

Abstract
Parkinson's disease is a debilitating movement disorder characterized by a generalized dysfunction of the nervous system, with a particularly prominent decline in the nigrostriatal dopaminergic pathway. Although there is currently no cure, drugs targeting the dopaminergic system provide major symptomatic relief. As well, agents directed to other neurotransmitter systems are of therapeutic benefit. Such drugs may act by directly improving functional deficits in these other systems, or they may restore aberrant motor activity that arises as a result of a dopaminergic imbalance. Recent research attention has focused on a role for drugs targeting the nicotinic cholinergic systems. The rationale for such work stems from basic research findings that there is an extensive overlap in the organization and function of the nicotinic cholinergic and dopaminergic systems in the basal ganglia. In addition, nicotinic acetylcholine receptor (nAChR) drugs could have clinical potential for Parkinson's disease. Evidence for this proposition stems from studies with experimental animal models showing that nicotine protects against neurotoxin-induced nigrostriatal damage and improves motor complications associated with l-DOPA, the “gold standard” for Parkinson's disease treatment. Nicotine interacts with multiple central nervous system receptors to generate therapeutic responses but also produces side effects. It is important therefore to identify the nAChR subtypes most beneficial for treating Parkinson's disease. Here we review nAChRs with particular emphasis on the subtypes that contribute to basal ganglia function. Accumulating evidence suggests that drugs targeting α6β2* and α4β2* nAChR may prove useful in the management of Parkinson's disease.
I. Introduction—Parkinson's Disease and Links to the Nicotinic Cholinergic System
Parkinson's disease is the second most common neurodegenerative disorder after Alzheimer's disease, and affects 2% of people over the age of 60 (Mayeux, 2003). It is a neurodegenerative movement disorder characterized by postural instability, bradykinesia and a generally asymmetric onset of tremor and rigidity (Lang, 2009; Poewe, 2009; Quik et al., 2009; Schapira, 2009; Feng and Maguire-Zeiss, 2010; Obeso et al., 2010). These motor symptoms are a consequence of degeneration of the nigrostriatal dopaminergic pathway, which is the most severely affected neurotransmitter system in Parkinson's disease. In addition, accumulating evidence shows that there is a generalized neuronal loss in the central and peripheral nervous system in this disorder (Braak et al., 2002, 2003). Numerous CNS1 neurotransmitter systems degenerate, such as the adrenergic, cholinergic, serotonergic, glutamatergic, and GABAergic pathways, although to a lesser degree than the nigrostriatal dopaminergic pathway (Curzon, 1977; Haber, 1986; Dubois et al., 1990; Poewe, 2009). Damage to these other systems may contribute to the motor problems and also underlie the nonmotor symptoms associated with Parkinson's disease, including deficits in cognition/memory, affect, sleep/wakefulness, and autonomic function (Lang, 2009; Poewe, 2009; Quik et al., 2009; Schapira, 2009; Calabresi et al., 2010; Feng and Maguire-Zeiss, 2010; Obeso et al., 2010).
The etiology of Parkinson's disease is currently uncertain and has been attributed to a complex interplay between genetic and environmental factors (Schapira, 2009; Bekris et al., 2010; Obeso et al., 2010). A small minority of cases (∼5%) is genetic (familial), with Mendelian inheritance. Gene mutations linked to Parkinson's disease include PARK1 to PARK 18, which seem to be responsible for approximately 50% of familial cases and ∼2% of sporadic forms (Schapira, 2009; Obeso et al., 2010). Of these, the most well studied include PARK1/4, which involves point mutations and multiplications in the α-synuclein gene. Deletions or point mutations in the PARK2 gene, which encodes parkin, are linked to autosomal recessive juvenile-onset parkinsonism. Recessive mutations in PARK6 or PINK1 (which encodes a mitochondrial kinase) are responsible for a familial form of early-onset parkinsonism. Recessively inherited missense and exonic deletion mutations in PARK 7 or DJ1 have also been reported although these are very rare. The most common mutations in either familial or sporadic Parkinson's disease involve mutations in PARK8 or LRRK2 (encoding leucine-rich repeat kinase 2). The LRRK2 protein contains both Rab GTPase and kinase enzymatic activities, which have been implicated in multiple neuronal functions under physiological conditions. In addition to genetic mutations, environmental factors have also been linked to the occurrence of Parkinson's disease. The greatest positive risk factor is pesticide exposure, whereas tobacco use has consistently been linked to a decreased incidence of Parkinson's disease (Quik et al., 2009).
The most effective current treatment for Parkinson's disease motor symptoms is dopamine replacement therapy with l-DOPA and/or dopamine agonists. These drugs are particularly beneficial for improving motor deficits in Parkinson's disease; however, side effects commonly arise and drug effectiveness diminishes with disease progression (Lang, 2009; Poewe, 2009; Quik et al., 2009; Schapira, 2009; Feng and Maguire-Zeiss, 2010; Obeso et al., 2010). Moreover, the nonmotor symptoms linked to Parkinson's disease, such as dementia, sleep deficits, depression, and others, are not improved with these pharmacotherapies. There is therefore a critical need to develop improved treatments for Parkinson's disease, ideally to halt disease progression but also to provide better symptomatic relief of the motor and nonmotor symptoms. The focus of this review is on a potential role for the nicotinic cholinergic system, based on the following rationale: a considerable literature demonstrates an extensive anatomical and functional overlap between the nicotinic cholinergic and dopaminergic systems in the nigrostriatal pathway, which plays a pivotal role in Parkinson's disease. In addition, accumulating studies suggest that drugs that interact at nAChRs, such as nicotine, may protect against nigrostriatal damage. Moreover, nicotine and nAChR drugs alleviate some of the motor side effects associated with dopamine replacement therapy. Finally, the emerging procognitive and antidepressant effects of nAChR drugs may offer therapeutic benefit for the dementia and depressive symptoms observed in Parkinson's disease.
II. Inter-Relationship between Nicotinic Cholinergic and Dopaminergic Systems
A. Striatum
The subcortical region, referred to as the striatum because of its striated or striped appearance, consists of the caudate nucleus and putamen. In rodents, the caudate and the putamen are merged, but in primates, these structures are separated by the internal capsule. The striatum is divided into dorsal and ventral territories. The dorsal striatum primarily receives dopaminergic innervation from the substantia nigra (SN) pars compacta, with little contribution from the ventral tegmental area (VTA) (Björklund and Dunnett, 2007) (Fig. 1). It is this nigrostriatal pathway that selectively degenerates in Parkinson's disease (Fig. 1) (Obeso et al., 2000; Parent et al., 2000; Smith and Kieval, 2000). Dopaminergic innervation of the ventral striatum (also known as the nucleus accumbens core) is from the VTA, with some input from the dorsal SN. Likewise, primate dorsal striatum primarily receives projections from the SN. However, there is a more pronounced “intermingling” of the pathways from the SN and VTA in the ventral striatum or nucleus accumbens in primates (Björklund and Dunnett, 2007). In Parkinson's disease, the dorsal striatum is affected to the greatest degree, reflecting the degeneration of the nigrostriatal pathway, whereas the mesolimbic projection from the VTA is relatively spared (Obeso et al., 2000; Parent et al., 2000; Smith and Kieval, 2000). Unless specified, the term “striatum” will henceforth be used to denote the striatal areas compromised in Parkinson's disease.
Fig. 1.

Schematic representation of the nigrostriatal dopaminergic systems in the rat brain and its links to the pedunculopontine (PPT) nucleus. a, sagittal section, stained with cresyl violet, illustrating the nigrostriatal pathway (green) that projects from cell bodies in the SN pars compacta of the midbrain to the caudate-putamen (CPu; dorsal striatum) of the rat forebrain. Note its “striated” appearance. The ventral striatum, below the CPu, corresponds to the nucleus accumbens (NAc). The SN receives cholinergic (red) and glutamatergic (blue) inputs from the PPT in the brain stem. The CPu receives glutamatergic (blue) inputs from the somatosensory and association cortices. Cc, corpus callosum; Cer, cerebellum; MFB, medial forebrain bundle. b, transverse sections at the level of the dashed lines in a. Bregma coordinates indicate distance anterior (+) and posterior (−) to this landmark on the skull. [Reprinted from Rice ME, Avshalumov MV, and Patel JC (2007) Hydrogen peroxide as a diffusible messenger: evidence from voltammetric studies of dopamine release in brain slices, in Electrochemical Methods for Neuroscience (Michael AC and Borland LM eds) pp 205–232, CRC Press. Copyright © 2007 CRC Press. Used with permission.].
The striatum boasts some of the highest levels of dopamine and acetylcholine (ACh) in the brain (Fig. 2). Historically these two transmitters were viewed as having antagonistic roles (Calabresi et al., 2000; Cragg, 2006), reflecting the beneficial effects of muscarinic antagonists in Parkinson's disease (Langmead et al., 2008). The terminal fields of the dopaminergic afferents have extensive arborizations (Wilson and Groves, 1980). Thus, each dopaminergic afferent contacts a large area of the striatum to exert a coordinated influence (Fig. 2). The principal target is the GABAergic medium spiny projection neurons, which constitute more than 90% of the neuronal population in the striatum. These neurons form the direct and indirect output pathways to the basal ganglia whereby motor function is moderated (Bolam et al., 2000; Obeso et al., 2000; Parent et al., 2000; Smith and Kieval, 2000). In the direct pathway, information from the striatum is transmitted directly to the output structures of the basal ganglia. These include the SN pars reticulata and the entopeduncular nucleus in rodents (the latter corresponds to the internal segment of globus pallidus in primates), and thence to the brainstem (for the control of head, neck, and eye movements involved in gaze and focus) or the thalamus and motor cortex, respectively (Fig. 3). The indirect pathway proceeds via the globus pallidus (the external segment of globus pallidus in primates) and subthalamic nucleus before reaching the SN pars reticulata and the entopeduncular nucleus. The dopaminergic input to the striatum from the SN represents a substantial feedback component of this circuitry (Bolam et al., 2000).
Fig. 2.
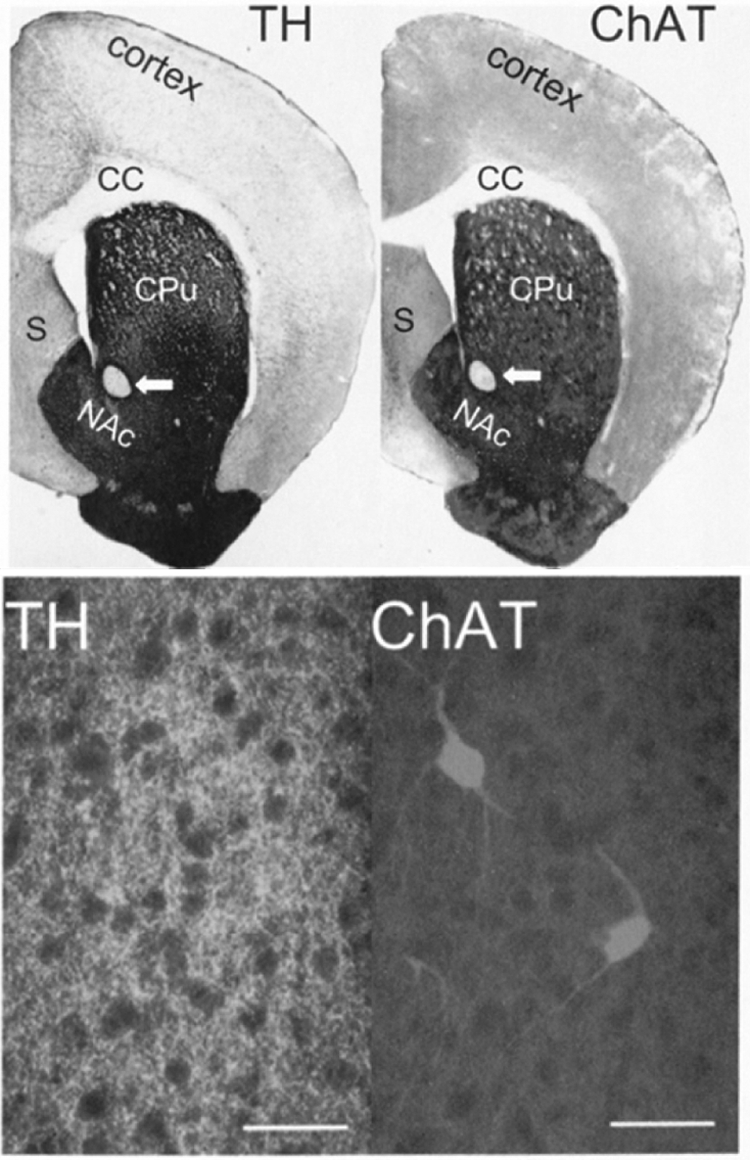
Dense and overlapping distribution of ACh and dopamine in the rat striatum. Top, bright-field photomicrographs show tyrosine hydroxylase (TH) and choline acetyltransferase (ChAT) antibody staining of forebrain sections. Arrows, anterior commissure; CC, corpus callosum; CPu, caudate putamen; NAc, nucleus accumbens; S, septum. Bottom, higher magnification immunofluorescence images of striatum double labeled for TH (left) and ChAT (right), revealing sparse cholinergic interneurons and dense fiber tracts for both transmitters. Scale bars, 50 μm. [Reproduced from Zhou FM, Liang Y, and Dani JA (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 4:1224–1229. Copyright © 2001 Nature Publishing Group. Used with permission.].
Fig. 3.

Schematic of the nigrostriatal pathway in relation to the basic circuitry of the basal ganglia. Dopaminergic neurons of the SN pars compacta and corticostriatal glutamatergic neurons converge on the medium spiny neurons of the striatum. These are the principal output neurons of the “direct” (D) or “indirect” (I) pathways. The direct pathway (heavy shaded lines) projects directly to the entopeduncular nucleus (EPN; internal segment of the globus pallidus in primates) or the SN pars reticulata (SNr), and thence to the thalamus or brain stem, respectively. The indirect pathway (heavy dashed shaded lines) makes synaptic connections in the globus pallidus (GP; external segment of the globus pallidus in primates) and subthalamic nucleus (STN) en route to the EPN and SNr. Some additional connections are shown as dotted lines. See Bolam et al. (2000) for details.
The dopamine axons form symmetric synapses onto the shafts of the dendritic spines of medium spiny neurons. Here they are well placed to modulate the incoming activity from corticostriatal glutamatergic afferents that form asymmetric synapses onto the heads of the spines (Smith and Bolam, 1990) (see Fig. 4a). There is reciprocal modulation of glutamate and dopamine inputs at the presynaptic level, by transmitter spillover from the synapse acting via dopamine D2 and metabotropic glutamate receptors, respectively (Wang and Pickel, 2002; Zhang and Sulzer, 2003). Indirect glutamatergic influence mediated by diffusible messengers has also been proposed (Avshalumov et al., 2008). It is at this presynaptic level that nicotine exerts its major influence in the striatum.
Fig. 4.

Cellular localization of nAChR subtypes in striatum (a) and SN (b). a, in the striatum, the nigrostriatal and corticostriatal afferents converge on the shafts and heads, respectively, of the spines of medium spiny projection neurons. The nigrostriatal dopaminergic terminals (DA) bear a variety of α4β2* and α6β2* nAChR subtypes. α7 nAChRs are proposed to reside on the glutamatergic terminals (Glu). Other neuronal elements in the striatum, the GABAergic and cholinergic interneurons and serotonergic afferents from the raphe nucleus, are also indicated. GABAergic terminals express α4β2* nAChRs: these may arise from interneurons, as illustrated, or from axon collaterals, medium spiny neurons, or globus pallidus neurons (not shown). A fast-spiking subpopulation of GABAergic interneurons may express an unidentified subtype of nAChR, possibly α7 (gray receptor). Evidence for the presence of nAChRs on cholinergic interneurons and serotonergic afferents is inconclusive (gray receptor). The subunit composition of nAChR subtypes expressed in the striatum is illustrated in the right panel. Two agonist-binding sites are indicated at the interface between α and β2 subunits in heteromeric nAChRs, whereas α7 nAChRs have five putative binding sites. b, in the SN, the dopaminergic neurons (DA) that project to the striatum are modulated by glutamatergic, cholinergic, and GABAergic afferents, as well as GABAergic interneurons. The dopamine neurons bear α4β2* and α6β2* nAChR subtypes that may be distinct from those expressed on the striatal terminals. Proposed subunit combinations are illustrated on the right. α7 nAChRs are also present on a proportion of these cell bodies, in contrast to the striatal dopaminergic terminals. GABAergic interneurons express heteromeric nAChRs; α4β2* nAChRs may also be present on GABAergic afferents, for example from the substantia nigra pars reticulata. In contrast to the VTA, glutamatergic afferents may bear both α7 and non-α7 nAChRs, but cholinergic afferents are apparently devoid of nAChRs.
ACh in the striatum is derived from a population of giant, aspiny cholinergic interneurons, whose diameter can reach 40 μm (Fig. 2). Although they represent less than 2% of the total neuronal population of the striatum, these large cells have an extensive network of processes, enabling them to affect activity throughout the striatum (Wilson and Groves, 1980; Calabresi et al., 2000). It has been estimated that there are only 40,000 cholinergic interneurons in each rat striatum, but each of these neurons forms half a million varicosities within a territory of up to 1 mm in diameter (Zhou et al., 2002; Tepper and Bolam, 2004). Thus throughout the striatum there is extensive overlap with the dopaminergic arborization that facilitates their cross-talk (Fig. 2). There is evidence for cholinergic synapses on distal dendrites and dendritic spines within the striatum, but ACh is also released nonsynaptically from varicosities to exert effects by volume transmission (Descarries et al., 1997). The cholinergic interneurons are tonically active, firing action potentials at a regular, slow rate (3–10/s) that results in a continuous pulsatile release of ACh under basal conditions (Wilson et al., 1990). The high levels of acetylcholinesterase present in the striatum facilitate the rapid hydrolysis of ACh, enabling extracellular ACh to reflect its pulsatile release, thus minimizing receptor desensitization (Zhou et al., 2001). This contrasts with the concentration profile achieved by exogenous drugs such as nicotine.
The dopamine afferents also exhibit a tonic activity at rest (the regular firing of single action potentials that constitutes a rhythmic pacemaker function). This results in a continuous “drip feed” of dopamine that maintains a tonic concentration of 10 to 20 nM in the striatum (Goto et al., 2007). Thus, spillover of dopamine and ACh released under resting conditions facilitates their interaction. Activation of midbrain dopamine neurons switches their activity to a “bursting” firing pattern, a phasic pattern of bursts of action potentials (Grace et al., 2007). The consequence is substantially greater dopamine release in the striatum, transiently achieving high micromolar or even millimolar levels (Goto et al., 2007). Burst firing of dopamine neurons is accompanied by silencing of the cholinergic interneurons, so ACh release ceases when dopamine release increases. This coordinated reciprocal response, which relies on both nigrostriatal and thalamic inputs to the striatum, emphasizes the complex inter-relationship between dopamine and cholinergic systems in this region (Zhou et al., 2002; Cragg, 2006). The dopamine terminals have a rich array of nAChR subtypes, whereas there is no consistent evidence for nicotinic autoreceptors on the cholinergic interneurons (see section III.A.3). The striatum also receives serotonergic afferents from the raphe nucleus that have elicited recent interest with respect to their role in motor control in the normal brain and in Parkinson's disease (Di Matteo et al., 2008).
