactivities of the generated radioligands.
Experimental Section
Materials
3-Fluoro-5-((2-methylthiazol-4-yl)ethynyl)benzonitrile (2a) [6], 2-(fluoromethyl)-4-((3-fluorophenyl)ethynyl)thiazole (2b) [3], 3-fluoro-5-((2-(fluoromethyl)thiazol-4-yl)ethynyl)benzonitrile (2c) [3], 2-methyl-4-((trimethylsilyl)ethynyl)thiazole (3a) [18], 2-(fluoromethyl)-4-((trimethylsilyl)ethynyl)thiazole (3b) [3], and 1-(diacetoxyiodo)-4-methoxybenzene [19] were synthesized as previously reported. [Hydroxy(tosyloxy)iodo]benzene (Koser’s reagent), 1,3-di iodobenzene (98%) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) were purchased from Sigma-Aldrich (Milwaukee, WI). 3,5-Diiodobenzonitrile (98%) was supplied by Spectra Group Ltd. (Millbury, OH). 3-Bromo-5-fluorobenzonitrile and 1-bromo-3-fluorobenzene were purchased from Oakwood Products, Inc. (West Columbia, SC). Other reagents and solvents were purchased from Sigma-Aldrich or Alfa Aesar (Ward Hill, MA) and used without further purification unless otherwise indicated. HPLC-grade MeCN was purchased from Burdick & Jackson (Morristown, NJ). MP-1 or QMA anionic resin cartridges were supplied by ORTG (Oakdale, TN).
General Methods
1H- (400 MHz), 13C- (100 MHz) and 19F- (376.5 MHz) NMR spectra were acquired on an Advance-400 spectrometer (Bruker, Billerica, MA). For 1H-NMR spectra, chemical shifts of residual deuterated solvents were used as internal standards. Chemical shifts (δ) for proton and carbon resonances are reported in parts per million (ppm) downfield from tetramethylsilane (TMS, δ = 0 ppm). 19F-NMR spectra were referenced to external CFCl3 (δ = 0 ppm) in the indicated solvent.
All reactions were monitored with either thin layer chromatography (TLC) or KI/starch paper. Plastic-backed silica gel layers with fluorescent indicator (250 μm, type 60, UV254; Alltech, Deerfield, IL) were used for TLC. The absence of a violet color when a reaction sample was exposed to KI/starch paper dipped in aqueous acetic acid (50% v/v) was taken to confirm complete consumption of iodine(III) reagent. Flash chromatography was performed on silica gel (200 mesh; EM Science, Gibbstown, NJ) with a hexane-ethyl acetate mixture as mobile phase. The purities of compounds 5a–e, 7a–c and 8a–c were determined by HPLC with a Gold 126 solvent module and 166 UV detector (Beckman Coulter) equipped with a Luna C18 column (5 μm, 4.6 × 250 mm, Phenomenex, Torrance, CA), eluted with HCOONH4 (10 mM) in MeCN:H2O (60:40 v/v) at 1.75 mL/min for 5a–e and at 1.1 mL/min for 7a–c and 8a–c. HPLC eluates were monitored for absorbance at 254 nm.
All iodonium salts were stored in amber vials under argon in a refrigerator (4–5 °C). Melting points were recorded on a Mel-Temp apparatus (Thermo Fisher Scientific Corp., Waltham, MA) and are uncorrected. LC-MS was performed on a LCQ Deca instrument (Thermo Fisher Scientific) equipped with a Luna C18(2) column (3 μm, 2 × 50 mm) eluted at 200 μL/min with a gradient of A (H2O-MeOH-AcOH; 90:10:0.5 v/v) and B (MeOH-AcOH; 100:0.5 v/v), initially composed of 20% B and linearly reaching 80% B over 3 min. High resolution mass spectra (HRMS) were obtained at the Mass Spectrometry Laboratory, University of Illinois (Urbana, IL).
Syntheses of aryl halides through Sonogashira cross-coupling reactions [3]
3-Iodo-5-((2-methylthiazol-4-yl)ethynyl)benzonitrile (5a)
Argon was bubbled into a stirred mixture of 3a (0.98 g, 5.0 mmol), 3,5-diiodobenzonitrile (4b, 2.13 g, 6.0 mmol), Pd(PPh3)4 (194 mg, 168 μmol), CuI (64 mg, 340 μmol) and Et3N (3.5 mL, 25.1 mmol) in anhydrous 1,2-dimethoxyethane (DME, 25 mL) while the solution was heated to 80 °C. After 10 min, (n-Bu)4NF in THF (1 M; 10 mL) was added dropwise to the reaction mixture under argon over 10 min. The resulting dark solution was stirred at 80 °C until TLC analysis showed complete consumption of starting material (~ 15 min). The reaction mixture was cooled to r.t., filtered through a pad of Celite and evaporated to dryness. The dark residue was dissolved in dichloromethane (50 mL) and then washed successively with water (2 × 50 mL) and brine (1 × 50 mL). The organic phase was dried over MgSO4, filtered and the solvent evaporated off under reduced pressure. The residue was purified on a silica gel column eluted successively with 10, 15 and 25% (v/v) EtOAc in hexane to give 5a as a white solid (0.72 g, 41%); mp 207–210 °C; 1H-NMR (CDCl3): δ 8.11 (t, J = 1.6 Hz, 1H), 7.93 (t, J = 1.6 Hz, 1H), 7.67 (t, J = 1.6 Hz, 1H), 7.45 (s, 1H), 2.75 (s, 3H); 13C-NMR (CDCl3): δ 166.3, 144.3, 139.9, 135.7, 133.9, 125.7, 124.0, 116.4, 114.3, 93.5, 87.1, 84.8, 19.3; GC-MS (M+): 350; HRMS (ESI, [M + H]+): calc’d for C13H8IN2S 350.9453; found, 350.9447; HPLC: tR, 8.3 min; purity, > 99%.
2-(Fluoromethyl)-4-((3-iodophenyl)ethynyl)thiazole (5b)
Use of the procedure described for 5a with 3b (1.0 g, 4.7 mmol) and 1,3-diiodobenzene (4a, 1.85 g, 5.62 mmol) gave 5b as a white solid (0.67 g, 42%); mp 81–82 °C; 1H-NMR (CDCl3): δ 7.92 (t, J = 1.6 Hz, 1H), 7.72–7.68 (m, 1H), 7.60 (s, 1H), 7.51 (dt, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.08 (t, J = 8.0 Hz, 1H), 5.64 (d, J = 46.8 Hz, 2H, CH2); 13C-NMR (CDCl3): δ 164.8 (d, J = 25.2 Hz), 140.3, 137.9, 137.3, 130.8, 129.9, 124.17, 124.15, 93.7, 87.7, 84.0, 80.6 (d, J = 170.0 Hz); 19F-NMR (CDCl3, 1H-decoupled): δ −211.3; GC-MS [M+]: 343; HRMS (ESI, [M + H]+): calc’d for C12H8FINS, 343.9406; found, 343.9412; HPLC: tR, 11.3 min; purity, > 99%.
3-((2-(Fluoromethyl)thiazol-4-yl)ethynyl)-5-iodobenzonitrile (5c)
Use of the procedure described for 5a with 3b (1.0 g, 4.7 mmol) and 3,5-diiodobenzonitrile (4b, 2.13 g, 6.0 mmol) gave 5c as a white solid (0.55 g, 32%); mp 184–186 °C; 1H-NMR (CDCl3): δ 8.12 (t, J = 1.6 Hz, 1H), 7.95 (t, J = 1.6 Hz, 1H), 7.78 (t, J = 1.6 Hz, 1H), 7.68 (s, 1H), 5.65 (d, J = 46.8 Hz, 2H, CH2); 13C-NMR (CDCl3): δ 165.3 (d, J = 25.2 Hz), 144.3, 140.1, 136.5, 133.9, 125.4 (d, J = 1.0 Hz), 125.3 116.3, 114.4, 93.6, 86.4, 85.4, 80.6 (d, J = 170.0 Hz); 19F-NMR (CDCl3, 1H-decoupled): δ −211.8; GC-MS [M+]: 368; HRMS (ESI, [M + H]+): calc’d for C13H7FIN2S, 368.9359; found, 368.9374; HPLC: tR, 8.7 min; purity > 99%.
3-Bromo-5-((2-(methyl)thiazol-4-yl)ethynyl)benzonitrile (5d)
Compound 3a (1.14 g, 5.7 mmol), 3,5-dibromobenzonitrile (4c, 1.5 g, 5.7 mmol), CuI (219 mg, 1.14 mmol), Pd(PPh3)2Cl2 (420 mg, 0.57 mmol), and Et3N (1.65 mL, 12 mmol) were placed in a 50-mL two-necked round-bottom flask with DME (10 mL) under argon. Argon was bubbled into the resulting dark solution for 30 min while it was heated to 80 °C. A solution of (n-Bu)4NF (1.0 M) in THF (6.5 mL) was added via syringe over 15 min. The reaction mixture was stirred at 80 °C. After 2 h, TLC showed that no starting material was present. The reaction mixture was then cooled to r.t. and poured into aqueous NH4OH (1.0 M; 100 mL) and extracted with methyl tert-butyl ether (3 × 35 mL). The combined organic fractions were dried over MgSO4, and evaporated to dryness in vacuo. Chromatography of the residue on silica gel (EtOAc-hexane, 10:90 to 50:50, v/v) gave the desired product as a white powder (955 mg; 55%); mp 128–130 °C; 1H-NMR: δ 7.90 (t, J = 1.6 Hz, 1H), 7.77–7.73 (m, 2H), 7.46 (s, 1H), 2.75 (s, 3H); 13C-NMR: δ 166.3, 138.6, 135.6, 134.3, 133.5, 125.9, 124.1, 122.8, 116.6, 114.4, 87.2, 84.9, 19.3; LC-MS [M + H]+, m/z: 303.1 and 305.1; HRMS (EI, M+): calc’d for C13H779BrN2S, 301.9513; found, 301.9504; HPLC: tR, 7.7 min; purity, > 99%.
3-Bromo-5-((2-(fluoromethyl)thiazol-4-yl)ethynyl)benzonitrile (5e)
Use of the procedure described for 5a with 3b (0.21 g, 1.0 mmol) and 3,5-dibromobenzonitrile (4c, 0.31 g, 1.2 mmol) gave 5e as a white solid (0.095 g, 30%); mp 118.5–120.5 °C; 1H-NMR (CDCl3): δ 7.92 (s, 1H), 7.76 (d, J = 1.6 Hz, 2H), 7.68 (s, 1H), 5.65 (d, J = 46.8 Hz, 2H); 13C-NMR (CDCl3): δ 165.3 (d, J = 24.1 Hz), 138.6, 136.5, 134.5, 133.5, 125.5, 125.4 (d, J = 1.0 Hz), 122.9, 116.5, 114.5, 86.4, 85.5, 80.6 (d, J = 171.1 Hz); 19F-NMR (CDCl3, 1H-decoupled): δ −211.7; GC-MS (M+): 320; HRMS (ESI, [M + H]+): calc’d for C13H779BrFN2S 320.9497; found, 320.9498; HPLC: tR, 7.9 min; purity, > 99%.