Although outside the scope of this review, it is important to recognize that ACh has another target receptor, the muscarinic receptor, that greatly outnumbers nAChRs (Conn et al., 2009). The G-protein-coupled muscarinic receptor exerts a modulatory influence via facilitatory M1-type receptors and inhibitory M2-type receptors, located both presynaptically on corticostriatal and nigrostriatal afferents and somatodendritically on the medium spiny neurons. Inhibitory muscarinic autoreceptors are also present on the cholinergic interneurons to regulate ACh release (Calabresi et al., 2000). In Parkinson's disease, muscarinic antagonists were one of the first treatments and are still sometimes used in a secondary role (Langmead et al., 2008; Conn et al., 2009). Their efficacy is attributed to a reduction in 1) the overactivity of the cholinergic interneurons and 2) the hyperactivity of corticostriatal glutamate neurotransmission that ensues after nigrostriatal denervation. Although they provide some benefit, these drugs are not without side effects, including cognitive impairment. Hence, there is a need for new and improved therapeutics. Any nicotinic agonist therapy will differ from the actions of ACh in its selectivity for nAChRs as well as in its extended pharmacokinetics compared with the highly regulated release of ACh.
B. Substantia Nigra
The dopaminergic neurons projecting to the dorsal striatum are primarily responsible for modulating motor functions and also cognitive aspects of motor learning (Kreitzer and Malenka, 2008). These arise from the A9 group of midbrain dopaminergic neurons, corresponding to the SN pars compacta (Fig. 1). The presence of neuromelanin in these cells gives them their eponymous dark coloration. The number of dopaminergic neurons in each SN pars compacta is estimated at ∼7000 per side in the mouse, ∼12,000 in the rat, up to 100,000 in monkeys, and more than 200,000 in young humans (Björklund and Dunnett, 2007). In addition to the predominant striatal innervation, some SN neurons innervate cortical and limbic areas. Dopamine is also released locally within the SN and VTA; this somatodendritic release can be modulated by nAChRs (Cheramy et al., 1981; Rahman et al., 2004a). The dendritic arborization extends into the underlying SN pars reticulata, populated with GABAergic neurons. Thus, dendritically released dopamine can influence the activity of dopamine projection neurons themselves via somatodendritic autoreceptors and GABAergic activity within the SN pars reticulata, which forms the output pathway of the direct and indirect circuits (Fig. 3) (Robertson, 1992; Zhou et al., 2009).
The SN pars compacta contains a population of GABAergic interneurons, as well as GABAergic afferents from the striatum, globus pallidus, and SN pars reticulata, that act as a brake on dopaminergic cell activation. Both the GABAergic and dopamine neurons bear nAChRs (Misgeld, 2004) (see section III.A.2; Fig. 4b). As already noted, midbrain dopamine neurons exhibit two distinct firing patterns, tonic single-spike activity and burst spike firing (Goto et al., 2007). The switch to burst firing is dependent on glutamatergic innervation, which comes from the subthalamic nucleus and the pedunculopontine nucleus (PPT) (Lee and Tepper, 2009). These glutamatergic nerve terminals may also be endowed with nAChRs (Keath et al., 2007).
C. Pedunculopontine Nucleus
The cholinergic input that provides the ACh to interact with nAChRs in the SN pars compacta comes from the PPT situated in the pons (Figs. 1 and 3). Analogous cholinergic innervation of the VTA is from the related laterodorsal tegmental nucleus. The PPT provides both cholinergic and glutamatergic inputs that synapse onto nigral dopamine neurons (Mena-Segovia et al., 2008) (Figs. 3 and 4B). The convergence of these inputs has raised the possibility that both neurotransmitters may be coreleased from the same terminals, with some evidence that this does occur in the squirrel monkey but not in the rat (Lavoie and Parent, 1994; Wang and Morales, 2009). Stimulation of the PPT elicits burst firing in SN pars compacta dopamine neurons (Lokwan et al., 1999; Floresco et al., 2003) and evokes dopamine release in the striatum, which is inhibited by application of either nicotinic or glutamatergic receptor antagonists into the SN pars compacta (Futami et al., 1995; Forster and Blaha, 2003). Indeed, it has been suggested that cholinergic activation is critical for promoting burst firing (Kitai et al., 1999). This idea resonates with a proposal for the VTA (based on studies with knockout mice lacking particular nAChR subunits) that cholinergic nicotinic activation of dopamine cell bodies serves as a gate that facilitates the switch to burst firing, enabling the dopamine neurons to respond to glutamatergic signals (Maskos, 2008, 2010).
III. Neuronal Nicotinic Acetylcholine Receptors
A. Structure and Heterogeneity
nAChRs are pentameric ligand-gated cation channels, permeable to Na+, K+, and, to varying degrees, Ca2+. The five membrane-spanning subunits create a central pore or channel that is opened in response to binding ACh or exogenous agonist. Muscle-type nAChRs, found in skeletal muscle and in Torpedo spp. electric tissues, have provided detailed insights into the structure and function of the receptor (Unwin, 2003). However, a distinct set of genes coding for neuronal nAChR subunits is expressed in neurons and some non-neuronal cells, including glia (Albuquerque et al., 2009; Millar and Gotti, 2009). To date, nine neuronal nAChR subunit genes have been shown to be expressed in various mammalian CNS neurons (α2–α7; β2–β4). The most abundant and widespread of these are β2, α4, and α7. Others have a more restricted distribution; for example, expression of α6 and β3 subunits is largely limited to catecholaminergic neurons. In addition, α8 has been found only in avian systems, and α9 and α10 are limited to cochlear hair cells, sensory neurons, and some non-neuronal cells. In theory, these subunits could combine to give a huge array of nAChR subtypes. However, the diversity of native nAChR subtypes is more limited, and assembly into viable nAChRs seems to be constrained by a set of presently poorly understood rules.
The simplest subunit combination is a pentamer of identical subunits (Albuquerque et al., 2009; Millar and Gotti, 2009). Of the subunits expressed in the mammalian brain, only the α7 subunit is able to form homomeric nAChRs in expression systems. Most, if not all, native α7 nAChRs are also homomers (Séguéla et al., 1993). In contrast, α2-α6 subunits are incapable of forming homomeric receptors and require β subunits (and additional α subunits in the case of α5) for formation of functional nAChRs (Albuquerque et al., 2009; Millar and Gotti, 2009). The agonist binding site occurs primarily on α subunits (“principal binding site”) but is formed at the interface with the adjacent β subunit that also contributes complementary binding residues (“complementary binding site”) (Corringer et al., 2000; Celie et al., 2004). For heteromeric neuronal nAChRs, α2, α3, α4, and α6 subunits can pair with β2 or β4 subunits to create an agonist binding site (see Fig. 4). As demonstrated for muscle-type nAChR, it is assumed that two binding sites per nAChR must be occupied for effective opening of the ion channel. In contrast to β2 and β4, the β3 subunit does not contribute to binding sites but is regarded as an “accessory subunit” that occupies the fifth position in the nAChR, analogous to the β1 subunit in muscle nAChR. The α5 subunit also functions exclusively as an accessory subunit, in that it lacks key residues critical for agonist binding (Kuryatov et al., 2008). Other α and β subunits can occupy the fifth position and form αβ agonist binding pairs (Fig. 4). Subunit composition determines nAChR properties, including channel open time, ion permeability and selectivity, and rate of desensitization, in addition to agonist sensitivity (Albuquerque et al., 2009; Millar and Gotti, 2009).
The structural diversity of nAChRs is paralleled by the diversity of their localization (at both cellular and tissue levels). Although this review is focused on nAChRs within the basal ganglia, it is important to emphasize that nAChR subtypes occur throughout the central and peripheral nervous systems, as well as on some non-neuronal cells. Thus nAChRs influence many physiological mechanisms, including pain, inflammation, cognition, and others (Bacher et al., 2009; Buckingham et al., 2009; McIntosh et al., 2009; Poorthuis et al., 2009; Sarter et al., 2009; Changeux, 2010a; Mineur and Picciotto, 2010; Philip et al., 2010). As a consequence, systemic nicotine and other agonists have many and varied biological effects, beyond the modulation of motor control.
1. Pharmacological Tools to Study Nicotinic Acetylcholine Receptor Subtypes in the Nigrostriatal System
A host of experimental approaches has been used to identify and study the different nAChR subtypes described in the previous section, including mRNA work, immunoprecipitation with selective nAChR subunit-directed antibodies, and the use of genetically engineered mice. In addition, pharmacological tools have assisted in identification and in characterization of distribution and function. Some of the more common drugs used as investigational tools are described below.
A variety of different agonists and antagonists have been used to study CNS α4β2* nAChRs (* signifies the possible presence of other subunits in the nAChR complex). However, emerging studies suggest that many of these compounds also act at other nAChR subtypes, particularly α6β2* nAChRs. Thus, mecamylamine and dihydro-β-erythroidine, two antagonists frequently used to investigate α4β2* nAChR-mediated function, also block α6β2* nAChRs (Exley et al., 2008; Meyer et al., 2008; Perez et al., 2008). With respect to α4β2* nAChR-directed agonists, 3-[(2S)-2-azetidinylmethoxy]-5-iodopyridine dihydrochloride (5-iodo-A-85380) was initially reported to selectively bind to α4β2*, with much lower affinity for α3β4* and α7 nAChRs (Mukhin et al., 2000). However, it was subsequently shown to act with similar potency at both α4β2* and α6β2* nAChRs (Kulak et al., 2002b). Varenicline, a partial α4β2* nAChR agonist, also interacts with α3β4*, α6β2*, and α7 nAChRs (Coe et al., 2005; Gonzales et al., 2006; Jorenby et al., 2006; Rollema et al., 2007a,b; M. Quik, unpublished observations). Sazetidine-A, another agent initially reported as selective for α4β2* receptors, binds with high affinity to α6β2* nAChRs and stimulates both α4β2* and α6β2* nAChR-mediated dopamine release (Xiao et al., 2006; Cucchiaro et al., 2008; Zwart et al., 2008; M. Quik, unpublished observations). In addition, the agonist ABT-089 (pozanicline) (Sullivan et al., 1997) has activity at both α4β2*and α6β2* receptors (Marks et al., 2009). Numerous other drugs in the literature have also been reported to interact with α4β2* nAChRs, including undesignated compounds from Abbott Laboratories (Abbott Park, IL), Targacept, Inc. (Winston-Salem, NC), SIBIA Neurosciences (La Jolla, CA), and University of Bath (Bath, UK). However, at this point, their selectivity is uncertain because their interaction with α6β2* nAChRs and/or other nAChR subtypes is not known (Bencherif et al., 1996; Cosford et al., 1996; Donnelly-Roberts et al., 1996, 1998; Bencherif et al., 2000; Sharples et al., 2000; Buccafusco et al., 2005; Gotti et al., 2006b; Lippiello et al., 2006; Wang et al., 2006; Dwoskin and Bardo, 2009). All together, these observations indicate that the majority of available α4β2* nAChR drugs may interact with other nAChR subtypes, notably α6β2* nAChRs; at best these agents should be regarded as only β2-selective. However, general β2-selective agonists may have therapeutic advantages, as discussed in section VIII.B (Huang et al., 2011).
A toxin that has proved invaluable in elucidating the nature and function of α6β2* nAChRs is α-conotoxinMII (McIntosh et al., 2004; Quik and McIntosh, 2006). This 16-amino acid peptide, originally isolated from the venom of the marine snail Conus magus, selectively interacts at α3β2* and α6β2* nAChRs (Cartier et al., 1996; Champtiaux et al., 2002). Because there is little evidence for the existence of α3β2* nAChRs in mouse brain (Whiteaker et al., 2002), α-conotoxinMII provides information specifically concerning α6β2* nAChR in the rodent nigrostriatal pathway. By contrast, α6β2* and a small population of α3β2* nAChRs are present in monkey striatum; thus, α-conotoxinMII would interact at both subtypes (Quik et al., 2005). Another toxin, α-conotoxinPIA from Conus purpurascens, can discriminate between α6* and α3* nAChRs (Dowell et al., 2003; Gotti et al., 2010) but it is not readily accessible. No other toxins, drugs, or agents presently exist that selectively interact with α3β2* and/or α6β2* nAChRs. Thus, discrimination of native nAChRs that have minor differences in subunit composition is currently not possible.
Because selective agonists for α7 nAChRs were not available until more recently, antagonists have been particularly important for the study of these receptors. A key antagonist used to study α7 nAChRs is α-bungarotoxin. This toxin, isolated from the venom of Bungarus multicinctus, binds to α1*, α7, and α9/10 nAChRs (Albuquerque et al., 2009). Because α1* receptors are present only in skeletal muscle, and α9/10 receptors are not found in the brain, α-bungarotoxin has proved to be a very selective tool for α7 nAChRs in the CNS. It is used primarily for in vitro studies because it does not cross the blood-brain barrier, although it can be injected intracerebrally. Methyllycaconitine is a small-molecular-weight α7 nAChR antagonist that readily enters the brain when given systemically (Macallan et al., 1988; Ward et al., 1990). However, although selective for α7 nAChRs at low nanomolar concentrations, it interacts with other nAChR subtypes, notably α6β2* nAChRs, at higher concentrations (Mogg et al., 2002). Small-molecular-weight α7 nAChR agonists include 3-[(3E)-3-[(2,4-dimethoxyphenyl)methylidene]-5,6-dihydro-4H-pyridin-2-yl]pyridine (GTS-21), (4-bromophenyl)-1,4-diazabicyclo[3.2.2]nonane-4-carboxylate (SSR180711), N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-4-chlorobenzamide (PNU-282987), and (2S)-2′H-spiro[4-azabicyclo[2.2.2]octane-2,5′-[1,3]oxazolidin]-2′-one (AR-R17779) (Levin et al., 1999; Simosky et al., 2002; Martin et al., 2004; Hansen et al., 2007; Söderman et al., 2011). Choline, the breakdown product of ACh, is also found to selectively activate α7 nAChRs at millimolar concentrations (Alkondon et al., 1997). Ambient levels of choline, especially in areas such as striatum, where there are tonically active cholinergic neurons, might be sufficient to maintain α7 nAChRs in a desensitized state. The recent generation of allosteric potentiators, some specific for α7 nAChRs, are valuable additions to the toolbox for enhancing or revealing the contribution of α7 nAChRs (Bertrand and Gopalakrishnan, 2007).
2. Nicotinic Acetylcholine Receptor Subtypes in the Substantia Nigra
Determining the cellular expression of nAChR subunits and the subcellular localization of defined nAChR subtypes poses a major challenge. However, this has proved somewhat easier to address for the nigrostriatal dopamine pathway than for other brain systems, because dopamine afferents are highly localized to this ascending pathway, which can be selectively lesioned by dopaminergic neurotoxins. As well, the striatum and SN are relatively homogeneous with respect to their neurochemical makeup and neuronal composition.
In rodents, mRNA for five nAChR subunits, (α4, α5, α6, β2, β3) is expressed at high levels in the SN pars compacta, with lower levels of α7 mRNA (Wada et al., 1989; Marks et al., 1992; Cui et al., 2003). The dopamine neurons and GABAergic interneurons that populate the SN pars compacta can be distinguished by expression of tyrosine hydroxylase or glutamate decarboxylase, respectively, and by their characteristic electrophysiological properties, such as intrinsic membrane potential and firing properties (Grace and Bunney, 1983a,b,c; Lacey et al., 1989). Single-cell reverse transcription-polymerase chain reaction or double-labeling in situ hybridization in combination with these indices of neuronal identity showed that most dopamine neurons express mRNA corresponding to α4, α5, α6, β2, and β3. At a lower level and in a lower proportion of cells, α3 nAChR mRNA was also detected (Klink et al., 2001; Azam et al., 2002). However, α3 subunit expression decreased during development in the rat; this decrease coincided with an increase in α6 subunit expression (Azam et al., 2007). In contrast, SN GABA neurons displayed a simpler expression pattern largely restricted to α4, β2, and α3. In addition, approximately 40% of both dopamine and GABA neurons of the SN expressed mRNA for α7 nAChRs (Klink et al., 2001). β4 mRNA expression was generally low, but higher in nondopaminergic neurons (Klink et al., 2001; Azam et al., 2002). Overall, the pattern of nAChR transcript expression in nonhuman primates resembles that in rodents, except for the α2 mRNA, which is absent in the rodent but present in the primate SN (Han et al., 2000; Quik et al., 2000a,b).
The next challenges were to determine 1) the subunit combinations forming native, functional nAChRs in these neurons and 2) their subcellular disposition (Fig. 4b). Klink et al. (2001) used pharmacological tools and transgenic mice lacking α4 or α7 subunits to analyze nicotinic currents elicited from cell bodies of SN dopamine neurons. They interpreted their data in favor of two predominant somatodendritic subtypes: α4α6(β2)2α5 (sensitive to blockade by α-conotoxinMII) and (α4)2(β2)2α5 (insensitive to α-conotoxinMII). The former subtype is distinct from any of those identified in striatum (see below), although more recent immunoprecipitation studies found no evidence for the association of α5 with α6 subunits in midbrain; thus, α4α6(β2)2β3 may be a more likely subunit composition (Gotti et al., 2010). However, Gotti et al. (2010) noted the different complement of nAChR subunit combinations identified in midbrain compared with striatum, suggesting the occurrence of some different nAChR subtypes in somatodendritic and terminal compartments. In addition, typical α7 nAChR-mediated currents could be elicited by choline in the lower proportion of dopaminergic neurons (Klink et al., 2001; Keath et al., 2007).
There is little functional evidence that addresses the subunit composition of nAChR on the sparse GABA neurons in the SN pars compacta, although both α7 and non-α7 nAChRs have been localized to the soma and proximal dendrites of GABAergic neurons of the SN pars reticulata (Poisik et al., 2008). In the VTA, the absence of functional responses (other than α7 nAChR responses) in GABAergic neurons in α4 or β2 knockout mice led to the proposition that non-α7 nAChRs are α4β2 and possibly α3α4β2 subtypes (Klink et al., 2001). However, in SN GABA neurons, nAChR subunit expression is more diverse (Klink et al., 2001), and nAChRs on GABA neurons exhibit a different pharmacological profile compared with VTA (Keath et al., 2007), leaving some uncertainty about the exact subunit composition and stoichiometry of the nAChRs present. The conclusion that GABA interneurons in the SN pars compacta, as well as the projection neurons of the SN pars reticulata, are endowed with heteromeric nAChRs that have distinct properties compared with those expressed on dopamine cell bodies is consistent with functional studies described in section III.B.1.
In summary, a large portfolio of nAChR subunits is expressed in the SN pars compacta, which contains the dopaminergic cell bodies. Multiple complex subtypes of nAChR seem to exist in these dopamine neurons, whereas GABA interneurons exhibit a somewhat simpler expression pattern (Fig. 4b). The presence of distinct nAChR subtypes on dopaminergic and GABAergic neurons in the SN has functional implications, in that it may allow nAChR drugs with differing pharmacodynamic profiles to interact preferentially with one or other of these cell types.
3. Nicotinic Acetylcholine Receptor Subtypes in the Striatum
Identification of nAChR subtypes present on dopamine axon terminals projecting to the striatum could not be undertaken at the single cell level. Instead, neurochemical assays of nAChR function have commonly been employed (Grady et al., 2007). These studies have also relied on subtype-selective pharmacological tools and knockout mice, complemented by immunoprecipitation assays using subtype-specific antibodies to pull down nAChR complexes labeled with a radioligand, typically [3H]epibatidine (Gotti et al., 2007). Selective lesioning of the nigrostriatal pathway using 6-hydroxydopamine or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to eliminate dopamine terminals has been used to distinguish this population (Zoli et al., 2002; Quik et al., 2007b). As a whole, this substantial body of work has generated a comprehensive and coherent picture of nAChR subtypes expressed on dopamine terminals, and the heterogeneity is surprising.
First, α3 and α7 subunits do not contribute to presynaptic nAChRs on dopamine terminals in the rodent striatum, despite their expression by some SN pars compacta neurons and their contribution to the population of nAChRs on SN cell bodies (Whiteaker et al., 2002). Conversely, β3 has been credited with contributing only to presynaptic α6* nAChRs (Zoli et al., 2002; Cui et al., 2003; Salminen et al., 2004) and may be important for targeting or trafficking nAChRs that include it (Tumkosit et al., 2006). Immunoprecipitation studies found modest amounts of α4α6β2β3 nAChRs in SN, as discussed above (Gotti et al., 2010); it is not known whether these represent functional somatodendritic receptors or nAChRs destined to be trafficked to the striatum. Absence of the β3 subunit in null mutant mice reduces but does not eliminate α6* nAChRs (Gotti et al., 2005). Although this observation suggests that β3 is not an obligatory partner of α6, the two subunits have been seen to decrease in parallel after lesions of the nigrostriatal pathway, which would suggest that they are tightly correlated (Zoli et al., 2002; Quik et al., 2003). A parsimonious explanation is that β3 normally combines with α6 for the most efficient assembly, stabilization, and/or trafficking of α6β3* nAChRs, and in its absence only low levels of inefficiently processed α6* nAChRs are maintained. The asterisk signifies the possible presence of other subunits in the nAChR complex (see Fig. 4a).
The subunits that are present in presynaptic nAChRs on dopaminergic terminals in rodent striatum include α4, α5, α6, β2, and β3. These comprise 5 subtypes that have been subdivided into two main categories based on their interaction with the snail toxin α-conotoxinMII (McIntosh et al., 1999; Whiteaker et al., 2000) (Fig. 4a). These include those containing the α6 subunit (α4α6β2β3, α6β2β3, α6β2), which are termed α-conotoxinMII-sensitive because they bind α-conotoxinMII. The other class (α4β2, α4α5β2 subtypes) does not contain α6, does not bind α-conotoxinMII, and is designated α-conotoxinMII-insensitive (Zoli et al., 2002; Salminen et al., 2004). Functional measurements have suggested a greater proportion of α6β2* nAChRs in the nucleus accumbens compared with striatum (caudate putamen) (Exley and Cragg, 2008).
nAChR subtype heterogeneity could be further increased by α4β2* and α6β2* nAChRs existing in two stoichiometric forms, as has been shown for heterologously expressed α4β2* nAChRs (Nelson et al., 2003; Moroni et al., 2006) (Fig. 4a). These alternate forms have distinct agonist sensitivities depending on whether α4 or β2 occupies the fifth, accessory position; that is, (α4)2(β2)3 displays higher affinity for ACh and nicotine than (α4)3(β2)2. With regard to the regulation of dopamine release from striatal terminals, pharmacological evidence in favor of the high-sensitivity (α4)2(β2)3 form (Anderson et al., 2009) or both high- and low-sensitivity forms (Grady et al., 2010a) has been presented. The interpretation of data for native subtypes is complicated by the high affinity shown by (α4)2(β2)2α5 nAChRs that are also present on dopamine terminals (Grady et al., 2010a). The occurrence and functional significance of alternative stoichiometries of α6β2 nAChRs has not yet been elucidated.
The proportion of α-conotoxinMII-sensitive to insensitive nAChR subtypes is ∼30:70 in rodents (Cartier et al., 1996; Kulak et al., 1997; Grady et al., 2007) but this ratio is ∼50:50 in monkey striatum (Kulak et al., 2002). This might reflect the additional presence of α-conotoxinMII-sensitive α3β2* nAChR on nigrostriatal terminals in monkey brain (Quik et al., 2005). After nigrostriatal lesions with either MPTP or 6-hydroxydopamine, α6 and β3 subunit-containing nAChRs decline in parallel with the loss of dopaminergic markers such as the dopamine transporter (Zoli et al., 2002; Champtiaux et al., 2003; Cui et al., 2003; Quik et al., 2003, 2005). By contrast, nAChRs composed of α4 and β2 (but not α6) subunits, which can be detected by their ability to bind agonist with high affinity in the presence of α-conotoxinMII, are much less affected by lesioning in both rodents and primates, such that up to 70% of α4β2* nAChRs may be spared (Quik et al., 2001; Kulak et al., 2002a; Zoli et al., 2002; Champtiaux et al., 2003; Quik et al., 2003). An explanation of this anomaly is that expression of the α6 and β3 subunits is restricted to catecholaminergic neurons, whereas α4β2* nAChRs also occur on nondopaminergic components of the striatum. However, the high level of residual α4β2* nAChRs is surprising. The paucity of in situ hybridization signals for expression of nAChR subunits in striatum or caudate putamen is striking (Wada et al., 1989; Marks et al., 1992; Le Novère et al., 1996) and reinforces the notion that the majority of nAChRs in this region is located presynaptically on afferents rather than expressed by intrinsic cells. There is neurochemical evidence for α7 nAChRs on glutamate afferents (Kaiser and Wonnacott, 2000; Marchi et al., 2002). nAChRs accounting for 30% of the striatal population of [3H]ACh binding sites (that equate with α4β2* nAChRs) were reported to occur on serotonin terminals (Schwartz et al., 1984), but this could not be reproduced in a subsequent study (Pradhan et al., 2002). However, nAChRs with a novel pharmacological profile were ascribed to serotonin terminals based on transmitter release studies from synaptosomes (Reuben and Clarke, 2000). The relationship between nAChRs and serotonergic terminals warrants clarification in view of recent evidence for the sprouting of these afferents in l-DOPA-induced dyskinesia (Rylander et al., 2010).
Medium spiny projection neurons seem to be devoid of nAChRs (Jones et al., 2001; Zhou et al., 2002) but fast-spiking GABA interneurons (which comprise a tiny proportion of the total neuronal population) were shown to respond to the application of ACh or carbachol in a manner consistent with somatodendritic (possibly extrasynaptic) nAChRs (Koós and Tepper, 2002). In an elegant study, photoactivation of optogenetically engineered cholinergic interneurons of the nucleus accumbens resulted in an increased frequency of inhibitory currents in medium spiny neurons, and this response was sensitive to mecamylamine (Witten et al., 2010). The network connections mediating the increased firing rate are unclear, but it is plausible that GABAergic interneurons bearing nAChRs are involved, rather than a direct cholinergic action on medium spiny neurons.
There is also evidence for functional α4β2* (including α4α5β2) nAChRs on GABAergic terminals from striatum (Grilli et al., 2009; McClure-Begley et al., 2009). These could arise from GABA interneurons, axon collaterals from the medium spiny projection neurons or collateral projections from the GABAergic neurons in the globus pallidus (Bolam et al., 2000).
Cholinergic interneurons might express β2 subunit mRNAs, because β2-like immunoreactivity was reported in “sparsely distributed large neurons” in the rat striatum that might correspond to cholinergic interneurons (Hill et al., 1993; Azam et al., 2003). On the other hand, Jones et al. (2001) did not detect any β2 immunolabeling of cell bodies in striatum. Functional studies support the presence of muscarinic but not nAChRs on the cell bodies of cholinergic interneurons (Calabresi et al., 1998; Windels and Kiyatkin, 2003). Moreover, there are conflicting reports of presynaptic nicotinic modulation of ACh release in striatum with positive results reported by some (Sandor et al., 1991; Yu and Wecker, 1994) but not others (Araujo et al., 1988).
One study reported MLA-sensitive nicotine-evoked currents, consistent with α7 nAChRs, in a proportion of medium spiny neurons, fast-spiking interneurons, and cholinergic interneurons in mouse striatum (Xiao et al., 2009). This is consistent with the finding of α7 subunit mRNA in some rat striatal cholinergic interneurons (Azam et al., 2003). However, levels of 125I-α-bungarotoxin binding in striatum are generally low, although higher in mouse than rat, indicative of only low levels of this nAChR subtype (Marks et al., 1986).
In summary, dopamine terminals in the striatum express a remarkable diversity of nAChRs, with the α4β2* and α6β2* nAChR subtypes studied most extensively. The α6β2* nAChRs are unique to dopamine terminals in striatum, whereas α4β2* nAChRs are more promiscuous and are localized to dopamine terminals and other neuronal elements. Much less is known about the latter category of α4β2* nAChRs with respect to their subunit composition and cellular localization, but their resistance to nigrostriatal degeneration makes it important to understand this population better. There is also little information about striatal α7 nAChRs. In addition, the role of α3β2* nAChRs, present on nigrostriatal terminals in primates but not found in rodent striatum, remains to be identified.
B. Nicotinic Acetylcholine Receptor Modulation of Dopaminergic Function
1. Substantia Nigra
As mentioned in section II.B, SN dopamine neurons exhibit a tonic pacemaker activity, with regular firing of action potentials (Grace and Bunney, 1984a,b). Cholinergic inputs from the PPT regulate the activity of dopamine neurons (Mena-Segovia et al., 2008). Early studies in rats recognized that systemic nicotine increased the firing rate of SN pars compacta neurons (Lichtensteiger et al., 1982; Clarke et al., 1985) and promoted burst firing (Grenhoff et al., 1986). These findings have been recapitulated in a recent study (Zhang et al., 2009b). Iontophoretic application of ACh in the SN pars compacta also enhanced firing, and this was inhibited by dihydro-β-erythroidine but not atropine, implicating a nicotinic, rather than muscarinic, receptor-mediated response (Lichtensteiger et al., 1982). These studies suggested a direct excitatory action of nicotine on SN dopamine neurons, mimicking the endogenous cholinergic innervation from the PPT (Clarke et al., 1987). However, the excitability of dopamine neurons is restrained by GABAergic inputs and influenced by glutamatergic afferents (Misgeld, 2004; Lee and Tepper, 2009). The presence of nAChRs on these elements as well as on dopamine cell bodies and dendrites (Fig. 4b) presents a more complex scenario for the nicotinic regulation of dopamine cell activity.
The nicotinic regulation of SN pars compacta dopamine cell firing has been studied much less extensively than that in the VTA, reflecting interest in the mesolimbic “reward” pathway with respect to nicotine dependence. This observation also marks another limitation in the field, in that there has been greater emphasis on the actions of the exogenous drug nicotine than on the endogenous cholinergic mechanisms governing physiological function. Nevertheless, given the comparable firing patterns (Zhang et al., 2009b) and similarities in the neurochemical interactions and disposition of nAChRs in the VTA and SN (Livingstone and Wonnacott, 2009) and comparable, albeit pharmacologically distinct, nicotinic modulation in the VTA and SN (Keath et al., 2007), it can be argued that similar mechanisms are likely to operate in the two regions. With these caveats, we can propose a model for the SN pars compacta analogous to that proposed for the VTA (McKay et al., 2007), in which activation of β2* nAChRs on dopamine neurons increases firing rates. Concomitant desensitization of distinct β2* nAChRs on GABA interneurons, with a resultant decrease in inhibitory input onto the dopamine cells, would enhance the activation of dopamine neurons.
The propensity of β2* nAChRs on GABA interneurons to desensitize more rapidly than those on dopamine cell bodies (Yin and French, 2000) is presumed to reflect subtle differences in their subunit composition, local environment, or cell-specific regulatory mechanisms (Dani et al., 2000). As discussed in section III.A.1, nAChRs on dopamine neurons are more complex in subunit composition and diversity (Fig. 4b). In particular, expression of the α6 nAChR subunit is limited to catecholaminergic neurons, and α6β2* nAChRs occur in dopamine but not GABA neurons of the SN (Klink et al., 2001). Indeed, mice with a gain-of-function mutation in the channel-forming M2 segment (L9′S) of the α6 nAChR subunit are hyperactive (Drenan et al., 2008). This behavior is attributed to midbrain dopamine neurons' being rendered hypersensitive to cholinergic activation by endogenous ACh or exogenous nicotine. As this phenotype is also dependent on the presence of α4 nAChR subunits, α6α4β2* nAChRs may be the dominant α6β2* receptor subtype in dopamine neurons (Drenan et al., 2010). Thus α6β2* nAChRs could account, at least in part, for the differential responses of GABA and dopamine neurons.
Midbrain α7 nAChRs are present on a proportion of dopamine neurons and are also proposed to reside on glutamate terminals (Fig. 4b). In SN pars compacta, activation of these presynaptic α7 nAChR receptors, in concert with non-α7 nAChRs, also presumed to be on glutamate afferents, increases the frequency of spontaneous EPSCs recorded from dopamine neurons (Keath et al., 2007). Only α7 nAChRs have been implicated on glutamate afferents to the VTA (Mansvelder and McGehee, 2000; Placzek et al., 2009), and this distinction may reflect differences in the glutamatergic innervation of the two regions (Misgeld, 2004). Coincident activation of presynaptic α7 nAChRs and postsynaptic depolarization as a consequence of activating somatodendritic β2* nAChR can induce long-term potentiation in VTA dopamine neurons in vitro (Mansvelder and McGehee, 2000). Although α7 nAChRs commonly display fast desensitization, this depends on agonist concentration, and low levels of agonist can elicit more sustained α7 nAChR activity (Papke and Porter Papke, 2002). Moreover, α7 nAChRs exhibit very high relative permeability to Ca2+, and brief Ca2+ transients arising from opening of the α7 nAChR can be augmented (spatially and temporally as well as in magnitude), by promoting Ca2+-induced Ca2+ release from internal stores (Tsuneki et al., 2000; Dajas-Bailador and Wonnacott, 2004; Fucile, 2004). Such features equip α7 nAChR for a role in synaptic plasticity (Mansvelder and McGehee, 2000; McKay et al., 2007). Glutamate release, and its enhancement via α7 nAChRs, has been implicated in the switch to burst firing in the VTA, mediated by NMDA receptors (Chergui et al., 1993; Overton and Clark, 1997; Schilström et al., 2003). Indeed, the absence of burst firing in slice preparations signifies the importance of afferent inputs for this phenomenon (Grace and Onn, 1989). The SN pars compacta of rodents has relatively fewer α7 nAChRs than the VTA and smaller choline-evoked currents (Wooltorton et al., 2003; Keath et al., 2007), raising some questions over the role of α7 nAChRs in the SN. However, the glutamate afferents to the SN pars compacta, arising principally from the subthalamic nucleus, as well as the PPT, do induce burst firing, mainly via NMDA receptors, in an fashion analogous to that of the prefrontal cortex inputs to the VTA (Lee and Tepper, 2009).
The relative contributions of α7 and β2* nAChRs in the VTA were explored in an elegant study by Mameli-Engvall et al. (2006). In this study, extracellular single unit recordings were made from the VTA of anesthetized wild-type and null mutant mice lacking the β2 or α7 subunit, and interpretations were confirmed by lentiviral re-expression of the absent β2 subunit. Spontaneous firing rate and firing pattern in wild-type animals could be classified into four categories: low firing/low bursting, low firing/high bursting, high firing/low bursting, or high firing/high bursting. β2 subunits (and hence β2* nAChRs) were essential for high frequency rates of firing and/or bursting. It is suggested that β2* nAChRs could provide sufficient depolarization for activation of NMDA receptors, functioning as a “switch” or “gate” between basal and excited states (Mameli-Engvall et al., 2006; Ungless and Cragg, 2006). α7 nAChRs seemed to have a more subtle role, as both low-firing/low-bursting and high-firing/high-bursting states were observed in α7 knockout mice, but the intermediate states were absent (Mameli-Engvall et al., 2006). How presynaptic α7 nAChRs on glutamate afferents versus somatodendritic α7 nAChRs on dopamine neurons differentially shape these responses is unknown.
In summary, the activity of dopamine neurons in the SN pars compacta is driven, in part, by cholinergic input from the PPT. Evidence from the VTA suggests that somatodendritic β2* nAChRs (notably α6β2* nAChRs that are confined to the dopaminergic neurons) mediate the principal effects of ACh, and play a critical, permissive role with respect to facilitating responses to glutamatergic inputs (Maskos, 2008). α7 nAChRs exert a more subtle influence. Presynaptic α7 nAChRs on glutamate afferents may contribute to burst firing and synaptic plasticity. The significance of somatodendritic α7 nAChRs on a proportion of dopamine neurons is presently unclear. The activity of dopamine neurons is constrained by GABA afferents and local interneurons; nicotine (but not tonically released ACh) preferentially desensitizes α4β2* nAChRs on GABA interneurons, relieving this inhibition. In addition, bursting in SN pars compacta neurons may also be influenced by the autoinhibitory actions of dendritically released dopamine (Pucak and Grace, 1994), which is also subject to nicotinic modulation (Reuben and Clarke, 2000; Rahman et al., 2004a; Rahman et al., 2004b).
2. Striatum
The consequence of SN pars compacta dopamine neuron activity is the release of dopamine in the striatum. The tonic firing of single action potentials (typically at a frequency of 1–5 Hz) maintains low, nanomolar concentrations of extracellular dopamine (Goto et al., 2007). Burst firing, which accompanies behaviorally salient stimuli, produces proportionately much greater release of dopamine, achieving local transient concentrations in the micromolar to millimolar range. As discussed in section II.C, cholinergic input from the PPT is a determinant of burst firing, and direct stimulation of the PPT elicits striatal dopamine release via nigral nicotinic and glutamate receptors (Forster and Blaha, 2003). Thus, physiologically, striatal dopamine release is largely driven by action potentials generated in the cell bodies of the SN pars compacta. However, levels of extracellular dopamine also depend on terminal release efficiency, diffusion and spillover from the synapse versus reuptake by dopamine transporters, and regulation via autoreceptors and other inputs (Zhang et al., 2009a). It is in this context that striatal nAChRs contribute.
Dopaminergic terminals in the striatum are well endowed with a distinct population of β2* nAChRs (Fig. 4a), and much attention has focused on nAChR-mediated dopamine release in vitro. The use of striatal synaptosome or chopped tissue preparations clearly demonstrated the ability of presynaptic nAChRs to promote Ca2+-dependent dopamine release in the absence of any other depolarizing stimulus (Wonnacott, 1997; Grady et al., 2007). Using knockout mice lacking specific subunits and pharmacological tools, at least five subtypes of β2* nAChRs were found to contribute to striatal dopamine release (Fig. 4a). The α4α6β2β3 subtype is deduced to have the highest sensitivity (EC50 for nicotine-evoked [3H]dopamine release was 230 nM) (Salminen et al., 2004, 2007). The presynaptic nicotinic modulation of dopamine release demonstrated by using radiolabeled transmitter has been corroborated for endogenous dopamine using fast-scan cyclic voltammetry (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004; Exley et al., 2008; Meyer et al., 2008; Perez et al., 2008).
Superfusion of isolated nerve terminals (that precludes neurochemical cross talk between boutons) has generally failed to demonstrate any contribution from α7 nAChRs that are considered to be absent from these terminals. However, in rat striatal slice preparations some local anatomical integrity is preserved that permits neurochemical cross talk, α7 nAChRs have been shown to enhance [3H]dopamine release (Kaiser and Wonnacott, 2000). This action is absent in striatal tissue from α7 knockout mice (Quarta et al., 2009) and is blocked by inhibitors of ionotropic glutamate receptors, consistent with the localization of α7 nAChRs on glutamate afferents (Kaiser and Wonnacott, 2000; Marchi et al., 2002; Livingstone and Wonnacott, 2009). There are elaborate reciprocal interactions between dopamine and glutamate afferents in the striatum (Calabresi et al., 1998; Avshalumov et al., 2008). However, no contribution from α7 nAChRs was detected in measurements of endogenous dopamine release evoked by low- or high-frequency stimulation in mouse striatal slices (Zhou et al., 2001; Exley and Cragg, 2008).