Syntheses of tributylstannanes (6a–c) [13]
3-((2-Methylthiazol-4-yl)ethynyl)-5-(tri(n-butyl)stannyl)benzonitrile (6a)
Argon was bubbled into a stirred mixture of 5a (0.38 g, 1.1 mmol), hexa-n-butylditin (1.27 g, 2.2 mmol) and Pd(PPh3)4 (0.13 g, 0.11 mmol) in toluene (20 mL) while the mixture was heated to 115 °C (~ 15 min). Stirring and heating were then continued until TLC analysis showed complete consumption of 5a (~ 24 h). The reaction mixture was cooled to r.t., filtered through a column of Celite and evaporated to dryness. Unreacted hexa-n-butylditin was removed from the dark residue on silica gel column with hexane as mobile phase. Further elution of the column with hexane-EtOAc (95:5 v/v) gave 6a as a pale yellow oil (0.22 g, 39%). 1H-NMR (CDCl3): δ 7.87 (s, 1H), 7.73 (s, 1H), 7.67 (s, 2H), 7.44 (s, 1H), 2.75 (s, 3H), 1.64–1.41 (m, 6H), 1.33 (sextet, J = 7.2 Hz, 6H), 1.21–1.02 (m, 6H), 0.90 (t, J = 7.2 Hz, 9H); 13C-NMR (CDCl3): δ 166.0, 144.8, 143.2, 139.1, 136.3, 134.2, 123.2, 123.1, 118.7, 112.3, 87.0, 85.5, 28.9, 27.3, 19.3, 13.7, 9.9.
2-(Fluoromethyl)-4-((3-(tri(n-butyl)stannyl)phenyl)ethynyl)thiazole (6b)
Use of the procedure described for 6a with 5b (0.43 g, 1.26 mmol) and hexa-n-butylditin (1.46 g, 2.52 mmol) gave 6b as a colorless oil (0.32 g, 51%). The reaction time was 16 h. 1H-NMR (CDCl3): δ 7.66 (s, 1H), 7.58 (s, 1H), 7.52–7.43 (m, 2H), 7.33–7.28 (m, 1H), 5.64 (d, J = 46.8 Hz, 2H, CH2), 1.64–1.43 (m, 6H), 1.33 (sextet, J = 7.2 Hz, 6H), 1.18–0.98 (m, 6H), 0.89 (t, J = 7.2 Hz, 9H); 13C-NMR (CDCl3): δ 164.6 (d, J = 24.1 Hz), 142.4, 139.6, 138.1, 136.8, 131.4, 127.6, 123.4, 121.8, 90.2, 82.7, 80.7 (d, J = 172.1 Hz), 29.1, 27.4, 13.7, 9.6; 19F-NMR (CDCl3, 1H-decoupled): δ −211.3.
3-((2-(Fluoromethyl)thiazol-4-yl)ethynyl)-5-(tri(n-butyl)stannyl)benzonitrile (6c)
Use of the procedure described for 6a with 5c (0.28 g, 0.75 mmol) and hexa-n-butylditin (0.87 g, 1.5 mmol) gave 6c as colorless oil (0.23 g, 57%). The reaction time was 17 h. 1H-NMR (CDCl3): δ 7.86–7.81 (m, 1H), 7.74 (t, J = 1.6 Hz, 1H), 7.70–7.68 (m, 1H), 7.65 (s, 1H), 5.65 (d, J = 46.8 Hz, 2H, CH2), 1.59–1.48 (m, 6H), 1.33 (sextet, J = 7.2 Hz, 6H), 1.16–1.08 (m, 6H), 0.90 (t, J = 7.2 Hz, 9H); 13C-NMR (CDCl3): δ 165.1 (d, J = 26.2 Hz), 145.0, 143.2, 139.4, 137.2, 134.3, 124.6 (d, J = 2.0 Hz), 122.8, 118.6, 112.4, 87.7, 84.8, 80.6 (d, J = 171.1 Hz), 28.9, 27.3, 13.6, 9.9; 19F-NMR (CDCl3, 1H-decoupled): δ −211.5.
Syntheses of diaryliodonium tosylates (7a–c) through treatment of 6a–c with Koser’s reagent
(3-Cyano-5-((2-methylthiazol-4-yl)ethynyl)phenyl)(phenyl)iodonium tosylate (7a)
Koser’s reagent (0.084 g, 0.21 mmol) was added to a solution of 6a (0.11 g, 0.21 mmol) in dichloromethane (3 mL). The mixture was stirred under argon at r.t. until TLC analysis and KI/starch paper test showed reaction was complete (48 h). Evaporation of solvent gave a yellow oil. Diethyl ether (15 mL) was added to this oil and the resulting mixture was sonicated for 5 min. The precipitate was filtered off and recrystallized, first from MeCN-Et2O and then from MeOH-Et2O, to give 7a as a white solid (17 mg, 14%). mp 217–220 °C (dec.); 1H-NMR (CD3OD): δ 8.66 (t, J = 1.6 Hz, 1H), 8.63 (t, J = 1.6 Hz, 1H), 8.27 (d, J = 7.2 Hz, 2H), 8.21 (t, J = 1.6 Hz, 1H), 7.82 (s, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.59 (t, J = 8.0 Hz, 2H), 7.22 (d, J = 7.6 Hz, 2H), 2.73 (s, 3H), 2.36 (s, 3H); 13C-NMR (d6-DMSO): δ 166.4, 145.7, 141.6, 138.2, 137.9, 137.5, 135.3, 134.0, 132.4, 131.9, 128.0, 126.7, 125.4, 125.3, 117.1, 116.7, 116.2, 114.4, 88.2, 84.3, 20.7, 18.7; LC-MS (ESI, [M–OTs]+): 426.9; HRMS (ESI, [M–OTs]+): calc’d for C19H12IN2S, 426.9766; found, 426.9759; HPLC: tR, 2.9 min; purity > 99%.