Presynaptic β2* nAChRs seem to act by depolarizing the terminal bouton. This leads to activation of voltage-operated Ca2+ channels and influx of Ca2+ (in addition to some Ca2+ entry through the nAChR channels), and dopamine release occurs by classic exocytosis (Soliakov and Wonnacott, 1997; Kulak et al., 2001). The voltage dependence and inward rectification of neuronal nAChRs means that these receptors generate the biggest responses at resting or hyperpolarized membrane potentials (Mulle et al., 1992). Given that dopamine neurons are tonically active, the responsiveness of nAChRs during this pacemaker activity may be compromised. However, local application of nicotine into the striatum of conscious freely moving rats provokes dopamine overflow, consistent with the ability of presynaptic nAChRs to exert a positive effect in vivo (Toth et al., 1992; Marshall et al., 1997). Nevertheless, it is clear from the previous section that midbrain nAChRs drive burst firing, and under bursting conditions (which result in greater terminal depolarization), it may be anticipated that presynaptic nAChRs will show diminished responsiveness to agonist.
Most of the in vitro studies cited so far have focused on the actions of exogenous agonists, notably nicotine, on pharmacologically defined nAChR subtypes in the striatum, with the goal of exploring the actions of psychomotor stimulants. However, in the context of understanding Parkinson's disease, it is important to comprehend the physiological modus operandi of nAChRs. In the striatum, nAChRs will respond to ACh released from cholinergic interneurons; these are tonically active when dopamine neurons are quiescent (tonic firing) but shut down when dopamine neurons switch to burst firing mode (Cragg, 2006). To better understand the physiological impact of presynaptic nicotinic modulation, dopamine release has been explored in slice preparations in which the endogenous firing patterns of dopamine neurons can be simulated by electrical simulation (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004; Exley et al., 2008; Meyer et al., 2008; Perez et al., 2008). In these studies, detection of dopamine using carbon fiber microelectrodes and fast-scan cyclic voltammetry combines sensitivity with high temporal and spatial resolution. At low-frequency stimulation, dopamine release as a result of the activation of presynaptic nAChRs by endogenous ACh was indicated by the inhibitory effects of vesamicol, to deplete ACh stores, or nAChR antagonists (Zhou et al., 2001). The decreased amounts of dopamine found in response to ambenonium, an acetylcholinesterase inhibitor, were presumed to reflect nAChR desensitization by the sustained concentration of ACh under these conditions. In agreement with this model, bath-applied agonist (including ACh) decreased evoked release. This distinction is important: it emphasizes that although the cholinergic interneurons are tonically active, the frequency of release events is such that ACh signals are terminated by acetylcholinesterase to create a pulsatile delivery, thus avoiding receptor desensitization. This profile is not mimicked by drug application, either experimentally or therapeutically.
The observation by Zhou et al. (2001) that under conditions of low frequency stimulation, nicotine seems to act like a blocker has been confirmed and extended by others (Rice and Cragg, 2004; Zhang and Sulzer, 2004; Exley and Cragg, 2008). Comparison of the effects of nicotine or mecamylamine at a range of stimulation frequencies revealed that although both drugs suppressed release at tonic frequencies (<10 Hz), they enhanced release by phasic bursts (>25 Hz) (Rice and Cragg, 2004; Zhang and Sulzer, 2004; Exley and Cragg, 2008). Similar effects were reported for primate striatal slices (Perez et al., 2009). Thus, inhibition of nAChRs (by desensitization or antagonist) serves to enhance the contrast between levels of dopamine evoked by phasic versus tonic stimulation. This unexpected result has been explained by the occurrence of use-dependent, short-term depression of dopamine release probability at rapidly successive pulses (Cragg, 2003; Rice and Cragg, 2004; Zhang and Sulzer, 2004). At low-frequency stimulation, endogenous ACh increases release probability by activating nAChRs. However, this leads to short-term depression and less release per pulse in response to high-frequency stimulation (bursts). Reducing nAChR activation (by desensitization or antagonist) decreases short-term depression, permitting a greater response to emerge at higher frequencies. What is missing from this picture is the endogenous behavior under bursting conditions, when cholinergic interneurons are coordinately regulated to cease firing (referred to as a “pause”). Thus, ACh will not be released and nAChRs will not be activated. The consequent decrease in nicotinic stimulation is also predicted to decrease short-term depression and enhance the contrast between the two firing patterns. This mechanism has been referred to as a “cholinergic filter” (Rice and Cragg, 2004; Zhang and Sulzer, 2004) that may represent a unique nicotinic mechanism, because inhibiting dopamine autoreceptors or transporters failed to alter the ratio of phasic/tonic dopamine responses despite increasing dopamine release (Zhang et al., 2009a). A potential limitation of this in vitro model is that it is presently uncertain how cholinergic activity changes in response to manipulation of firing patterns experimentally in slice preparations and whether this replicates the “pause” observed in vivo.
The β2-selective antagonist dihydro-β-erythroidine mimics the effects seen with mecamylamine, confirming that β2* nAChRs account for this phenomenon (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004). The contribution of the α6β2* nAChR subpopulation was dissected using the α6β2*-selective antagonist α-conotoxinMII (Exley et al., 2008; Meyer et al., 2008; Perez et al., 2008). In mouse, α6β2* and α4(non-α6)β2* nAChRs were distinguishable by their sensitivity to stimulus intensity, responding to low and high stimulus strengths, respectively. This result prompted the speculation that these nAChR subtypes may be localized to separate dopaminergic fibers, although it is possible they are expressed on the same fibers but have different sensitivity to acetylcholine such that weak stimulation elicits less acetylcholine release than strong stimulation (Meyer et al., 2008). The contributions of α4(non α6)β2* and α6β2* nAChRs were found to vary with experimental parameters, the latter appearing to have a lesser impact under phasic stimulation conditions (Exley et al., 2008; Meyer et al., 2008; Perez et al., 2008). Comparison of responses across the mouse striatum indicated that α6β2* nAChRs exerted a more prominent effect on the nicotinic “filter” that modulates dopamine release in nucleus accumbens (ventral striatum) than in caudate putamen (dorsal striatum) (Exley et al., 2008). In primates, α6/α3β2* nAChRs seemed equally effective in both striatal regions when dopamine release was evoked by a single stimulus, accounting for at least 80% of the nicotinic modulation (Perez et al., 2009). In contrast, when stimulation was delivered as a train of four pulses to simulate a burst, dopamine release with α6/α3β2* nAChR blockade was overcome in the dorsal but not ventral striatum from monkeys (Perez et al., 2009). These data demonstrate that α6β2* nAChRs differentially control dopamine release in these two regions.
In a comparison of dorsal and ventral striatum, and the nucleus accumbens shell, Zhang et al. (2009b)) showed that the dorsal striatum displays a higher probability of release in response to a single stimulus, representing tonic (low-frequency) stimulation in both primates (Cragg, 2003) and rodents (Zhang et al., 2009b). Consequently, this region showed less frequency-dependent facilitation (determined as the ratio of responses to 20 pulses/1 pulse, delivered at 20 Hz). Blockade of β2* nAChRs, dopamine transporters or D2 receptors indicated that nAChRs are primarily responsible for the differential frequency dependence of dopamine neurons in the dorsal striatum. Whereas these studies (Zhang et al., 2009b) did not consider the contribution of particular β2* nAChR subtypes, Drenan et al. (2010) found that a gain-of-function mutation in the α6 nAChR subunit changed the pattern of stimulus-dependent dopamine release to one that more closely resembled that found in ventral striatum. The mutant mice showed reduced synaptic depression and increased frequency-dependent facilitation (the ratio of responses to four pulses/one pulse, delivered at 100 Hz) in dorsal striatum slices, compared with results from wild-type mice. Moreover, the kinetics of the dopamine waveforms were altered. One interpretation of this study is that presynaptic α6α4β2* nAChRs have a major role in locally shaping dopamine release in the striatum, but alternative explanations are possible, and it should be remembered that the gain of function mutation creates a nAChR with considerably altered activity. Indeed, many factors could account for these observed changes, including adaptive alterations during development of the mutant animals.
The impact of α7 nAChRs (presumed to reside on glutamate afferents to the striatum) on nicotinic filtering of dopamine release has received less attention. Antagonism of α7 nAChRs had no effect on dopamine release in response to low- or high-frequency stimulation of mouse striatal slices (Zhou et al., 2001), but recent studies in rat striatal slices revealed a subtle contribution of α7 nAChRs to nicotinic modulation at high-frequency stimulation (Seipel and Yakel, 2010). The integration of multiple transmitter influences (including GABA, glutamate, and serotonin) that may be modulated by presynaptic nAChRs and their local impact on dopamine release is not yet understood.
In summary, although the firing pattern exhibited by dopamine neurons is the major determinant of dopamine release probability in the striatum, evidence is emerging that the multiplicity of heteromeric nAChR subtypes on dopamine terminals is important for locally shaping dopamine responses. Presynaptic nAChRs are proposed to act as filters to interpret dynamic ACh signals that are reciprocally coordinated with dopamine neuron firing. Thus nAChRs serve to discriminate tonic and phasic patterns of stimulation. Nicotinic drugs can amplify this discrimination, but they do this by nAChR desensitization or inhibition, such that nicotinic agonists achieve the same effect as antagonists. Both α4β2* and α6β2* nAChRs contribute to the regulation of striatal dopamine release, with the α6β2* nAChR population playing a dominant role in the nucleus accumbens and also making a significant contribution in the striatum. These observations have implications for the development of drugs with optimal benefit for Parkinson's disease.
C. Downstream Dopaminergic Signaling Mechanisms Linked to Nicotinic Acetylcholine Receptors
The concept that different populations of striatal medium spiny projection neurons are responsible for distinct aspects of motor control resulted in the classification of the “direct” and “indirect” pathways (Albin et al., 1989; Graybiel et al., 1990; Parent, 1990) (Fig. 3). These circuits act in an opposing fashion with the direct pathway, resulting in disinhibition of the thalamus or brain stem nuclei, whereas the indirect pathway exerts an inhibitory influence. The nigrostriatal dopaminergic inputs differentially regulate these two pathways through the segregation of dopamine D1 and D2 receptors to medium spiny neurons of the direct and indirect pathways, respectively (Gerfen et al., 1990). Thus, in simplistic terms, dopamine acting via stimulatory D1 receptors serves to enhance disinhibition, whereas its actions through inhibitory D2 receptors decrease inhibition. The net effect is the relief of the brake on thalamocortical drive to the motor cortex, favoring the initiation or smooth execution of motor function. This is consistent with the bradykinesia characteristic of Parkinson's disease when the ‘permissive’ nigrostriatal projection degenerates. Therapies predicted to boost dopamine release in the striatum, for example agonists targeting α4β2* or α6β2* nAChRs, would help to counteract the “brake.” Therefore, it is of interest to understand the impact of nAChR regulation on downstream dopamine receptor-linked mechanisms within the postsynaptic neurons.
In medium spiny neurons of the striatum an important site of signal integration that is potentially modulated by nAChRs is the protein DARPP-32. Multiple signaling pathways converge to regulate this “molecular switch,” identified as dopamine and cAMP regulated phosphoprotein with molecular weight 32 kDa (DARPP-32) (Svenningsson et al., 2004) (Fig. 5). The activity of DARPP-32 is promoted or reduced by dopamine acting via D1 or D2 receptors, respectively. DARPP-32 is expressed in both striatonigral (“direct”) and striatopallidal (“indirect”) neurons and its selective deletion in these subsets of neurons results in decreased and increased locomotor function, respectively. This is consistent with the proposed roles of the direct and indirect pathways and establishes a contribution of DARPP-32 signaling to this regulation (Bateup et al., 2010).
Fig. 5.

Pivotal role of DARPP-32 in postsynaptic signaling in medium spiny neurons, illustrating the potential for nicotinic modulation. DARPP-32 integrates inputs from multiple systems to regulate PP-1; only dopamine and glutamate receptors are shown for clarity. Postsynaptic dopamine D1 and D2 receptors are largely segregated to the “direct” (striatonigral) and “indirect” (striatopallidal) projection pathways, respectively. Other components shown are presumed to be common to all medium spiny neurons. DARPP-32 via PP-1 influences the activity of numerous target proteins, including receptors, ion channels and transcription factors. nAChRs that modulate dopamine release, notably α4β2* and α6β2* subtypes (shown here on nigrostriatal terminals but also present on dopaminergic cell bodies in the SN) can also affect postsynaptic excitability and synaptic plasticity through these mechanisms.
DARPP-32 is a key determinant of medium spiny neuron excitability by virtue of its ability to regulate the phosphorylation status of various targets (including receptors, ion channels, and transcription factors) through the inhibition of the multifunctional protein phosphatase 1 (PP-1). D1 receptor stimulation is reported to increase currents through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors and L-type Ca2+ channels in striatal neurons, and this depends upon DARPP-32 inhibition of PP-1 (Surmeier et al., 1995; Yan et al., 1999). DARPP-32 knockout mice lack the ability to produce D1 receptor-mediated potentiation of NMDA-evoked currents (Flores-Hernández et al., 2002). Selective deletion of DARPP-32 in striatonigral or striatopallidal neurons results in functional deficits in long-term potentiation in both neuronal populations (Bateup et al., 2010).
nAChR stimulation is predicted to influence the excitability of postsynaptic striatal neurons via increases in dopamine release (Fig. 5). Indeed, in vivo administration of psychomotor stimulants, including nicotine, that promote dopamine release have been shown to increase DARPP-32 phosphorylation and activation of extracellular signal-regulated kinase in a subset of striatal medium spiny neurons (Valjent et al., 2005). Nicotine increased the phosphorylation of striatal DARPP-32 at multiple sites in mice given systemic injections of nicotine (Zhu et al., 2005). In rat striatal slices in vitro, nicotine has been reported to modify DARPP-32 phosphorylation in a concentration- and time-dependent manner, by acting at β2* and α7 nAChRs (Hamada et al., 2004; Hamada et al., 2005). Sensitivity to dopamine receptor antagonists supports the view that nicotine-evoked changes in DARPP-32 phosphorylation depend on dopamine release. It is unclear whether the same or different changes occur in striatonigral and striatopallidal neurons, although the observation of distinct dopamine D1 and D2 receptor-mediated responses is consistent with their segregation.
The therapeutic efficacy of l-DOPA in Parkinson's disease relies on the preservation of normal postsynaptic signaling mechanisms. The evidence from animal models of the disease, as well as neurochemical assessment of post mortem human brain tissue from patients, suggests that levels of DARPP-32 are unchanged after nigrostriatal denervation (Raisman-Vozari et al., 1990; Nishino et al., 1993). However, functional changes in DARPP-32 may still ensue; for example, increased phosphorylation of DARPP-32 at the inhibitory Thr75 site (with no change in phosphorylation of the activating Thr34 site, compared with healthy controls) has been reported (Brown et al., 2005). DARPP-32 signaling is also implicated in the development of l-DOPA-induced dyskinesia (Santini et al., 2007) (see section VIII.A below).
In summary, the control of motor function via the balance of activity in the direct and indirect pathways projecting from the striatum is influenced by dopamine acting at dopamine D1 and D2 receptors, respectively. The ability of dopamine to modify activity within the medium spiny neurons is achieved, at least in part, via shifts in the balance of phosphorylation and dephosphorylation manipulated by the key regulator protein DARPP-32, an inhibitor of PP-1. DARPP-32 integrates signals from nigrostriatal and other inputs (notably the corticostriatal afferents that release glutamate onto medium spiny neurons) to regulate excitability and longer lasting functions, including synaptic plasticity in the projection neurons. Nicotinic stimulation (studies to date have mostly focused on nicotine) can influence DARPP-32 signaling, predominantly by promoting dopamine release via α4β2* and α6β2* nAChRs, although α7 nAChR-mediated glutamate release has also been implicated in vitro, at higher nicotine concentrations (Hamada et al., 2004). More studies are needed to elaborate the effects of selective nAChR activation on postsynaptic molecular mechanisms. The ability of nAChRs to influence signaling within the direct and indirect pathways is compatible with their therapeutic potential for treating Parkinson's disease.
IV. Long-Term Regulation of Nicotinic Acetylcholine Receptors Expression and Function
A. Effect of Long-Term Nicotine Administration
Before nicotine can be used therapeutically for neurodegenerative disorders, it is important to understand the long-term consequences of its administration on nAChR subtype expression and function. As mentioned earlier, short-term exposure results in numerous biological responses because nicotine acts in diverse tissues throughout the body and interacts with different nAChR subtypes at a given site. Repeated administration adds an additional layer of complexity because of receptor desensitization, tolerance, sensitization, up-regulation, and down-regulation. (Corringer et al., 2006; Quik and McIntosh, 2006; Picciotto et al., 2008; Walsh et al., 2008; Buccafusco et al., 2009). A comprehensive knowledge of these changes is important for a clear understanding of the mechanisms whereby nicotine protects against nigrostriatal damage and/or improves l-DOPA-induced dyskinesias.
As discussed earlier, one of the most widely distributed CNS nAChRs is the α4β2* subtype. Long-term nicotine administration, delivered in multiple forms (injection, minipump, drinking water, or self-administration), increases the numbers of α4β2* binding sites throughout the brain in experimental animal models and humans (Marks et al., 1983; Schwartz and Kellar, 1983; Benwell et al., 1988; Perry et al., 1999; Staley et al., 2006; Xiao et al., 2009; Moretti et al., 2010). There was some initial controversy concerning α4β2* receptor up-regulation in the striatum, because increases were observed in some studies but not others. This discrepancy now seems to have arisen because the radioligands originally used for receptor identification bound to multiple nAChR subtypes that were differentially up- and down-regulated with nicotine treatment. This problem has now been circumvented through the use of α-conotoxinMII, which binds to α6β2* nAChRs. It thus allows for the selective identification of α4β2* receptor subtypes by a subtractive process. Studies with α-conotoxinMII show that striatal α4β2* receptors are indeed up-regulated by nicotine treatment. Current evidence suggests that two α4β2* nAChR populations exist in striatum (i.e., the α4β2 and the α4α5β2 subtypes) (Grady et al., 2010b; Moretti et al., 2010) (Fig. 4a). These are differentially regulated by nicotine, with an increase in the α4β2 subtype but no change in the α4α5β2 receptor (Table 1) (Moretti et al., 2010). These findings underscore the complex changes that arise in α4β2* receptor-mediated responsiveness with drug treatment.
TABLE 1.