(3-((2-(Fluoromethyl)thiazol-4-yl)ethynyl)phenyl)(phenyl)iodonium tosylate (7b)
Use of the procedure described for 7a with 6b (0.13 g, 0.26 mmol) and Koser’s reagent (0.10 g, 0.26 mmol) with a reaction time of 24 h gave 7b as a white solid (55 mg, 36%) after two crystallizations, first from MeCN-Et2O and then from MeOH-Et2O. mp 175–177 °C (dec.); 1H-NMR (CD3OD): δ 8.41 (t, J = 1.6 Hz, 1H), 8.27–8.16 (m, 3H), 8.00 (s, 1H), 7.84 (dt, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.75–7.67 (m, 3H), 7.60–7.52 (m, 3H), 7.22 (d, J = 8.0 Hz, 2H), 5.65 (d, J = 46.8 Hz, 2H, CH2), 2.36 (s, 3H); 13C-NMR (CD3OD): δ 167.1 (d, J = 23.1 Hz), 143.6, 141.7, 138.9, 137.8, 136.6, 136.4, 133.9, 133.3, 133.1, 130.9, 129.9, 127.6 (d, J = 2.0 Hz), 127.2, 127.0, 116.3, 115.9, 87.3, 86.6, 81.2 (d, J = 168.0 Hz), 21.3; 19F-NMR (CD3OD, 1H-decoupled): δ −211.6; LC-MS (ESI, [M–OTs]+): 419.9, HRMS (ESI, [M–OTs]+): calc’d for C18H12FINS, 419.9719; found, 419.9724; HPLC: tR 3.0 min; > 99% purity.
(3-Cyano-5-((2-(fluoromethyl)thiazol-4-yl)ethynyl)phenyl)(phenyl)iodonium tosylate (7c)
Use of the procedure described for 7a with 6c (0.55 g, 1.04 mmol) and Koser’s reagent (0.41 g, 1.04 mmol), with a reaction time of 48 h in dichloromethane (15 mL), gave 7c as a white solid (118 mg, 18%) after crystallization from MeCN-Et2O and then from MeOH-Et2O. mp 205–208 °C (dec.); 1H-NMR (CD3OD): δ 8.69 (t, J = 1.6 Hz, 1H), 8.65 (t, J = 1.6 Hz, 1H), 8.27 (dt, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 8.23 (t, J = 1.2 Hz, 1H), 8.07 (s, 1H), 7.75 (t, J = 7.2 Hz, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.59 (t, J = 7.6 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 5.65 (d, J = 46.8 Hz, 2H, CH2), 2.36 (s, 3H); 13C-NMR (d6-DMSO): δ 165.0 (d, J = 22.1 Hz), 145.7, 141.7, 138.5, 138.0, 137.6, 135.4, 134.9, 132.4, 132.0, 128.6 (d, J = 2.0 Hz), 128.1, 125.5, 125.1, 117.2, 116.8, 116.2, 114.4, 87.4, 84.8, 80.1 (d, J = 165.0 Hz), 20.8; 19F-NMR (CD3OD, 1H-decoupled): δ −211.9; LC-MS (ESI, [M–OTs]+): 444.9. HRMS (ESI, [M–OTs]+): calc’d for C19H11FIN2S, 444.9672; found, 444.9689; HPLC: tR, 2.9 min; purity, > 99%.
Syntheses of diaryliodonium tosylates (8a–c) through treatment of 6a–c with in situ generated [4-(hydroxy(tosyloxy))iodo]anisole
(3-Cyano-5-((2-methylthiazol-4-yl)ethynyl)phenyl)(4-methoxyphenyl)iodonium tosylate (8a)
A solution of p-toluenesulfonic acid monohydrate (0.027 g, 0.14 mmol) in MeCN (1 mL) was added to a solution of 1-(diacetoxyiodo)-4-methoxybenzene (0.053 g, 0.14 mmol) in MeCN (1 mL) under argon. The resulting pale yellow solution was stirred at r.t. A pale yellow solid started to precipitate within 5 min. A solution of 6a (0.071 g, 0.14 mmol) in dichloromethane (3 mL) was added dropwise to the mixture, which was then stirred under argon at r.t. until TLC analysis and KI/starch paper test indicated completion of reaction (24 h). Solvent removal under reduced pressure gave a yellow oil. Diethyl ether (15 mL) was added to this oil and the resulting mixture was sonicated for 5 min. The precipitate was collected by filtration and recrystallized, first from MeCN-Et2O and then MeOH-Et2O, to give 8a as a white solid (19 mg, 22%). mp 206 °C (dec.); 1H NMR (CD3OD): δ 8.59 (t, J = 1.2 Hz, 1H), 8.55 (t, J = 1.2 Hz, 1H), 8.21–8.13 (m, 3H), 7.82 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 7.6 Hz, 2H), 7.09 (d, J = 9.2 Hz, 2H), 3.86 (s, 3H), 2.73 (2, 3H), 2.36 (s, 3H); 13C NMR (CD3OD): δ 169.1, 164.9, 143.5, 142.2, 141.8, 139.2, 139.0, 138.7, 136.1, 129.9, 128.1, 127.1, 127.0, 119.2, 116.9, 116.8, 116.6, 105.0, 89.0, 85.3, 56.5, 21.4, 18.8; LC-MS (ESI, [M–OTs]+): 456.9; HRMS (ESI, [M–OTs]+): calc’d for C20H14IN2OS, 456.9872; found, 456.9872; HPLC: tR, 3.0 min; purity, > 99%.
(3-((2-(Fluoromethyl)thiazol-4-yl)ethynyl)phenyl)(4-methoxyphenyl)iodonium tosylate, (8b)
Use of the procedure described for 8a with 6b (0.26 g, 0.5 mmol) and in situ generated [hydroxy(tosyloxy)iodo]arene (HTIA; 0.5 mmol) gave 8b as a white solid (155 mg, 50%) after recrystallization from MeCN-Et2O and MeOH-petroleum ether. The reaction time was 24 h. mp 161–164 °C (dec.); 1H-NMR (CD3OD): δ 8.33 (s, 1H), 8.20–8.08 (m, 3 H), 8.00 (s, 1 H), 7.80 (d, J = 7.6 Hz, 1H), 7.68 (d, J = 8.0 Hz, 2H), 7.52 (t, J = 8.0 Hz, 1H), 7.20 (d, J = 8.0 Hz, 2H), 7.06 (d, J = 8.8 Hz , 2H), 5.64 (d, J = 46.8 Hz, 2H, CH2), 3.84 (s, 3H), 2.34 (s, 3H); 13C-NMR (CD3OD): δ 167.0 (d, J = 23.1 Hz), 164.7, 143.6, 141.7, 138.8, 138.4, 137.8, 136.2, 136.16, 133.0, 129.9, 127.6 (d, J = 2.0 Hz), 127.1, 127.0, 119.0, 116.4, 104.7, 87.4, 86.5, 81.2 (d, J = 168.0 Hz), 56.4, 21.4; 19F-NMR (CD3OD, 1H-decoupled): δ −211.7; LC-MS (ESI, [M–OTs]+): 449.9; HRMS (ESI, [M–OTs]+): calc’d for C19H14FINOS, 449.9825; found, 449.9825; HPLC: tR, 3.2 min; purity, 98.9%.
(3-Cyano-5-((2-(fluoromethyl)thiazol-4-yl)ethynyl)phenyl)(4-methoxyphenyl)iodonium tosylate (8c)
Use of the procedure described for 8a with 6c (0.23 g, 0.43 mmol) and in situ generated HTIA (0.43 mmol) gave 8c as a white solid (64 mg, 23%) after double recrystallization from MeCN-Et2O. The reaction time was 24 h. mp 196–198 °C (dec.); 1H-NMR (CD3OD): δ 8.61 (t, J = 1.6 Hz, 1H), 8.57 (t, J = 1.6 Hz, 1H), 8.23–8.14 (m, 3H), 8.07 (s, 1H), 7.68 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz , 2H), 7.14–7.06 (m, 2H), 5.65 (d, J = 46.8 Hz, 2H, CH2), 3.86 (s, 3H), 2.36 (s, 3H); 13C-NMR (d6-DMSO): δ 164.9 (d, J = 22.1 Hz), 162.1, 145.5, 141.4, 138.2, 137.8, 137.5, 137.3, 134.9, 128.5, 128.0, 125.4, 124.9, 117.6, 117.2, 116.2, 114.2, 106.0, 87.2, 84.8, 80.0 (d, J = 164.0 Hz), 55.6, 20.7; 19F-NMR (CD3OD, 1H-decoupled): δ −211.8; LC-MS (ESI, [M–OTs]+): 475.0; HRMS (ESI, [M–OTs]+) calc’d for C20H13FIN2OS, 474.9777; found, 474.9765; HPLC: tR, 3.1 min; purity, > 99%.
Radiochemistry
For radiation safety to personnel, radiochemistry was performed in a lead-shielded hot-cell. Radioactivity was measured with a Atomlab 300 ionization calibrator (Biodex Medical Systems, Shirley, NY), and corrected for background and physical decay. Radiofluorinations were performed on either a NanoTek microfluidic apparatus [8] (Advion, Ithaca, NY) or in an in-house modified Synthia module, equipped with a microwave cavity.[11]
Production of [18F]Fluoride Ion Reagents
No-carrier-added (NCA) [18F]fluoride ion was obtained through the 18O(p,n)18F nuclear reaction by irradiating [18O]water (95 atom %) for 90–120 min with a proton beam (17 MeV; 20 μA) generated with a PETrace cyclotron (GE Medical Systems, Milwaukee, WI).
For reactions in the microfluidic apparatus, the [18F]fluoride ion (50–200 mCi) was adsorbed from the [18O]H2O (90–400 μL) onto anion resin (either MP-1 or QMA) held within a small cartridge, and then released with a solution of K2CO3 (1.24 mg, 9 μmol) plus K 2.2.2 (6.77 mg, 18 μmol) in MeCN-H2O (450 μL, 9:1 v/v) into a ∨-vial (2-mL, Alltech). The solution was dried by two cycles of MeCN (0.5 mL) addition and evaporation. The dry [18F]F−-K+-K 2.2.2 reagent was then dissolved in DMF (400 μL) and loaded into a storage loop (315 μL) in the apparatus.