Alterations in striatal nAChR subtype expression with chronic nicotine treatment
| nAChR Subtype | Effect of Nicotine Treatment | System | References |
|---|---|---|---|
| α6β2* | ↓ or No change | Rats, mice, monkeys | Lai et al., 2005; Mugnaini et al., 2006; Perry et al., 2007; Moretti et al., 2010; but see Parker et al., 2004 |
| α6α4β2β3 | ↓ | Rats, mice | Perez et al., 2008; Huang et al., 2009 |
| α6β2β3 | ↑ | Mice, cultured cells | Tumkosit et al., 2006; Perez et al., 2008; Walsh et al., 2008 |
| α6β2β3 | ↓ | Rats | Huang et al., 2009 |
| α3β2 | No change | Rats | Moretti et al., 2010 |
| α4β2 | ↑ | Rats, mice, monkeys | Marks et al., 1983; Schwartz and Kellar, 1983; Benwell et al., 1988; Perry et al., 1999; McCallum et al., 2006; Staley et al., 2006; Xiao et al., 2009; Moretti et al., 2010 |
| α4α5β2 | No change | Rats | Moretti et al., 2010 |
| α7 | ↑ or No change | Rats, mice, monkeys | Pauly et al., 1991; Quik et al., 2000a; Lai et al., 2005; Moretti et al., 2010 |
Nicotine exposure differentially controls α6β2* nAChR subtype expression compared with that of α4β2*. Nicotine administration for several day either decreases or does not change striatal α6β2* nAChR levels (Nguyen et al., 2003; Lai et al., 2005; McCallum et al., 2006; Mugnaini et al., 2006; Perry et al., 2007; Moretti et al., 2010). To add to the complexity, the α6β2* nAChR subtypes present in striatum (Fig. 4a) are themselves differently affected by nicotine treatment. Studies with the novel α-conotoxinMII analog E11A, which discriminates between α4α6β2β3 and α6β2β3 subtypes, show that nicotine treatment selectively decreased the former but increased the latter population in mouse striatum (Perez et al., 2008). This up-regulation of α6β2β3 nAChRs is in agreement with expression studies in culture that also show that α6β2 or α6β2β3 receptors are up-regulated with long-term nicotine exposure (Tumkosit et al., 2006; Walsh et al., 2008). It should be noted, however, that these differential nicotine-induced changes in mouse α6α4β2β3 and α6β2β3 subtypes did not occur in rat striatum, suggesting it may be species-dependent or possibly related to differences in treatment paradigms between rats and mice (Perez et al., 2008; Huang et al., 2009).
Another nAChR present in striatum is the α7 subtype (Fig. 4a). Its functional role is less clear because studies are confounded by the very low expression of α7 nAChRs in rat, monkey, and human striatum. However, it is readily detected in mouse striatum, where it may be increased or not affected by nicotine administration (Pauly et al., 1991; Lai et al., 2005; Moretti et al., 2010). These variable results may reflect a requirement for higher concentrations of nicotine to up-regulate α7 nAChRs compared with α4β2* nAChRs.
The molecular basis for the differential regulation of various striatal nAChR subtypes is an area actively under investigation, the α4β2* nAChR subtype being most extensively investigated to date (Govind et al., 2009; Lester et al., 2009). There is a consensus that up-regulation of α4β2* nAChRs is primarily controlled at the post-transcriptional level, with little change in α4 or β2 nAChR mRNA levels (Marks et al., 1992). Various mechanisms have been proposed to explain nicotine-mediated up-regulation (Picciotto et al., 2008; Buccafusco et al., 2009; Benowitz, 2010; Changeux, 2010b; Mao and McGehee, 2010). One hypothesis is that the initial receptor desensitization contributes to up-regulation, possibly via receptor phosphorylation (Léna and Changeux, 1993; Swope et al., 1999; Wiesner and Fuhrer, 2006). More recently, it has been proposed that nicotine acts as a chaperone to promote maturation of α4 nAChR subunit precursors that might otherwise be degraded by currently unidentified endogenous molecular components (Sallette et al., 2005; Corringer et al., 2006; Tumkosit et al., 2006; Kuryatov et al., 2008; Walsh et al., 2008; Lester et al., 2009; Jones et al., 2010). The enhanced incorporation of α4 subunits into the α4β2 nAChR may subsequently influence agonist-mediated regulation of other nAChR subtypes. For instance, the enhanced formation of α4β2 receptors may reduce free α4 subunit levels, resulting in a decline in the α6α4β2β3 receptor subtype. By contrast, α6β2β3 receptor levels may still increase because they do not require the α4 subunit. The presence of the β3 subunit in α6β2* nAChRs also increases receptor up-regulation (Tumkosit et al., 2006). Receptor up- and down-regulation is not only controlled by the nature of the subunits in the nAChR complex but is also influenced by chaperone proteins, such as RIC-3, neurexin1β, VLIP-1, and others (Wanamaker and Green, 2007; Millar, 2008; Cheng et al., 2009; Zhao et al., 2009). As well, work suggests that the ubiquitin-proteasome system regulates the stability of neuronal nAChRs with a resultant receptor up-regulation (Rezvani et al., 2010). Numerous mechanisms thus exist to modulate receptor expression and responsiveness with long-term nAChR drug administration.
A balance between these differing molecular mechanisms would ultimately determine the short- and long-term consequences of nicotinic drugs on nAChR subtype expression. This in turn would control nAChR-mediated behaviors linked to Parkinson's disease, such as locomotor activity.
B. Effect of Nigrostriatal Damage
As briefly indicated (section III.A.3), damage to the nigrostriatal dopaminergic pathway has a major impact on nAChR expression in striatum in parkinsonian animal models (Schwartz et al., 1984; Quik et al., 2001, 2003; Kulak et al., 2002a; Zoli et al., 2002; Champtiaux et al., 2003) and in Parkinson's disease cases (Aubert et al., 1992; Perry et al., 1995; Court et al., 2000; Quik et al., 2004; Bohr et al., 2005; Gotti et al., 2006a). There seems to be a particularly pronounced decline in α6β2* nAChRs, losses paralleling those in the dopamine transporter, a well established marker of striatal dopaminergic terminals (Table 2) (Quik et al., 2001, 2003; Zoli et al., 2002). These data provide one of the pieces of evidence that α6β2* nAChR are primarily located on incoming dopaminergic afferents to the striatum (Fig. 4a). Further work done with α-conotoxin E11A, a toxin that discriminates between α6β2* nAChR subtypes, showed that the α4α6β2β3 receptor is the first to be lost with nigrostriatal damage, with decreases in the α6β2β3 receptor subtype only with more severe lesioning (Bordia et al., 2007). This pattern of decline is observed across species (mice, rats, monkeys, humans), highlighting the possibility that the α4α6β2β3 receptor subtype may identify a particularly vulnerable population of dopaminergic afferents to the striatum.
TABLE 2.
Decline in nAChR subtype expression with nigrostriatal damage
Moderate lesion is a ≥80% decline in the dopamine transporter; a severe lesion is ≥95% decline in the dopamine transporter.
| nAChR subtype | Present on Striatal Dopamine Terminals | Severity of Lesion |
Reference | |
|---|---|---|---|---|
| Moderate | Severe | |||
| α6β2* | Yes | ↓↓ | ↓↓↓ | Quik et al., 2001, 2005; Zoli et al., 2002 |
| α6α4β2β3 | Yes | ↓↓ | ↓↓↓ | Bordia et al., 2007 |
| α6β2β3 | Yes | − | ↓↓ | Bordia et al., 2007 |
| α3β2 | Yes | N.D. | ↓↓ | Quik et al., 2005 |
| α4β2 | Yes | ↓ | ↓↓ | Zoli et al., 2002; Quik et al., 2005 |
| α4α5β2 | Yes | ↓ | ↓↓ | Zoli et al., 2002 |
| α4β2* | No | − | − | |
| α7 | No | − | − | Zoli et al., 2002; Quik et al., 2005 |
↓, decline; −, no change; N.D., not determined.
By contrast to the dramatic declines in α6β2* nAChRs with nigrostriatal damage, the decrease in the α4β2* nAChR population that does not include α6 is much less severe (Table 2). The reason for the apparently smaller decline in the α4β2* receptor subtype most likely relates to its more diverse localization, with receptors present on dopamine and GABAergic terminals and possibly on some GABAergic interneurons and serotonin afferents in the striatum (Kulak et al., 2002a; Zoli et al., 2002; Champtiaux et al., 2003; Quik et al., 2003, 2004; Bohr et al., 2005) (see section.III.A.2; Fig. 4a). An almost complete dopaminergic lesion leads to only a 30 to 50% decline in striatal α4β2* nAChRs; this would suggest that 50 to 70% of the α4β2* receptors are present on dopaminergic terminals (Kulak et al., 2002a; Zoli et al., 2002; Champtiaux et al., 2003; Quik et al., 2003). Studies using synaptosomal preparations, which primarily contain nerve terminals, suggest that striatal presynaptic α4β2* nAChRs consist of both the α4β2 and the α4α5β2 subtypes and are present on dopaminergic as well as other neurons (McClure-Begley et al., 2009; Grady et al., 2010b). Further evidence for this stems from studies showing that the α5 subunit declines by ∼90% with nigrostriatal damage whereas the α4 subunit is decreased by only ∼50% (Zoli et al., 2002). These data suggest that the greater proportion of α4α5β2 nAChRs are present on dopaminergic terminals in the striatum, whereas the α4β2 receptors are distributed on various neuronal types (Fig. 4a). Thus, with near complete denervation, there would be a major loss of the α4α5β2 but only a partial decline in the α4β2 receptor subtype.
Studies to evaluate effects of nigrostriatal damage on striatal α7 nAChRs are difficult because of their low expression levels in rat, monkey, and human striatum. However, where such studies have been done, α7 receptor expression seems not to change with lesioning (Quik et al., 2000a, 2003). These findings are consistent with the concept that α7 nAChRs are present on nondopaminergic terminals, including notably glutamatergic afferents, in the striatum (Fig. 4a) (Kaiser and Wonnacott, 2000).
Although there has been a general focus on the striatum, it is also important to consider the fate of nAChRs in the SN pars compacta with nigrostriatal damage. Work in this brain region is much more limited; however, studies in rats and monkeys have shown that nAChR binding is decreased with lesioning (Clarke and Pert, 1985; Quik et al., 2002, 2010). With respect to subtype selectivity, there is a more pronounced decline in α6β2* compared with α4β2* nAChR expression in the SN pars compacta with nigrostriatal damage (Quik et al., 2002, 2010). Because the decrease in α6β2* nAChRs correlates with that in the dopamine transporter, these data are consistent with the localization of α6β2* nAChR to somatodendritic sites on dopaminergic neurons (Klink et al., 2001). By contrast, α4β2* nAChRs are present on dopaminergic neurons and GABAergic interneurons and afferents from the SN pars reticulata. Thus, residual somatodendritic nAChRs left after partial lesions or in the early stages of Parkinson's disease also offer credible therapeutic targets for nicotinic drugs, especially in view of their importance in driving burst firing and striatal dopamine release (see section III B.1). This identifies another area that merits further study.
To conclude, the nAChRs most affected by nigrostriatal damage are those present on dopaminergic neurons in the nigrostriatal pathway. The most pronounced losses are observed in the α6β2* and α4α5β2 subtypes; smaller declines are found in α4β2 receptors, and there is no change in the α7 receptor subtype. These data, summarized in Fig. 6, suggest that drugs that target α6β2* and α4β2* nAChRs may prove most useful in ameliorating nAChR-mediated function with nigrostriatal damage.
Fig. 6.

Schematic diagram depicting the effects of partial and near-complete dopaminergic lesioning on expression of nAChR subtypes in striatum. In intact striatum (left), α6β2* nAChRs are primarily expressed on nigrostriatal dopaminergic terminals (DA). α4β2* nAChRs are also located on dopaminergic terminals and comprise 30 to 50% of the striatal α4β2* population. The remaining 70 to 50% of α4β2* nAChRs, which are likely to be predominantly composed only of the α4 and β2 subunits, are present on other neuronal or non-neuronal elements (other). α7 nAChRs are inferred to be located on glutamatergic (Glu) afferents. In rats with a partial striatal lesion (middle), the α6α4β2β3 nAChR subtype disappears first, and thus the effects of nicotine or nicotinic drugs would be mediated by residual α6β2* subtypes and by α4β2* and possibly α7 nAChR subtypes. With a near-complete lesion (right), all presynaptic nAChRs are lost from nigrostriatal dopaminergic terminals. However, α4β2 nAChRs are preserved on other elements and, possibly together with α7 nAChRs, will contribute to local nAChR-mediated effects. Enhanced glutamatergic signaling is indicated. See Huang et al. (2011) for further details.
V. Role for Nicotine and Nicotinic Acetylcholine Receptor Ligands in Parkinson's Disease
Parkinson's disease is frequently considered a disorder with a primary dysfunction of the nigrostriatal dopaminergic pathway. However, as discussed, the dopaminergic system is integrated with numerous other CNS systems, including the nicotinic cholinergic system (Calabresi and Di Filippo, 2008; Exley and Cragg, 2008; Barik and Wonnacott, 2009). The close functional interaction between the cholinergic and dopaminergic systems forms the basis for the hypothesis that nAChR drugs may be useful for Parkinson's disease therapy. Accumulating evidence suggests that nicotine and nAChR ligands may influence Parkinson's disease motor symptoms via two distinct mechanisms: 1) they may protect against nigrostriatal damage to improve motor control over the long term. 2) In addition, nAChR ligands may directly stimulate the dopaminergic system to acutely ameliorate motor-related symptoms.
In the following sections, we describe the evidence that nAChR drugs may be valuable as a disease-modifying therapy for neuroprotection against nigrostriatal degeneration and may also be helpful for alleviating treatment-related side effects, specifically l-DOPA-induced dyskinesias.
VI. Smoking, Nicotine, and Neuroprotection against Nigrostriatal Damage
A. Epidemiological Studies and Smoking
Over a half-century of studies show overwhelmingly that smoking is inversely associated with Parkinson's disease (Morens et al., 1995; Gorell et al., 1999; Hernán et al., 2001; Allam et al., 2004; Alves et al., 2004; Ritz et al., 2007; Thacker et al., 2007; Elbaz and Moisan, 2008; Morozova et al., 2008; Chen et al., 2010; Ritz and Rhodes, 2010) and other Lewy body-related pathologic conditions (Tsuang et al., 2010). A prime question is whether this decreased incidence of Parkinson's disease in those with a history of smoking is due to the increased mortality risk associated with smoking. This appears not to be the case as data from several epidemiological studies, including a large prospective cohort, showed that mortality in patients with Parkinson's disease was not influenced by smoking status; that is, mortality was similar in nonsmoking and smoking patients with Parkinson's disease (Alves et al., 2004; Driver et al., 2008). The inverse correlation between smoking and Parkinson's disease is dose-dependent, with a decreased disease incidence with both increased smoking intensity (number of cigarettes smoked per day) and number of years of smoking (Morens et al., 1995; Gorell et al., 1999; Hernán et al., 2001; Allam et al., 2004; Alves et al., 2004; Ritz et al., 2007; Thacker et al., 2007; Elbaz and Moisan, 2008; Morozova et al., 2008; Chen et al., 2010; Ritz and Rhodes, 2010). The reduced Parkinson's disease incidence seems to be independent of sex and education, is more pronounced in current than in former smokers, and also occurs with other forms of tobacco, such as cigars, pipes, chewing tobacco, or snuff (O'Reilly et al., 2005; Ritz et al., 2007).
The findings that the lower risk of Parkinson's disease is inversely correlated with smoking duration, intensity, and recentness but is not linked to increased mortality suggests a true biologic effect. These data may constitute an important clue in identifying drugs that protect against the neurodegenerative changes in Parkinson's disease. It may be relevant that α4β2* nAChRs are up-regulated in current but not former smokers (Breese et al., 1997).
B. Nicotine Neuroprotection in Parkinsonian Animal Models
An important question is the identity of the agent(s) in tobacco that contributes to this putative neuroprotective effect of smoking, because its use may reduce Parkinson's disease progression. This would represent a milestone in Parkinson's disease treatment, because current therapies provide only symptomatic relief. Certainly there are thousands of chemicals in tobacco smoke, any of which may have therapeutic value. These include nicotine and components that inhibit monoamine oxidase; however, there has been a focus on nicotine because it is present in fairly high concentrations in tobacco and stimulates dopamine release, as discussed earlier.
One approach to evaluate a role for nicotine is to investigate its neuroprotective properties against nigrostriatal damage in parkinsonian animal models (Belluardo et al., 2000; O'Neill et al., 2002; Quik et al., 2007b; Picciotto and Zoli, 2008; Bencherif, 2009). A frequently used rat model involves unilateral administration of 6-hydroxydopamine into the striatum, SN pars compacta, or medial forebrain bundle or hemisection of the nigrostriatal dopaminergic pathway (Figs. 1 and 6) (Cenci and Lundblad, 2007). These treatment protocols result in unilateral declines in striatal dopaminergic function, with contralateral deficits in motor activity. Bilateral models are not used because these are incompatible with animal survival (Cenci and Lundblad, 2007). In rats, nicotine pretreatment before lesioning reduces neuronal damage as assessed using markers of striatal dopaminergic integrity, such as dopamine and metabolite levels, tyrosine hydroxylase, the dopamine transporter, and the vesicular monoamine transporter. As expected, the degree of protection against nigrostriatal damage is dependent on lesion size, with optimal effectiveness against moderate lesioning. Nicotine dosing is also an important variable. Nicotine exhibits a U-shaped dose-response curve; that is, maximal protection is seen with intermediate nicotine dosing regimens (Costa et al., 2001; Ryan et al., 2001; Soto-Otero et al., 2002). Studies using MPTP-lesioned monkeys, a model that more closely mimics features of Parkinson's disease, also show that a 2-month nicotine pretreatment period protects against nigrostriatal damage (Quik et al., 2006a,b). As reviewed earlier, the ability of nicotine to protect against dopaminergic damage in parkinsonian mice has been inconsistent with positive results in some studies but not others (Quik et al., 2007b). A likely explanation for this variability in mice may relate to their rapid rate of nicotine metabolism (t1/2 = 5–10 min in mice compared with 60 min in rats and monkeys) (Matta et al., 2007).
Although nicotine is clearly protective against toxin-induced nigrostriatal damage in parkinsonian rats and monkeys, it should be noted that nicotine does not restore function if administered to parkinsonian animals when nigrostriatal damage is complete (Huang et al., 2009). A 2- to 3-week nicotine regimen given before nigrostriatal damage improved dopamine transporter levels in the striatum of lesioned rats compared with animals not receiving nicotine. However, there was no change in transporter levels when nicotine was administered to rats after lesioning and subsequently given nicotine for the same time interval (Huang et al., 2009). These observations suggest that nicotine's primary action is protection against ongoing degeneration rather than restoration of damaged neurons.
In addition to nicotine, smoke contains numerous other components, any of which may play a synergistic and/or additive role in protection against nigrostriatal damage. As mentioned earlier, this includes agents that modulate metabolic enzymes such as monoamine oxidase (Castagnoli and Murugesan, 2004). The rationale for this possibility stemmed from studies showing reduced levels of monoamine oxidase activity in the brains of smokers (Fowler et al., 1996a,b). In addition, other work showed that tobacco components that inhibit monoamine amine oxidase activity protected against nigrostriatal damage in mouse striatum (Castagnoli et al., 2001). These data provide support for the idea that monoamine oxidase inhibitors in tobacco may decrease enzymic activity in the brains of patients with Parkinson's disease, thereby reducing the synthesis of endogenous toxins that cause nigrostriatal damage in addition to prolonging the availability of dopamine. Nicotine and/or other agents in tobacco may also act by modifying brain expression of cytochrome P450 (P450). This possibility stemmed from work showing that enzymic activity is higher in smokers and is induced by nicotine in experimental animals. Enhanced P450 expression may contribute to neuroprotection by increasing the breakdown of toxic agents that damage the nigrostriatal dopaminergic system (Miksys and Tyndale, 2006).
In summary, epidemiological data show that smoking represents the largest, most consistent negative risk factor for Parkinson's disease. Experimental studies with parkinsonian animals demonstrate that nicotine significantly reduces nigrostriatal damage, with an average 30% protection in different animal models. These combined data support the hypothesis that nicotine treatment may yield a therapeutic strategy to reduce Parkinson's disease progression. Motor symptoms generally only arise in Parkinson's disease when nigral dopaminergic neurons are reduced by >50% and striatal dopamine by >70% (Olanow, 2004; Samii et al., 2004). Therefore, a 30% protection against nigrostriatal damage may allow for normal function for many years before Parkinson's disease symptoms arise. Although studies have been done to evaluate the symptomatic benefits of short-term nicotine administration, as detailed in section VII.A, nicotine's potential to protect against neuronal damage in Parkinson's disease is not known. A recent Michael J. Fox Foundation-funded international clinical trial vetting the potential of a nicotine skin patch to modify the progression of Parkinson's disease should provide some answers to this important question.