For high activity radiofluorinations, such as those for radiotracer production, the [18F]fluoride ion was dried by microwave heating in a modified Synthia apparatus.[11] Thus, a solution of K2CO3 (0.28 mg, 2 μmol) plus K 2.2.2 (1.50 mg, 4 μmol) in MeCN-H2O (100 μL, 9:1 v/v) and MeCN (200–400 μL) was added to a ∨-vial (1-mL, Alltech) containing [18F]fluoride ion (160–300 mCi) in [18O]H2O (200–500 μL). The vial was then placed in the microwave cavity and heated with microwaves (90 W in 2 × 2 min pulses) under N2 gas flow (200 mL/min) to remove water-MeCN azeotrope. Three further cycles of MeCN (650 μL) addition and evaporation were then performed. The dried [18F]F−-K+-K 2.2.2 reagent was dissolved in a solvent of choice, then loaded into a storage loop of the apparatus.
HPLC methods for the separation and analysis of radiofluorination products
HPLC systems were equipped with radioactivity and UV absorbance detectors (λmax = 310 nm).
Method A
The radioactive product was separated on a Luna C18 column (5 μm, 10 × 250 mm) eluted at 4 mL/min with a mixture of H2O (A) and MeCN (B). Mobile phase composition started at 45% B for 2 min, increased linearly to 75% B over 20 min and finally was held at 75% B for 30 min.
Method B
The radioactive product was separated on a Luna C18 column (5 μm, 10 × 250 mm), eluted at 3.5 mL/min with a mixture of 25 mM aqueous HCOONH4 (A) and MeCN:25 mM HCOONH4 (3:1 v/v; B). Mobile phase composition started at 68% B for 2 min, increased linearly to 78% B over 12 min and finally was held at 78% B for 20 min.
Method C
This method was similar to Method B, except that mobile phase composition started at 68% B for 35 min, increased linearly to 100% B over 2 min, and was then held at 100% B for 10 min.
Method D
The radioactive product was analyzed on a Luna C18 column (10 μm, 4.6 × 250 mm), eluted at 1.75 mL/min with a mixture of H2O (A) and MeCN (B). Mobile phase composition started at 60% B and increased linearly to 90% B over 7 min.
Method E
The radioactive product was analyzed on a Luna C18 column (5 μm, 4.6 × 250 mm) eluted at 1.75 mL/min with a mobile phase of HCOONH4 (10 mM) in MeCN:H2O (60:40 v/v).
Radiofluorination reactions in a micro-reactor
Details of the microfluidic apparatus and its operation have been given in our earlier publication.[8] Essentially, the apparatus is composed of a 4-m long coiled silica capillary glass reactor with an internal volume of 31.4 μL which may be heated to any set temperature. The reactor may be fed with two separate reagents from their respective storage loops at set flow rates (10–15 μL/min) controlled by motorized syringes. All reactions resulted in some adsorption of radioactivity to PEEK, silica glass tubing or syringe surfaces of the apparatus. RCYs were measured with HPLC and were corrected for adsorbed radioactivity i.e., the computed RCY is based on the decay-corrected activity of labeled product as a percentage of radioactivity infused into the reactor. The amount of radioactivity lost through absorption was variable and averaged 20% (n =32).
With separate infusion of halo precursor and [18F]fluoride reagent
Solutions of bromo precursor 5e (10 mM) in DMF and [18F]F−-K+-K 2.2.2 reagent in DMF (~ 0.4 mCi/μL) were loaded into their respective storage loops (315-μL each). The precursor solution (15 μL) and [18F]F−-K+-K 2.2.2 reagent (10 μL) were simultaneously infused into the micro-reactor at set flow rates of 6 and 4 μL/min, respectively, while the micro-reactor was kept at a fixed temperature of 180 °C. Reaction (residence) time was 188 s. The reactor outputs were quenched with H2O–MeCN (1:1 v/v, 1 mL). An aliquot of the reaction mixture was analyzed with HPLC (Method D), revealing [18F]2c with the same retention time as reference 2c. Reactions were repeated using different infusion volume, flow rate and temperature.
With separate infusion of iodonium salt precursor and [18F]fluoride reagent
DMF solutions of diaryliodonium salt precursor (10 mM) and [18F]F−-K+-K 2.2.2 reagent (0.3 mCi/μL) were loaded into the respective storage loops (315 μL each). Precursor solution (15 μL) and [18F]F−-K+-K 2.2.2 reagent (10 μL) were simultaneously infused into the micro-reactor at 4.5 and 3 μL/min, respectively with the micro-reactor held at 160 °C. The residence time was 251 s. Reaction outputs were quenched with H2O–MeCN (1:1 v/v, 1 mL). An aliquot of the reaction mixture was analyzed with HPLC (Method D). The radioactive products were identified by comparison of their HPLC retention times with those of reference 2a–c, fluorobenzene and 4-fluoroanisole. Each product fraction was also collected for confirmation of its identity with LC-MS or LC-MS/MS analysis of its associated carrier. In some radiofluorination reactions with precursors 7c and 8c, TEMPO (10 μmol) was included in the precursor solution. Reactions were repeated using different infusion volume, flow rate and temperature.
With separate infusion of iodonium salt precursor and [18F]fluoride reagent on production activity scale
DMF solutions of 7c (14 mM) and [18F]F−-K+-K 2.2.2 reagent (0.4 mCi/μL) were separately loaded into their respective storage loops. These solutions (254 μL) were simultaneously infused into the micro-reactor at a flow rate of 10 μL/min with the micro-reactor held at 180 °C. The reaction mixture was diluted with H2O (3 mL) and injected on to HPLC (Method B) ([18F]2c, tR = 22.5 min).
With infusion of premixed reagents
A DMF solution of precursor 7c (10 mM) was added to dried [18F]F−-K+-K 2.2.2 reagent (0.48 mCi/μL) and the solution was loaded into one storage loop. A portion of this solution (195 μL) was infused at 10 μL/min into the micro-reactor held at 180 °C. The reaction outputs were quenched with H2O (3 mL) and the product [18F]2c separated with HPLC (Method B).
Microwave-assisted radiofluorination reactions in a Synthia apparatus
Radiofluorination of bromo precursor 5d to produce [18F]2a
A solution of the bromo precursor 5d (3.0 mg) in DMSO (750 μL) was added to a closed glass ∨-vial (1-mL internal volume) containing dried [18F]F−-K+-K 2.2.2 reagent (94 mCi) and heated at 150 °C (100 W in five pulses of 2 min). The temperature reached ~ 150 °C during microwave irradiation and decreased to ~ 50 °C between microwave pulses. The reaction mixture was diluted with H2O (0.7 mL) and [18F]2a separated with HPLC (Method A, tR = 25.0 min).
Radiofluorination of bromo precursor 5e to produce [18F]2c was carried out in similar manner. [18F]2c was separated with HPLC (Method A, tR = 27.0 min).
Radiofluorination of diaryliodonium salt precursors
A solution of diaryliodonium salt (3–4 mg, ~ 5 μmol) and TEMPO (10 μmol) in DMF (0.5 mL) was added to a closed ∨-vial containing dried [18F]F−-K+-K 2.2.2 reagent (25–160 mCi). This solution was irradiated with microwaves (100 W, 2 min and 80 W, 2 min). The reaction temperature reached ~ 105 °C during irradiation and decreased to ~ 50 °C between pulses. The reaction mixture was diluted with H2O (0.7 mL) and the radioactive product was separated by HPLC (Method C; tR = 25.7, 35.9 and 29.2 min, for [18F]2a–2c), respectively).
Radioactive products [18F]2a–c were analyzed with HPLC Method E (tR = 5.8, 7.5 and 5.3 min, respectively). The carrier associated with each product was also analyzed with LC-MS or LC-MS/MS to confirm product identity.
Determination of radioligand specific radioactivity
The specific radioactivities of [18F]2a–c were determined with HPLC. For example, a purified [18F]2a sample of known radioactivity (~ 0.1 mCi) was analyzed with HPLC method E. The mass of carrier 2a in the injectate was determined from a pre-calibrated mass response curve obtained under identical HPLC conditions. Specific activities (mCi/μmol) are reported as the radioactivity of [18F]2a in the injected sample (mCi), divided by the mass of carrier 2a (μmol), corrected for physical decay to the time of end of radionuclide production.
Conclusions
Although diaryliodonium salts of simple molecules have been widely examined as precursors in radiofluorination reactions, examples of the radiofluorination of diaryliodonium salts to produce useful PET radiotracers are relatively rare.[20] This lack of application may stem from limitations in the syntheses and applications of diaryliodonium salts of more complex molecules. Here we have shown that appropriate diaryliodonium salt precursors may be readily synthesized, stored and applied to produce 18F-labeled mGluR5 radioligands in RCYs and specific radioactivities that are useful for molecular imaging with PET. Our results further demonstrate the potential of diaryliodonium salts as precursors for previously poorly accessible 18F-labeled targets.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes of Health (NIMH). The authors are grateful to the NIH Clinical Center PET Department (Chief, Dr. Peter Herscovitch) for the cyclotron production of fluorine-18.
Footnotes
Electronic supplementary information (ESI) available: Copies of 1H, 13C and 19F NMR spectra of 5a–e, 6a–c, 7a–c and 8a–c; preparative and analytical HPLC chromatograms for [18F]2a–c.
References
- 1.(a) Lasne M-C, Perrio C, Rouden J, Barré L, Roeda D, Dollé F, Crouzel C. Top. Curr. Chem. 2002;222:201–258. [Google Scholar]; (b) Dollé F, Roeda D, Kuhnast B, Lasne M-C. In: Fluorine and Health. Tresaaud A, Haufe G, editors. 2008. pp. 3–65. Ch 1. [Google Scholar]; (c) Cai L, Lu S, Pike VW. Eur. J. Org. Chem. 2008;17:2853–2873. [Google Scholar]; (d) Pimlott SL, Sutherland A. Chem. Soc. Rev. 2011;40:149–162. doi: 10.1039/b922628c. [DOI] [PubMed] [Google Scholar]; (e) Li ZB, Conti PS. Adv. Drug Delivery Rev. 2010;62:1031–1051. doi: 10.1016/j.addr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ruth TJ, Wolf AP. Radiochim. Acta. 1979;26:21–24. [Google Scholar]; (b) Guillaume M, Luxen A, Nebeling B, Argentini M, Clark JC, Pike VW. Appl. Radiat. Isot. 1991;42:749–762. [Google Scholar]; (c) Qaim SM, Clark JC, Crouzel C, Guillaume M, Helmeke HJ, Nebeling B, Pike VW, Stöcklin G. In: Radiopharmaceuticals for Positron Emission Tomography. Stöcklin G, Pike VW, editors. Kluwer Academic Publishers; Dordrecht, Netherlands: 1993. pp. 1–43. [Google Scholar]