VII. Nicotine and Symptomatic Improvement
A. Patients with Parkinson's Disease
A number of reports and small clinical trials over the last 2 decades show that nicotine treatment reduces motor symptoms in patients with Parkinson's disease (Ishikawa and Miyatake, 1993; Fagerström et al., 1994; Kelton et al., 2000; Mitsuoka et al., 2002; Hanagasi et al., 2007). However, these are counterbalanced by an approximately equal number of studies that demonstrate no improvement in clinical features with either nicotine or a nicotinic agonist (Clemens et al., 1995; Vieregge et al., 2001; Lemay et al., 2004; Parkinson Study Group, 2006) or even a slight worsening of symptoms (Ebersbach et al., 1999). A comparison of the different studies was done with the intent of identifying factors that may account for the positive outcomes (Table 3). Careful analyses suggested that improvement did not correlate with nicotine formulation, dosage, or treatment paradigm. Instead, the most obvious success indicator seemed to relate to study design. Improvement was observed in studies with only a small number of patients (1–15 patients), with no effect of nicotine on Parkinson's disease symptoms in the larger study groups (16–77 patients). In addition, improvement seems to correlate with an open label but not placebo-controlled protocol. Although initially unexpected, this latter outcome is consistent with work that demonstrates significant improvements in Parkinson disease symptoms with placebo treatments, with a 9 to 59% placebo effect across different trials (de la Fuente-Fernández and Stoessl, 2002; Goetz et al., 2008). A large, recent analysis involving 858 patients with Parkinson's disease on placebo demonstrated an overall placebo response rate of 16%, with a range of 0 to 55% (Goetz et al., 2008). These finding highlight the importance of including a placebo group in drug and other experimental trials.
TABLE 3.
Clinical improvement of Parkinsonism with nicotine treatment correlates best with study design
Parkinsonism was assessed by measurement of a variety of different outcomes, including testing of fine motor skills, tremor, rigidity, bradykinesia, posture, hand and finger dexterity, or by using the following rating scales: the Unified Parkinson's Disease Rating Scale, Hoehn and Yahr, Columbia University, and Schwab-England.
| Study Design | Improve Parkinsonism | Nicotine Formulation | No. of Patients | Treatment Regimen |
References | ||
|---|---|---|---|---|---|---|---|
| Duration of Dose Titration | Duration of Maintenance Dose | Maintenance Dose/Day | |||||
| Open-label | Yes | Smoking/ nicotine gum | 6 | Chronic smoker | Ishikawa and Miyatake, 1993 | ||
| Yes | Intravenous nicotine and patch | 15 | 2 weeks | 1–2 weeks | 14 mg | Kelton et al., 2000 | |
| Yes | Nicotine gum | 8 | N.D. | 1 day | N.A. | Mitsuoka et al., 2002 | |
| Yes | Smoking | 1 | Chronic smoker | Hanagasi et al., 2007 | |||
| Yes | Nicotine patch | 6 | 14 weeks | 4 weeks | Up to 105 mg | Villafane et al., 2007 | |
| No | Nicotine patch | 22 | 22 day | 3 day | 21 mg | Lemay et al., 2004 | |
| Double-blinded | No | Nicotine gum | 48 | N.D. | 1 day | 3 × 2 mg | Clemens et al., 1995 |
| No | Nicotine patch | 16 | N.D. | 12 h | 7 mg | Ebersbach et al., 1999 | |
| No | Nicotine patch | 32 | 1 week | 2 weeks | 14 mg | Vieregge et al., 2001 | |
| No | SIB-1508Y | 77 | 2 weeks | 2 weeks | 10 mg | Parkinson Study Group, 2006 | |
| Yes | Nicotine gum and patch | 2 | ≥7 months | 15 mg patch + 4 × 4 mg gum | Fagerström et al., 1994 | ||
N.D., not determined; N.A., not available.
B. Parkinsonian Animal Models
The clinical data demonstrating inconsistent effects of nicotine on Parkinson's disease motor symptoms are in agreement with results in parkinsonian animal models (Table 4). Work with unilateral 6-hydroxydopamine-lesioned rats or mice show that neither short- nor long-term nicotine treatment modified parkinsonism either with or without l-DOPA treatment (Bordia et al., 2008, 2010; Huang et al., 2011). In these experiments, the effect of nicotine on motor function was tested only after toxin-induced nigrostriatal damage was complete. In another study, nicotine pretreatment did improve dopamine agonist-induced turning behavior after lesioning; however, data interpretation is complicated by the fact that nicotine pretreatment may have protected against nigrostriatal damage (Meshul et al., 2002). Nicotine or nicotinic agonists have also been tested on motor deficits in nonhuman primate models with variable effects. Long-term nicotine administration did not alter parkinsonism in MPTP-lesioned monkeys (Quik et al., 2006b). However, in other monkey studies, short-term nicotine injection did improve some motor deficits (Domino et al., 1999), and nicotinic agonist treatment acted synergistically with l-DOPA to ameliorate parkinsonism (Schneider et al., 1998).
TABLE 4.
Variable effects of nicotine on parkinsonism in animal models with nigrostriatal damage
| Animal Model & Change in Parkinsonism | Drug | Study Duration | Reference |
|---|---|---|---|
| 6-OHDA-lesioned rat | |||
| None | Nicotine | Weeks | Bordia et al., 2008 |
| Improvement | Nicotine | Acute dose | Meshul et al., 2002 |
| 6-OHDA-lesioned mouse | |||
| None | Nicotine | Weeks | Huang et al., 2011 |
| MPTP-lesioned monkey | |||
| None | Nicotine | Weeks | Quik et al., 2006b |
| Improvement | Nicotine | Acute dose | Domino et al., 1999 |
| Synergistic with l-DOPA | SIB-1508Y | Acute dose | Schneider et al., 1998 |
6-OHDA, 6-hydroxydopamine.
In summary, the data from patients with Parkinson's disease and parkinsonian animal models have yielded conflicting outcomes concerning the efficacy of nicotine in improving motor problems linked to nigrostriatal damage. The discrepant nature of the results suggest that nicotine does not improve Parkinson's disease motor symptoms or that nicotine is of benefit only under specific conditions that remain to be defined.
VIII. Nicotine and l-DOPA-Induced Dyskinesias
A. l-DOPA Treatment for Parkinson's Disease Induces Dyskinesias
As stated earlier, the most prominent problem in Parkinson's disease is the motor deficits, which can effectively be treated with dopamine replacement therapies. The “gold standard” treatment is with the dopamine precursor l-DOPA, which greatly improves quality of life particularly in the early stages of the disease. However, long-term l-DOPA use results in the development of side effects such as dyskinesias (Fahn, 2008; Lang, 2009; Poewe, 2009; Schapira et al., 2009; Pezzoli and Zini, 2010). These abnormal involuntary movements of the head, trunk, and/or extremities interfere with functions of daily living and may become very debilitating. They are relatively common with approximately a 30% incidence after 2 years of l-DOPA treatment, 40% by 5 years, and 90% by 10 years (Ahlskog and Muenter, 2001). A variety of strategies have been proposed to minimize their onset, including a delay in the initiation of l-DOPA therapy, reduction in l-DOPA dose and/or treatment with dopamine agonists (Fahn, 2008; Lang, 2009; Poewe, 2009; Schapira et al., 2009; Pezzoli and Zini, 2010). However, there are drawbacks to these approaches, because modifications in l-DOPA treatment generally compromise the control of Parkinson's disease symptoms, whereas dopamine agonists fail to be as effective as l-DOPA and are associated with their own set of side effects. Amantadine, a drug with multiple CNS actions, including inhibition of NMDA glutamate receptors, is currently the only drug that is used clinically for the treatment for l-DOPA–induced dyskinesias, although its efficacy is limited. Continuous delivery therapies are under consideration to minimize the fluctuations in dopamine levels that may contribute to the development of dyskinesias. These include subcutaneous or intravenous dopamine agonist therapies, transdermal patches with dopamine agonists, or intraduodenal l-DOPA infusion. Other pharmacological treatments include concurrent l-DOPA treatment with catechol-O-methyl transferase and monoamine oxidase inhibitors to prolong l-DOPA effectiveness (Jankovic and Stacy, 2007; Fahn, 2008; Lang, 2009; Poewe, 2009; Schapira et al., 2009; Obeso et al., 2010). A surgical strategy that holds great promise in certain subsets of patients is deep-brain stimulation; however, its invasive nature is a limiting factor (Fahn, 2008; Benabid et al., 2009). Overall, the control of l-DOPA-induced dyskinesias is fairly limited. There is therefore a great unmet need to identify novel therapies.
Knowledge of the cellular and molecular mechanisms responsible for the occurrence of l-DOPA-induced dyskinesias would greatly facilitate in the development of targeted treatments. Indeed, this is an area under investigation in numerous laboratories. l-DOPA seems to mediate its initial effects via a dopamine receptor D1-mediated interaction. A current hypothesis is that dyskinesias arise because of an inability to turn down supersensitive signaling responses downstream of dopamine D1 receptors that arose in response to nigrostriatal dopaminergic denervation. DARPP-32 is one of the targets modified in l-DOPA-induced dyskinesia in rodents, with increased phosphorylation of its activating Thr34 site and several downstream targets including extracellular signal-regulated kinase and cFos (Fig. 5; see section III.C) (Picconi et al., 2003; Santini et al., 2007). The sustained activation of intracellular signaling pathways induced by multiple l-DOPA dosing leads to aberrant CNS activity that is behaviorally expressed as abnormal involuntary movements (Cenci and Konradi, 2010). This is compatible with the dependence of the molecular changes in l-DOPA-induced dyskinesia on supersensitive D1 receptor activation. This model implicates predominantly the “direct” output pathway that disinhibits the thalamus to promote motor commands (Fig. 3).
In addition to the dopaminergic system, evidence also suggests a prominent involvement of the serotonergic system in the occurrence of l-DOPA-induced dyskinesias. This idea is based on work showing that l-DOPA-induced dyskinesias are reduced with serotonergic denervation of the raphe nuclei or by treatment with selective serotonergic antagonists (Carta et al., 2007, 2008b; Eskow et al., 2007, 2009; Muñoz et al., 2008). Serotonergic inputs from the raphe to SN, and to a lesser extent to the striatum, the globus pallidus and the subthalamus are most likely involved (Di Matteo et al., 2008). Indeed, increased sprouting of serotonin terminals in the striatum is associated with the presence of l-DOPA-induced dyskinesias (Rylander et al., 2010). These data suggest that treatments that diminish serotonergic tone may be of clinical benefit in the management of l-DOPA-induced dyskinesias in patients with Parkinson's disease. The possibility of nicotinic modulation of this serotonergic system remains to be investigated.
In fact, research is in progress in numerous laboratories to investigate the antidyskinetic potential of drugs directed to various molecular targets linked to the nigrostriatal system, including the glutamatergic, GABAergic, adenosine, cannabinoid, noradrenergic, and other systems (Carta et al., 2008a; Fox et al., 2008; Jenner, 2008a,b; Cao et al., 2010; Lebel et al., 2010; Morin et al., 2010). Work is also being done to investigate the antidyskinetic potential of nicotine and nAChR-directed ligands in view of the extensive nicotinic cholinergic innervation in the nigrostriatal system (Quik et al., 2009).
B. Nicotine Decreases l-DOPA-Induced Dyskinesias in Parkinsonian Animal Models
Studies in several parkinsonian animal models show that nicotine treatment results in significant declines in l-DOPA-induced dyskinesias (Quik et al., 2007a; Bordia et al., 2008, 2010; Huang et al., 2011). Long-term nicotine dosing via several different modes of administration, including drinking water, minipump, and injection, decreased l-DOPA-induced dyskinetic movements in unilateral 6-hydroxydopamine-lesioned mice and rats and also in MPTP-lesioned monkeys, a model that bears many resemblances to Parkinson's disease. Tolerance did not seem to develop, with the effect of nicotine persisting with prolonged treatment time (months) (Quik et al., 2007a; Bordia et al., 2008, 2010; Huang et al., 2011).
An important question that arises is the subtype and location of nAChRs that underlie nicotine's ability to reduce l-DOPA-induced dyskinesias, because this may allow for the development of selective pharmacotherapies with minimal side effects. As summarized in the schematic in Fig. 6, the most relevant current targets seem to be the α6β2* and α4β2* nAChRs. We therefore tested the effect of agonists directed to these subtypes for their antidyskinetic potential in unilateral 6-hydroxydopamine-lesioned rats (Huang et al., 2011). Varenicline, an agonist that interacts with multiple nAChR subtypes (Mihalak et al., 2006; Rollema et al., 2007b), reduced l-DOPA-induced dyskinetic-like movements by 50% in parkinsonian rats. A similar decline was obtained with 5-iodo-A-85380, a nAChR agonist that interacts preferentially with α4β2* and α6β2* nAChRs (see section III.A.1). These latter two nAChR populations may thus be involved in the antidyskinetic effect. The effectiveness of these novel nAChR agonists, and also of nicotine, seemed dependent on the degree of nigrostriatal damage, with reductions in dyskinesias with moderate (∼30%) but not near-complete (∼99%) nigrostriatal lesions (Huang et al., 2011). None of the drugs reduced the antiparkinsonian action of l-DOPA, suggesting that the mechanisms by which nicotine modulates l-DOPA-induced dyskinesias are distinct from those involved in motor functions linked to parkinsonism.
In summary, nicotine and CNS agonists targeting α4β2* and α6β2* nAChR subtypes reduced l-DOPA-induced dyskinesias in a rodent model, the magnitude of the decline depending on the integrity of the dopaminergic system. These data suggest that α4β2* and α6β2* nAChR agonists may be useful for the treatment of l-DOPA-induced dyskinesias in Parkinson's disease.
IX. Conclusion and Future Directions
There is a consensus among epidemiologists that smoking is associated with a reduced incidence of Parkinson's disease. These observations led to the hypothesis that the nicotine in tobacco may play a role in protection against nigrostriatal damage because nicotine is well known to interact with the dopaminergic system. This idea was substantiated by studies showing that nicotine treatment protected against neurotoxin-induced nigrostriatal damage in both rodent and nonhuman primate parkinsonian animal models. Converging evidence thus supports a clinical trial to determine whether nicotine reduces Parkinson's disease progression.
Studies have also been done to investigate short-term effects of nicotine on Parkinson's disease motor symptoms because nicotine is well known to modulate dopaminergic function in short-term studies. The results of this work are less clear with improvement in motor deficits in approximately half of the clinical trials and only in some studies involving parkinsonian animal models. These conflicting results may suggest that nicotine is ineffective or that it possibly attenuates symptoms only under specified experimental conditions that remain to be identified. Further work is necessary to resolve this controversy.
More recent developments indicate that nicotine administration may be useful as an adjunct therapy to minimize l-DOPA-induced dyskinesias, a troubling side effect of l-DOPA therapy. Evidence for this stems from animal studies showing that nicotine reduces l-DOPA-induced dyskinesias in several parkinsonian animal models including rats, mice, and monkeys. These experimental observations suggest that a clinical trial testing the effect of nicotine may be warranted. Several nicotine formulations are currently available as smoking cessation aids, including the nicotine patch, gum, lozenge, nasal spray, and nasal inhalant. One or more of these applications may have potential for reducing l-DOPA-induced dyskinesias in Parkinson's disease. The beneficial effects also seen with varenicline, a synthetic drug licensed for use in smoking cessation, against l-DOPA-induced dyskinesias in animal studies encourages the view that there is a therapeutic niche for new nicotinic ligands.
An outstanding gap at the present time is whether nicotine exerts its beneficial effects in Parkinson's disease by acting at select nAChR subtypes and whether the same subtypes mediate the neuroprotective and the antidyskinetic effects of nicotine. This knowledge is important as it may lead to directed therapies with optimal therapeutic benefits and minimal side effects. Initial studies suggest that drugs directed to α4β2* and α6β2* nAChR populations present on nigrostriatal dopamine terminal neurons, and notably on their afferents projecting to the striatum, may be most relevant both for symptomatic therapy and long-term protection against nigrostriatal damage. Further studies to identify the most relevant nAChR subtypes, and their signaling pathways may reveal novel drug targets to combat Parkinson's disease.
Another gap in current knowledge relates to the most effective nAChR drug formulation for Parkinson's disease management. nAChR agonists are well known to initiate a rapid receptor desensitization with a consequent functional blockade. The question that arises is whether nAChR agonists exert their beneficial effects via receptor activation, or by blocking receptor mediated activity, in which case antagonists may be more useful from a clinical standpoint. Alternatively, partial nAChR agonists (such as varenicline) may be more effective therapeutically. Continued research is required to address these possibilities.
Although not a focus of the current review, nonmotor symptoms are increasingly recognized as a significant problem in patients with Parkinson's disease (Hawkes, 2008; O'Sullivan et al., 2008; Schapira, 2009; Obeso et al., 2010). These include olfactory dysfunction, sleep disorders, cognitive declines, depression, and pain. To our knowledge, studies have not yet been done to evaluate whether nicotine may relieve such symptoms in Parkinsonian animal models or in patients with Parkinson's disease. However, nAChR drugs have been reported to modulate neuronal circuits involved in some of these deficits to facilitate cognitive performance, reduce pain, and alleviate depression (Bacher et al., 2009; Buckingham et al., 2009; McIntosh et al., 2009; Poorthuis et al., 2009; Sarter et al., 2009; Changeux, 2010a; Mineur and Picciotto, 2010; Philip et al., 2010). Thus, the use of nAChR drugs may not only protect against nigrostriatal damage and reduce l-DOPA-induced dyskinesias but also prove beneficial against nonmotor symptoms associated with Parkinson's disease. Future work is necessary to address this gap.
Acknowledgments
This work was supported by National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants NS42091, NS59910, NS65851]; the Michael J. Fox Foundation; the California Tobacco Related Disease Research Program [Grant 17RT-0119] (to M.Q.); and the Parkinson's Disease Society (to S.W.). We thank Sharon Grady (University of Colorado, Boulder, CO), and Stephanie Cragg (University of Oxford, Oxford, UK), for helpful comments concerning the manuscript.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Quik and Wonnacott.
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.003269.
- 5-iodo-A-85380
- 3-[(2S)-2-azetidinylmethoxy]-5-iodopyridine dihydrochloride
- ABT-089
- pozanicline
- ACh
- acetylcholine
- AR-R17779
- (2S)-2′H-spiro[4-azabicyclo[2.2.2]octane-2,5′-[1,3]oxazolidin]-2′-one
- CNS
- central nervous system
- DARPP-32
- 32-kDa dopamine- and cAMP-regulated phosphoprotein
- GTS-21
- 3-[(3E)-3-[(2,4-dimethoxyphenyl)methylidene]-5,6-dihydro-4H-pyridin-2-yl]pyridine
- MPTP
- 6-hydroxydopamine or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- nAChR
- nicotinic acetylcholine receptor
- NMDA
- N-methyl-d-aspartate
- P450
- cytochrome P450
- PNU-282987
- N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-4-chlorobenzamide
- PP-1
- protein phosphatase 1
- PPT
- pedunculopontine nucleus
- SN
- substantia nigra
- SSR180711
- (4-bromophenyl)-1,4-diazabicyclo[3.2.2]nonane-4-carboxylate
- VTA
- ventral tegmental area.
References
- Ahlskog JE, Muenter MD. (2001) Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 16:448–458 [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12:366–375 [DOI] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. (1997) Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci 9:2734–2742 [DOI] [PubMed] [Google Scholar]
- Allam MF, Campbell MJ, Hofman A, Del Castillo AS, Fernández-Crehuet Navajas R. (2004) Smoking and Parkinson's disease: systematic review of prospective studies. Mov Disord 19:614–621 [DOI] [PubMed] [Google Scholar]
- Alves G, Kurz M, Lie SA, Larsen JP. (2004) Cigarette smoking in Parkinson's disease: influence on disease progression. Mov Disord 19:1087–1092 [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Malysz J, Grønlien JH, El Kouhen R, Håkerud M, Wetterstrand C, Briggs CA, Gopalakrishnan M. (2009) Stimulation of dopamine release by nicotinic acetylcholine receptor ligands in rat brain slices correlates with the profile of high, but not low, sensitivity alpha4beta2 subunit combination. Biochem Pharmacol 78:844–851 [DOI] [PubMed] [Google Scholar]
- Araujo DM, Lapchak PA, Robitaille Y, Gauthier S, Quirion R. (1988) Differential alteration of various cholinergic markers in cortical and subcortical regions of human brain in Alzheimer's disease. J Neurochem 50:1914–1923 [DOI] [PubMed] [Google Scholar]
- Aubert I, Araujo DM, Cécyre D, Robitaille Y, Gauthier S, Quirion R. (1992) Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer's and Parkinson's diseases. J Neurochem 58:529–541 [DOI] [PubMed] [Google Scholar]
- Avshalumov MV, Patel JC, Rice ME. (2008) AMPA receptor-dependent H2O2 generation in striatal medium spiny neurons but not dopamine axons: one source of a retrograde signal that can inhibit dopamine release. J Neurophysiol 100:1590–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, Chen Y, Leslie FM. (2007) Developmental regulation of nicotinic acetylcholine receptors within midbrain dopamine neurons. Neuroscience 144:1347–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, Winzer-Serhan U, Leslie FM. (2003) Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience 119:965–977 [DOI] [PubMed] [Google Scholar]
- Azam L, Winzer-Serhan UH, Chen Y, Leslie FM. (2002) Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol 444:260–274 [DOI] [PubMed] [Google Scholar]
- Bacher I, Wu B, Shytle DR, George TP. (2009) Mecamylamine - a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opin Pharmacother 10:2709–2721 [DOI] [PubMed] [Google Scholar]
- Barik J, Wonnacott S. (2009) Molecular and cellular mechanisms of action of nicotine in the CNS. Handb Exp Pharmacol 192:173–207 [DOI] [PubMed] [Google Scholar]
- Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ, Fisone G, Nestler EJ, Greengard P. (2010) Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc Natl Acad Sci USA 107:14845–14850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Mata IF, Zabetian CP. (2010) The genetics of Parkinson disease. J Geriatr Psychiatry Neurol 23:228–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluardo N, Mudò G, Blum M, Fuxe K. (2000) Central nicotinic receptors, neurotrophic factors and neuroprotection. Behav Brain Res 113:21–34 [DOI] [PubMed] [Google Scholar]
- Benabid AL, Chabardes S, Mitrofanis J, Pollak P. (2009) Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson's disease. Lancet Neurol 8:67–81 [DOI] [PubMed] [Google Scholar]
- Bencherif M. (2009) Neuronal nicotinic receptors as novel targets for inflammation and neuroprotection: mechanistic considerations and clinical relevance. Acta Pharmacol Sin 30:702–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencherif M, Bane AJ, Miller CH, Dull GM, Gatto GJ. (2000) TC-2559: a novel orally active ligand selective at neuronal acetylcholine receptors. European journal of pharmacology 409:45–55 [DOI] [PubMed] [Google Scholar]
- Bencherif M, Lovette ME, Fowler KW, Arrington S, Reeves L, Caldwell WS, Lippiello PM. (1996) RJR-2403: a nicotinic agonist with CNS selectivity I. In vitro characterization. J Pharmacol Exp Ther 279:1413–1421 [PubMed] [Google Scholar]
- Benowitz NL. (2010) Nicotine addiction. N Engl J Med 362:2295–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ, Anderson JM. (1988) Evidence that tobacco smoking increases the density of (-)-[3H]nicotine binding sites in human brain. J Neurochem 50:1243–1247 [DOI] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. (2007) Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol 74:1155–1163 [DOI] [PubMed] [Google Scholar]
- Björklund A, Dunnett SB. (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci 30:194–202 [DOI] [PubMed] [Google Scholar]
- Bohr IJ, Ray MA, McIntosh JM, Chalon S, Guilloteau D, McKeith IG, Perry RH, Clementi F, Perry EK, Court JA, et al. (2005) Cholinergic nicotinic receptor involvement in movement disorders associated with Lewy body diseases. An autoradiography study using [125I]alpha-conotoxinMII in the striatum and thalamus. Exp Neurol 191:292–300 [DOI] [PubMed] [Google Scholar]
- Bolam JP, Hanley JJ, Booth PA, Bevan MD. (2000) Synaptic organisation of the basal ganglia. J Anat 196:527–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordia T, Campos C, Huang L, Quik M. (2008) Continuous and intermittent nicotine treatment reduces L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesias in a rat model of Parkinson's disease. J Pharmacol Exp Ther 327:239–247 [DOI] [PubMed] [Google Scholar]
- Bordia T, Campos C, McIntosh JM, Quik M. (2010) Nicotinic receptor-mediated reduction in l-DOPA-induced dyskinesias may occur via desensitization. J Pharmacol Exp Ther 333:929–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordia T, Grady SR, McIntosh JM, Quik M. (2007) Nigrostriatal damage preferentially decreases a subpopulation of α6β2* nAChRs in mouse, monkey and Parkinson's disease striatum. Mol Pharmacol 72:52–61 [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U. (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol 249 (Suppl 3):III/1–5 [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211 [DOI] [PubMed] [Google Scholar]
- Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, Leonard S. (1997) Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther 282:7–13 [PubMed] [Google Scholar]
- Brown AM, Deutch AY, Colbran RJ. (2005) Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur J Neurosci 22:247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccafusco JJ, Beach JW, Terry AV., Jr (2009) Desensitization of nicotinic acetylcholine receptors as a strategy for drug development. J Pharmacol Exp Ther 328:364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccafusco JJ, Letchworth SR, Bencherif M, Lippiello PM. (2005) Long-lasting cognitive improvement with nicotinic receptor agonists: mechanisms of pharmacokinetic-pharmacodynamic discordance. Trends Pharmacol Sci 26:352–360 [DOI] [PubMed] [Google Scholar]
- Buckingham SD, Jones AK, Brown LA, Sattelle DB. (2009) Nicotinic acetylcholine receptor signalling: roles in Alzheimer's disease and amyloid neuroprotection. Pharmacol Rev 61:39–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. (2000) Acetylcholine-mediated modulation of striatal function. Trends Neurosci 23:120–126 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, North RA, Bernardi G. (1998) Muscarinic IPSPs in rat striatal cholinergic interneurones. J Physiol 510:421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Di Filippo M. (2008) ACh/dopamine crosstalk in motor control and reward: a crucial role for alpha 6-containing nicotinic receptors? Neuron 60:4–7 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. (2010) Levodopa-induced dyskinesias in patients with Parkinson's disease: filling the bench-to-bedside gap. Lancet Neurol 9:1106–1117 [DOI] [PubMed] [Google Scholar]
- Cao X, Yasuda T, Uthayathas S, Watts RL, Mouradian MM, Mochizuki H, Papa SM. (2010) Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J Neurosci 30:7335–7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Kirik D, Björklund A. (2007) Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 130:1819–1833 [DOI] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Muñoz A, Kirik D, Björklund A. (2008a) Involvement of the serotonin system in l-dopa-induced dyskinesias. Parkinsonism Relat Disord 14 (Suppl 2):S154–S158 [DOI] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Muñoz A, Kirik D, Björklund A. (2008b) Serotonin-dopamine interaction in the induction and maintenance of L-DOPA-induced dyskinesias. Progress in brain research 172:465–478 [DOI] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. (1996) A new alpha-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. J Biol Chem 271:7522–7528 [DOI] [PubMed] [Google Scholar]
- Castagnoli K, Murugesan T. (2004) Tobacco leaf, smoke and smoking, MAO inhibitors, Parkinson's disease and neuroprotection; are there links? Neurotoxicology 25:279–291 [DOI] [PubMed] [Google Scholar]
- Castagnoli KP, Steyn SJ, Petzer JP, Van der Schyf CJ, Castagnoli N., Jr (2001) Neuroprotection in the MPTP Parkinsonian C57BL/6 mouse model by a compound isolated from tobacco. Chem Res Toxicol 14:523–527 [DOI] [PubMed] [Google Scholar]
- Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. (2004) Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41:907–914 [DOI] [PubMed] [Google Scholar]
- Cenci MA, Konradi C. (2010) Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res 183:209–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. (2007) Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson's disease in rats and mice. Curr Protoc Neurosci Chapter 9:Unit 9.25 [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Léna C, Clementi F, Moretti M, Rossi FM, Le Novère N, et al. (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 23:7820–7829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux JP. (2002) Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci 22:1208–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP. (2010a) Allosteric receptors: from electric organ to cognition. Annu Rev Pharmacol Toxicol 50:1–38 [DOI] [PubMed] [Google Scholar]
- Changeux JP. (2010b) Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci 11:389–401 [DOI] [PubMed] [Google Scholar]
- Chen H, Huang X, Guo X, Mailman RB, Park Y, Kamel F, Umbach DM, Xu Q, Hollenbeck A, Schatzkin A, et al. (2010) Smoking duration, intensity, and risk of Parkinson disease. Neurology 74:878–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SB, Amici SA, Ren XQ, McKay SB, Treuil MW, Lindstrom JM, Rao J, Anand R. (2009) Presynaptic targeting of alpha4beta 2 nicotinic acetylcholine receptors is regulated by neurexin-1beta. J Biol Chem 284:23251–23259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. (1981) Dendritic release of dopamine in the substantia nigra. Nature 289:537–542 [DOI] [PubMed] [Google Scholar]
- Chergui K, Charléty PJ, Akaoka H, Saunier CF, Brunet JL, Buda M, Svensson TH, Chouvet G. (1993) Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur J Neurosci 5:137–144 [DOI] [PubMed] [Google Scholar]
- Clarke PB, Hommer DW, Pert A, Skirboll LR. (1985) Electrophysiological actions of nicotine on substantia nigra single units. Br J Pharmacol 85:827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PB, Hommer DW, Pert A, Skirboll LR. (1987) Innervation of substantia nigra neurons by cholinergic afferents from pedunculopontine nucleus in the rat: neuroanatomical and electrophysiological evidence. Neuroscience 23:1011–1019 [DOI] [PubMed] [Google Scholar]
- Clarke PB, Pert A. (1985) Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain research 348:355–358 [DOI] [PubMed] [Google Scholar]
- Clemens P, Baron JA, Coffey D, Reeves A. (1995) The short-term effect of nicotine chewing gum in patients with Parkinson's disease. Psychopharmacology (Berl) 117:253–256 [DOI] [PubMed] [Google Scholar]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, et al. (2005) Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 48:3474–3477 [DOI] [PubMed] [Google Scholar]
- Conn PJ, Jones CK, Lindsley CW. (2009) Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci 30:148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corringer PJ, Le Novère N, Changeux JP. (2000) Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol 40:431–458 [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Sallette J, Changeux JP. (2006) Nicotine enhances intracellular nicotinic receptor maturation: a novel mechanism of neural plasticity? J Physiol Paris 99:162–171 [DOI] [PubMed] [Google Scholar]
- Cosford ND, Bleicher L, Herbaut A, McCallum JS, Vernier JM, Dawson H, Whitten JP, Adams P, Chavez-Noriega L, Correa LD, et al. (1996) (S)-(-)-5-ethynyl-3-(1-methyl-2-pyrrolidinyl)pyridine maleate (SIB-1508Y): a novel anti-parkinsonian agent with selectivity for neuronal nicotinic acetylcholine receptors. J Med Chem 39:3235–3237 [DOI] [PubMed] [Google Scholar]
- Costa G, Abin-Carriquiry JA, Dajas F. (2001) Nicotine prevents striatal dopamine loss produced by 6-hydroxydopamine lesion in the substantia nigra. Brain research 888:336–342 [DOI] [PubMed] [Google Scholar]
- Court JA, Piggott MA, Lloyd S, Cookson N, Ballard CG, McKeith IG, Perry RH, Perry EK. (2000) Nicotine binding in human striatum: elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson's disease and Alzheimer's disease and in relation to neuroleptic medication. Neuroscience 98:79–87 [DOI] [PubMed] [Google Scholar]
- Cragg SJ. (2003) Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J Neurosci 23:4378–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ. (2006) Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci 29:125–131 [DOI] [PubMed] [Google Scholar]
- Cucchiaro G, Xiao Y, Gonzalez-Sulser A, Kellar KJ. (2008) Analgesic effects of Sazetidine-A, a new nicotinic cholinergic drug. Anesthesiology 109:512–519 [DOI] [PubMed] [Google Scholar]
- Cui C, Booker TK, Allen RS, Grady SR, Whiteaker P, Marks MJ, Salminen O, Tritto T, Butt CM, Allen WR, et al. (2003) The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci 23:11045–11053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curzon G. (1977) The biochemistry of the basal ganglia and Parkinson's disease. Postgrad Med J 53:719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. (2004) Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 25:317–324 [DOI] [PubMed] [Google Scholar]
- Dani JA, Radcliffe KA, Pidoplichko VI. (2000) Variations in desensitization of nicotinic acetylcholine receptors from hippocampus and midbrain dopamine areas. Eur J Pharmacol 393:31–38 [DOI] [PubMed] [Google Scholar]
- de la Fuente-Fernández R, Stoessl AJ. (2002) The placebo effect in Parkinson's disease. Trends Neurosci 25:302–306 [DOI] [PubMed] [Google Scholar]
- Descarries L, Gisiger V, Steriade M. (1997) Diffuse transmission by acetylcholine in the CNS. Prog Neurobiol 53:603–625 [DOI] [PubMed] [Google Scholar]
- Di Matteo V, Pierucci M, Esposito E, Crescimanno G, Benigno A, Di Giovanni G. (2008) Serotonin modulation of the basal ganglia circuitry: therapeutic implication for Parkinson's disease and other motor disorders. Prog Brain Res 172:423–463 [DOI] [PubMed] [Google Scholar]
- Domino EF, Ni L, Zhang H. (1999) Nicotine Alone and in Combination with l-DOPA methyl ester or the D(2) agonist N-0923 in MPTP-induced chronic hemiparkinsonian monkeys. Exp Neurol 158:414–421 [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Puttfarcken PS, Kuntzweiler TA, Briggs CA, Anderson DJ, Campbell JE, Piattoni-Kaplan M, McKenna DG, Wasicak JT, Holladay MW, et al. (1998) ABT-594 [(R)-5-(2-azetidinylmethoxy)-2-chloropyridine]: a novel, orally effective analgesic acting via neuronal nicotinic acetylcholine receptors: I. In vitro characterization. J Pharmacol Exp Ther 285:777–786 [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Xue IC, Arneric SP, Sullivan JP. (1996) In vitro neuroprotective properties of the novel cholinergic channel activator (ChCA), ABT-418. Brain research 719:36–44 [DOI] [PubMed] [Google Scholar]
- Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A, Yoshikami D, Lindstrom JM, McIntosh JM. (2003) Alpha-conotoxin PIA is selective for alpha6 subunit-containing nicotinic acetylcholine receptors. J Neurosci 23:8445–8452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, McIntosh JM, Marks MJ, Miwa JM, Lester HA. (2010) Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J Neurosci 30:9877–9889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, et al. (2008) In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha 6 nicotinic acetylcholine receptors. Neuron 60:123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driver JA, Kurth T, Buring JE, Gaziano JM, Logroscino G. (2008) Parkinson disease and risk of mortality: a prospective comorbidity-matched cohort study. Neurology 70:1423–1430 [DOI] [PubMed] [Google Scholar]
- Dubois B, Pilon B, Lhermitte F, Agid Y. (1990) Cholinergic deficiency and frontal dysfunction in Parkinson's disease. Ann Neurol 28:117–121 [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Bardo MT. (2009) Targeting nicotinic receptor antagonists as novel pharmacotherapies for tobacco dependence and relapse. Neuropsychopharmacology 34:244–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersbach G, Stöck M, Müller J, Wenning G, Wissel J, Poewe W. (1999) Worsening of motor performance in patients with Parkinson's disease following transdermal nicotine administration. Mov Disord 14:1011–1013 [DOI] [PubMed] [Google Scholar]
- Elbaz A, Moisan F. (2008) Update in the epidemiology of Parkinson's disease. Curr Opin Neurol 21:454–460 [DOI] [PubMed] [Google Scholar]
- Eskow KL, Dupre KB, Barnum CJ, Dickinson SO, Park JY, Bishop C. (2009) The role of the dorsal raphe nucleus in the development, expression, and treatment of L-dopa-induced dyskinesia in hemiparkinsonian rats. Synapse 63:610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskow KL, Gupta V, Alam S, Park JY, Bishop C. (2007) The partial 5-HT(1A) agonist buspirone reduces the expression and development of l-DOPA-induced dyskinesia in rats and improves l-DOPA efficacy. Pharmacol Biochem Behav 87:306–314 [DOI] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ. (2008) alpha6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology 33:2158–2166 [DOI] [PubMed] [Google Scholar]
- Exley R, Cragg SJ. (2008) Presynaptic nicotinic receptors: a dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. Br J Pharmacol 153 (Suppl 1):S283–S297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerström KO, Pomerleau O, Giordani B, Stelson F. (1994) Nicotine may relieve symptoms of Parkinson's disease. Psychopharmacology (Berl) 116:117–119 [DOI] [PubMed] [Google Scholar]
- Fahn S. (2008) How do you treat motor complications in Parkinson's disease: Medicine, surgery, or both? Ann Neurol 64:S56–S64 [DOI] [PubMed] [Google Scholar]
- Feng LR, Maguire-Zeiss KA. (2010) Gene therapy in Parkinson's disease: rationale and current status. CNS Drugs 24:177–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. (2003) Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci 6:968–973 [DOI] [PubMed] [Google Scholar]
- Flores-Hernández J, Cepeda C, Hernández-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. (2002) Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol 88:3010–3020 [DOI] [PubMed] [Google Scholar]
- Forster GL, Blaha CD. (2003) Pedunculopontine tegmental stimulation evokes striatal dopamine efflux by activation of acetylcholine and glutamate receptors in the midbrain and pons of the rat. Eur J Neurosci 17:751–762 [DOI] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Wang GJ, Pappas N, Logan J, MacGregor R, Alexoff D, Shea C, Schlyer D, Wolf AP, et al. (1996a) Inhibition of monoamine oxidase B in the brains of smokers. Nature 379:733–736 [DOI] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Wang GJ, Pappas N, Logan J, Shea C, Alexoff D, MacGregor RR, Schlyer DJ, Zezulkova I, et al. (1996b) Brain monoamine oxidase A inhibition in cigarette smokers. Proc Natl Acad Sci USA 93:14065–14069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox SH, Chuang R, Brotchie JM. (2008) Parkinson's disease–opportunities for novel therapeutics to reduce the problems of levodopa therapy. Prog Brain Res 172:479–494 [DOI] [PubMed] [Google Scholar]