- 3.Siméon FG, Brown AK, Zoghbi SS, Patterson VM, Innis RB, Pike VW. J. Med. Chem. 2007;50:3256–3266. doi: 10.1021/jm0701268. [DOI] [PubMed] [Google Scholar]
- 4.(a) Brown AK, Kimura Y, Zoghbi SS, Siméon FG, Liow J-S, Kreisl WC, Taku A, Fujita M, Pike VW, Innis RB. J. Nucl. Med. 2008;49:2042–2048. doi: 10.2967/jnumed.108.056291. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kimura Y, Siméon FG, Hatazawa J, Mozley PD, Pike VW, Innis RB, Fujita M. Eur. J. Nucl. Med. Mol. Imaging. 2010;37:1943–1949. doi: 10.1007/s00259-010-1447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shetty HU, Zoghbi SS, Siméon FG, Liow J-S, Brown AK, Kannan P, Innis RB, Pike VW. J. Pharmacol. Exp. Therapeutics. 2008;327:727–735. doi: 10.1124/jpet.108.143347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamill TG, Krause S, Ryan C, Bonnefous C, Govek S, Seiders TJ, Cosford NDP, Roppe J, Kamenecka T, Patel S, Gibson RE, Sanabria S, Riffel K, Eng W, King C, Yang X, Green MD, O’Malley SS, Hargreaves R, Burns DH. Synapse. 2005;56:205–216. doi: 10.1002/syn.20147. [DOI] [PubMed] [Google Scholar]
- 7.(a) Pike VW, Aigbirhio FI. J. Chem. Soc., Chem. Commun. 1995:2215–2216. [Google Scholar]; (b) Shah A, Pike VW, Widdowson DA. J. Chem. Soc., Perkin Trans 1. 1998:2043–2046. [Google Scholar]; c) Ross TL, Ermert J, Hocke C, Coenen HH. J. Am. Chem. Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 8.(a) Chun J-H, Lu SY, Lee Y-S, Pike VW. J. Org. Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee Y-S, Hodošček M, Chun J-H, Pike VW. Chem. Eur. J. 2010;16:10418–10423. doi: 10.1002/chem.201000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chun J-H, Lu SY, Pike VW. Eur. J. Org. Chem. doi: 10.1002/ejoc.201100382. In press. DOI: 10.1002/ejoc.201100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Lu SY, Watts P, Chin FT, Hong J, Musachio JL, Briard E, Pike VW. Lab Chip. 2004;4:523–525. doi: 10.1039/b407938h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu SY, Giamis AM, Pike VW. Curr. Radiopharm. 2009;2:49–55. doi: 10.2174/1874471010902010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Bjurling P, Reineck R, Westerburg G, Gee AD, Sutcliffe J, Långström B. In: Link JM, Ruth TJ, editors. Proc. Sixth Workshop on Targetry and Target Chem.; TRIUMF, Vancouver. 1995.pp. 282–284. [Google Scholar]; (b) Lazarova N, Siméon FG, Musachio JL, Lu SY, Pike VW. J. Label. Compd. Radiopharm. 2007;50:463–465. [Google Scholar]
- 12.Pike VW, Butt F, Shah A, Widdowson DA. J. Chem. Soc., Perkin Trans. 1. 1999;3:245–248. [Google Scholar]
- 13.Azizian H, Eaborn C, Pidcock A. J. Organomet. Chem. 1981;215:49–58. [Google Scholar]
- 14.Katritzky AR, Gallos JK, Durst DH. Magn. Reson. Chem. 1989;27:815–822. doi: 10.1002/mrc.1260270902. [DOI] [PubMed] [Google Scholar]
- 15.(a) Wadsworth HJ, Widdowson DA, Wilson E, Carroll MA. 2005. WO 2005/061415 A1. [Google Scholar]; (b) Carroll MA, Nairne J, Smith G, Widdowson DA. J. Fluorine Chem. 2007;128:127–132. [Google Scholar]; (c) DiMagno S. 2010. WO 2010/048170 A2. [Google Scholar]
- 16.Irving H, Reid RW. J. Chem. Soc. 1960:2078–2081. [Google Scholar]
- 17.(a) Hacker NP, Leff DV, Dektar JL. J. Org. Chem. 1991;56:2280–2282. [Google Scholar]; (b) Yagci Y, Yilmaz F, Kiralp S, Toppare L. Macromol. Chem. Phys. 2005;206:1178–1182. [Google Scholar]; (c) He JH, Mendoza VS. J. Polym. Sci. Part A – Polym. Chem. 1996;34:2809–2816. [Google Scholar]; (d) He JH, Zhang JC. J. Polym. Sci. Part A – Polym. Chem. 1999;37:4521–4527. [Google Scholar]; (e) He JH, Zhang JC. J. Photochem. Photobiol. A Chem. 1997;107:249–252. [Google Scholar]; (f) Padon KS, Scranton AB. J. Polym. Sci. Part A – Polym. Chem. 2000;38:2057–2066. [Google Scholar]; (g) Merritt EA, Olofsson B. Angew. Chem. Int. Ed. 2009;48:9052–9070. doi: 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]
- 18.Cosford NDP, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA. J. Med. Chem. 2003;46:204–206. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 19.McKillop A, Kemp D. Tetrahedron. 1989;45:3299–3306. [Google Scholar]
- 20.(a) Zhang M-R, Kumata K, Suzuki K. Tetrahedron Lett. 2007;48:8632–8635. [Google Scholar]; (b) Lee BC, Dence CS, Zhou H, Parent EE, Welch MJ, Katzenellenbogen JA. Nucl. Med. Biol. 2009;36:147–153. doi: 10.1016/j.nucmedbio.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.