- Fucile S. (2004) Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 35:1–8 [DOI] [PubMed] [Google Scholar]
- Futami T, Takakusaki K, Kitai ST. (1995) Glutamatergic and cholinergic inputs from the pedunculopontine tegmental nucleus to dopamine neurons in the substantia nigra pars compacta. Neurosci Res 21:331–342 [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. (1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250:1429–1432 [DOI] [PubMed] [Google Scholar]
- Goetz CG, Wuu J, McDermott MP, Adler CH, Fahn S, Freed CR, Hauser RA, Olanow WC, Shoulson I, Tandon PK, et al. (2008) Placebo response in Parkinson's disease: comparisons among 11 trials covering medical and surgical interventions. Mov Disord 23:690–699 [DOI] [PubMed] [Google Scholar]
- Gonzales D, Rennard SI, Nides M, Oncken C, Azoulay S, Billing CB, Watsky EJ, Gong J, Williams KE, Reeves KR, et al. (2006) Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. Jama 296:47–55 [DOI] [PubMed] [Google Scholar]
- Gorell JM, Rybicki BA, Johnson CC, Peterson EL. (1999) Smoking and Parkinson's disease: a dose-response relationship. Neurology 52:115–119 [DOI] [PubMed] [Google Scholar]
- Goto Y, Otani S, Grace AA. (2007) The Yin and Yang of dopamine release: a new perspective. Neuropharmacology 53:583–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Guiducci S, Tedesco V, Corbioli S, Zanetti L, Moretti M, Zanardi A, Rimondini R, Mugnaini M, Clementi F, et al. (2010) Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci 30:5311–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Bohr I, Ziabreva I, Vailati S, Longhi R, Riganti L, Gaimarri A, McKeith IG, Perry RH, et al. (2006a) Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer's disease, Parkinson's disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol Dis 23:481–489 [DOI] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Clementi F, Riganti L, McIntosh JM, Collins AC, Marks MJ, Whiteaker P. (2005) Expression of nigrostriatal alpha 6-containing nicotinic acetylcholine receptors is selectively reduced, but not eliminated, by beta 3 subunit gene deletion. Mol Pharmacol 67:2007–2015 [DOI] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M. (2007) Heterogeneity and complexity of native brain nicotinic receptors. Biochem Pharmacol 74:1102–1111 [DOI] [PubMed] [Google Scholar]
- Gotti C, Riganti L, Vailati S, Clementi F. (2006b) Brain neuronal nicotinic receptors as new targets for drug discovery. Curr Pharm Des 12:407–428 [DOI] [PubMed] [Google Scholar]
- Govind AP, Vezina P, Green WN. (2009) Nicotine-induced upregulation of nicotinic receptors: underlying mechanisms and relevance to nicotine addiction. Biochem Pharmacol 78:756–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1983a) Intracellular and extracellular electrophysiology of nigral dopaminergic neurons–1. Identification and characterization. Neuroscience 10:301–315 [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1983b) Intracellular and extracellular electrophysiology of nigral dopaminergic neurons–2. Action potential generating mechanisms and morphological correlates. Neuroscience 10:317–331 [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1983c) Intracellular and extracellular electrophysiology of nigral dopaminergic neurons–3. Evidence for electrotonic coupling. Neuroscience 10:333–348 [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1984a) The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci 4:2877–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1984b) The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci 4:2866–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Floresco SB, Goto Y, Lodge DJ. (2007) Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci 30:220–227 [DOI] [PubMed] [Google Scholar]
- Grace AA, Onn SP. (1989) Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci 9:3463–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Drenan RM, Breining SR, Yohannes D, Wageman CR, Fedorov NB, McKinney S, Whiteaker P, Bencherif M, Lester HA, et al. (2010a) Structural differences determine the relative selectivity of nicotinic compounds for native alpha 4 beta 2*-, alpha 6 beta 2*-, alpha 3 beta 4*- and alpha 7-nicotine acetylcholine receptors. Neuropharmacology 58:1054–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ. (2007) The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol 74:1235–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Salminen O, McIntosh JM, Marks MJ, Collins AC. (2010b) Mouse striatal dopamine nerve terminals express alpha4alpha5beta2 and two stoichiometric forms of alpha4beta2*-nicotinic acetylcholine receptors. J Mol Neurosci 40:91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Ohta K, Roffler-Tarlov S. (1990) Patterns of cell and fiber vulnerability in the mesostriatal system of the mutant mouse weaver. I. Gradients and compartments. J Neurosci 10:720–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenhoff J, Aston-Jones G, Svensson TH. (1986) Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand 128:351–358 [DOI] [PubMed] [Google Scholar]
- Grilli M, Zappettini S, Raiteri L, Marchi M. (2009) Nicotinic and muscarinic cholinergic receptors coexist on GABAergic nerve endings in the mouse striatum and interact in modulating GABA release. Neuropharmacology 56:610–614 [DOI] [PubMed] [Google Scholar]
- Haber SN. (1986) Neurotransmitters in the human and nonhuman primate basal ganglia. Hum Neurobiol 5:159–168 [PubMed] [Google Scholar]
- Hamada M, Hendrick JP, Ryan GR, Kuroiwa M, Higashi H, Tanaka M, Nairn AC, Greengard P, Nishi A. (2005) Nicotine regulates DARPP-32 (dopamine- and cAMP-regulated phosphoprotein of 32 kDa) phosphorylation at multiple sites in neostriatal neurons. J Pharmacol Exp Ther 315:872–878 [DOI] [PubMed] [Google Scholar]
- Hamada M, Higashi H, Nairn AC, Greengard P, Nishi A. (2004) Differential regulation of dopamine D1 and D2 signaling by nicotine in neostriatal neurons. J Neurochem 90:1094–1103 [DOI] [PubMed] [Google Scholar]
- Han ZY, Le Novère N, Zoli M, Hill JA, Jr, Champtiaux N, Changeux JP. (2000) Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur J Neurosci 12:3664–3674 [DOI] [PubMed] [Google Scholar]
- Hanagasi HA, Lees A, Johnson JO, Singleton A, Emre M. (2007) Smoking-responsive juvenile-onset Parkinsonism. Mov Disord 22:115–119 [DOI] [PubMed] [Google Scholar]
- Hansen HH, Timmermann DB, Peters D, Walters C, Damaj MI, Mikkelsen JD. (2007) Alpha-7 nicotinic acetylcholine receptor agonists selectively activate limbic regions of the rat forebrain: an effect similar to antipsychotics. J Neurosci Res 85:1810–1818 [DOI] [PubMed] [Google Scholar]
- Hawkes CH. (2008) The prodromal phase of sporadic Parkinson's disease: does it exist and if so how long is it? Mov Disord 23:1799–1807 [DOI] [PubMed] [Google Scholar]
- Hernán MA, Zhang SM, Rueda-deCastro AM, Colditz GA, Speizer FE, Ascherio A. (2001) Cigarette smoking and the incidence of Parkinson's disease in two prospective studies. Ann Neurol 50:780–786 [DOI] [PubMed] [Google Scholar]
- Hill JA, Jr, Zoli M, Bourgeois JP, Changeux JP. (1993) Immunocytochemical localization of a neuronal nicotinic receptor: the beta 2-subunit. J Neurosci 13:1551–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LZ, Campos C, Ly J, Ivy Carroll F, Quik M. (2011) Nicotinic receptor agonists decrease l-dopa-induced dyskinesias most effectively in moderately lesioned parkinsonian rats. Neuropharmacology 60:861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LZ, Parameswaran N, Bordia T, Michael McIntosh J, Quik M. (2009) Nicotine is neuroprotective when administered before but not after nigrostriatal damage in rats and monkeys. J Neurochem 109:826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A, Miyatake T. (1993) Effects of smoking in patients with early-onset Parkinson's disease. J Neurol Sci 117:28–32 [DOI] [PubMed] [Google Scholar]
- Jankovic J, Stacy M. (2007) Medical management of levodopa-associated motor complications in patients with Parkinson's disease. CNS Drugs 21:677–692 [DOI] [PubMed] [Google Scholar]
- Jenner P. (2008a) Functional models of Parkinson's disease: a valuable tool in the development of novel therapies. Ann Neurol 64:S16–S29 [DOI] [PubMed] [Google Scholar]
- Jenner P. (2008b) Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci 9:665–677 [DOI] [PubMed] [Google Scholar]
- Jones AK, Buckingham SD, Sattelle DB. (2010) Proteins interacting with nicotinic acetylcholine receptors: expanding functional and therapeutic horizons. Trends Pharmacol Sci 31:455–462 [DOI] [PubMed] [Google Scholar]
- Jones IW, Bolam JP, Wonnacott S. (2001) Presynaptic localisation of the nicotinic acetylcholine receptor beta2 subunit immunoreactivity in rat nigrostriatal dopaminergic neurones. J Comp Neurol 439:235–247 [DOI] [PubMed] [Google Scholar]
- Jorenby DE, Hays JT, Rigotti NA, Azoulay S, Watsky EJ, Williams KE, Billing CB, Gong J, Reeves KR, and Varenicline Phase 3 Study Group (2006) Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. Jama 296:56–63 [DOI] [PubMed] [Google Scholar]
- Kaiser S, Wonnacott S. (2000) alpha-bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol 58:312–318 [DOI] [PubMed] [Google Scholar]
- Keath JR, Iacoviello MP, Barrett LE, Mansvelder HD, McGehee DS. (2007) Differential modulation by nicotine of substantia nigra versus ventral tegmental area dopamine neurons. J Neurophysiol 98:3388–3396 [DOI] [PubMed] [Google Scholar]
- Kelton MC, Kahn HJ, Conrath CL, Newhouse PA. (2000) The effects of nicotine on Parkinson's disease. Brain Cogn 43:274–282 [PubMed] [Google Scholar]
- Kitai ST, Shepard PD, Callaway JC, Scroggs R. (1999) Afferent modulation of dopamine neuron firing patterns. Curr Opin Neurobiol 9:690–697 [DOI] [PubMed] [Google Scholar]
- Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP. (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21:1452–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koós T, Tepper JM. (2002) Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci 22:529–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. (2008) Striatal plasticity and basal ganglia circuit function. Neuron 60:543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak JM, McIntosh JM, Quik M. (2002a) Loss of nicotinic receptors in monkey striatum after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment is due to a decline in alpha-conotoxin MII sites. Mol Pharmacol 61:230–238 [DOI] [PubMed] [Google Scholar]
- Kulak JM, McIntosh JM, Yoshikami D, Olivera BM. (2001) Nicotine-evoked transmitter release from synaptosomes: functional association of specific presynaptic acetylcholine receptors and voltage-gated calcium channels. J Neurochem 77:1581–1589 [DOI] [PubMed] [Google Scholar]
- Kulak JM, Nguyen TA, Olivera BM, McIntosh JM. (1997) Alpha-conotoxin MII blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. J Neurosci 17:5263–5270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak JM, Sum J, Musachio JL, McIntosh JM, Quik M. (2002b) 5-Iodo-A-85380 binds to alpha-conotoxin MII-sensitive nicotinic acetylcholine receptors (nAChRs) as well as alpha4beta2* subtypes. J Neurochem 81:403–406 [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Onksen J, Lindstrom J. (2008) Roles of accessory subunits in alpha4beta2(*) nicotinic receptors. Mol Pharmacol 74:132–143 [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. (1989) Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J Neurosci 9:1233–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A, Parameswaran N, Khwaja M, Whiteaker P, Lindstrom JM, Fan H, McIntosh JM, Grady SR, Quik M. (2005) Long-term nicotine treatment decreases striatal alpha6* nicotinic acetylcholine receptor sites and function in mice. Mol Pharmacol 67:1639–1647 [DOI] [PubMed] [Google Scholar]
- Lang AE. (2009) When and how should treatment be started in Parkinson disease? Neurology 72:S39–S43 [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Watson J, Reavill C. (2008) Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther 117:232–243 [DOI] [PubMed] [Google Scholar]
- Lavoie B, Parent A. (1994) Pedunculopontine nucleus in the squirrel monkey: projections to the basal ganglia as revealed by anterograde tract-tracing methods. J Comp Neurol 344:210–231 [DOI] [PubMed] [Google Scholar]
- Le Novère N, Zoli M, Changeux JP. (1996) Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 8:2428–2439 [DOI] [PubMed] [Google Scholar]
- Lebel M, Chagniel L, Bureau G, Cyr M. (2010) Striatal inhibition of PKA prevents levodopa-induced behavioural and molecular changes in the hemiparkinsonian rat. Neurobiol Dis 38:59–67 [DOI] [PubMed] [Google Scholar]
- Lee CR, Tepper JM. (2009) Basal ganglia control of substantia nigra dopaminergic neurons. J Neural Transm Suppl 73:71–90 [DOI] [PubMed] [Google Scholar]
- Lemay S, Chouinard S, Blanchet P, Masson H, Soland V, Beuter A, Bédard MA. (2004) Lack of efficacy of a nicotine transdermal treatment on motor and cognitive deficits in Parkinson's disease. Prog Neuropsychopharmacol Biol Psychiatry 28:31–39 [DOI] [PubMed] [Google Scholar]
- Léna C, Changeux JP. (1993) Allosteric modulations of the nicotinic acetylcholine receptor. Trends Neurosci 16:181–186 [DOI] [PubMed] [Google Scholar]
- Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Banghart MR, Dougherty DA, Goate AM, Wang JC. (2009) Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J 11:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Bettegowda C, Blosser J, Gordon J. (1999) AR-R17779, and alpha7 nicotinic agonist, improves learning and memory in rats. Behav Pharmacol 10:675–680 [DOI] [PubMed] [Google Scholar]
- Lichtensteiger W, Hefti F, Felix D, Huwyler T, Melamed E, Schlumpf M. (1982) Stimulation of nigrostriatal dopamine neurones by nicotine. Neuropharmacology 21:963–968 [DOI] [PubMed] [Google Scholar]
- Lippiello P, Letchworth SR, Gatto GJ, Traina VM, Bencherif M. (2006) Ispronicline: a novel alpha4beta2 nicotinic acetylcholine receptor-selective agonist with cognition-enhancing and neuroprotective properties. J Mol Neurosci 30:19–20 [DOI] [PubMed] [Google Scholar]
- Livingstone PD, Wonnacott S. (2009) Nicotinic acetylcholine receptors and the ascending dopamine pathways. Biochem Pharmacol 78:744–755 [DOI] [PubMed] [Google Scholar]
- Lokwan SJ, Overton PG, Berry MS, Clark D. (1999) Stimulation of the pedunculopontine tegmental nucleus in the rat produces burst firing in A9 dopaminergic neurons. Neuroscience 92:245–254 [DOI] [PubMed] [Google Scholar]
- Macallan DR, Lunt GG, Wonnacott S, Swanson KL, Rapoport H, Albuquerque EX. (1988) Methyllycaconitine and (+)-anatoxin-a differentiate between nicotinic receptors in vertebrate and invertebrate nervous systems. FEBS Lett 226:357–363 [DOI] [PubMed] [Google Scholar]
- Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP, Faure P. (2006) Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron 50:911–921 [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. (2000) Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27:349–357 [DOI] [PubMed] [Google Scholar]
- Mao D, McGehee DS. (2010) Nicotine and behavioral sensitization. J Mol Neurosci 40:154–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. (2002) Direct evidence that release-stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem 80:1071–1078 [DOI] [PubMed] [Google Scholar]
- Marks MJ, Burch JB, Collins AC. (1983) Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther 226:817–825 [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. (1992) Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 12:2765–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Stitzel JA, Romm E, Wehner JM, Collins AC. (1986) Nicotinic binding sites in rat and mouse brain: comparison of acetylcholine, nicotine, and alpha-bungarotoxin. Mol Pharmacol 30:427–436 [PubMed] [Google Scholar]
- Marks MJ, Wageman CR, Grady SR, Gopalakrishnan M, Briggs CA. (2009) Selectivity of ABT-089 for alpha4beta2* and alpha6beta2* nicotinic acetylcholine receptors in brain. Biochem Pharmacol 78:795–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall DL, Redfern PH, Wonnacott S. (1997) Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: comparison of naive and chronic nicotine-treated rats. J Neurochem 68:1511–1519 [DOI] [PubMed] [Google Scholar]
- Martin LF, Kem WR, Freedman R. (2004) Alpha-7 nicotinic receptor agonists: potential new candidates for the treatment of schizophrenia. Psychopharmacology (Berl) 174:54–64 [DOI] [PubMed] [Google Scholar]
- Maskos U. (2008) The cholinergic mesopontine tegmentum is a relatively neglected nicotinic master modulator of the dopaminergic system: relevance to drugs of abuse and pathology. Br J Pharmacol 153 (Suppl 1):S438–S445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos U. (2010) Role of endogenous acetylcholine in the control of the dopaminergic system via nicotinic receptors. J Neurochem 114:641–646 [DOI] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, et al. (2007) Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 190:269–319 [DOI] [PubMed] [Google Scholar]
- Mayeux R. (2003) Epidemiology of neurodegeneration. Annu Rev Neurosci 26:81–104 [DOI] [PubMed] [Google Scholar]
- McCallum SE, Parameswaran N, Bordia T, Fan H, Tyndale RF, Langston JW, McIntosh JM, Quik M. (2006) Increases in alpha4* but not alpha3*/alpha6* nicotinic receptor sites and function in the primate striatum following chronic oral nicotine treatment. J Neurochem 96:1028–1041 [DOI] [PubMed] [Google Scholar]
- McClure-Begley TD, King NM, Collins AC, Stitzel JA, Wehner JM, Butt CM. (2009) Acetylcholine-stimulated [3H]GABA release from mouse brain synaptosomes is modulated by alpha4beta2 and alpha4alpha5beta2 nicotinic receptor subtypes. Mol Pharmacol 75:918–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M. (2009) Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol 78:693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. (2004) Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol 65:944–952 [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Santos AD, Olivera BM. (1999) Conus peptides targeted to specific nicotinic acetylcholine receptor subtypes. Annu Rev Biochem 68:59–88 [DOI] [PubMed] [Google Scholar]
- McKay BE, Placzek AN, Dani JA. (2007) Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 74:1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena-Segovia J, Winn P, Bolam JP. (2008) Cholinergic modulation of midbrain dopaminergic systems. Brain Res Rev 58:265–271 [DOI] [PubMed] [Google Scholar]
- Meshul CK, Kamel D, Moore C, Kay TS, Krentz L. (2002) Nicotine alters striatal glutamate function and decreases the apomorphine-induced contralateral rotations in 6-OHDA-lesioned rats. Exp Neurol 175:257–274 [DOI] [PubMed] [Google Scholar]
- Meyer EL, Yoshikami D, McIntosh JM. (2008) The neuronal nicotinic acetylcholine receptors alpha 4* and alpha 6* differentially modulate dopamine release in mouse striatal slices. J Neurochem 105:1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalak KB, Carroll FI, Luetje CW. (2006) Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol 70:801–805 [DOI] [PubMed] [Google Scholar]
- Miksys S, Tyndale RF. (2006) Nicotine induces brain CYP enzymes: relevance to Parkinson's disease. J Neural Transm Suppl (70):177–180 [DOI] [PubMed] [Google Scholar]
- Millar NS. (2008) RIC-3: a nicotinic acetylcholine receptor chaperone. Br J Pharmacol 153 (Suppl 1):S177–S183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar NS, Gotti C. (2009) Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 56:237–246 [DOI] [PubMed] [Google Scholar]
- Mineur YS, Picciotto MR. (2010) Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis. Trends Pharmacol Sci 31:580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U. (2004) Innervation of the substantia nigra. Cell Tissue Res 318:107–114 [DOI] [PubMed] [Google Scholar]
- Mitsuoka T, Kaseda Y, Yamashita H, Kohriyama T, Kawakami H, Nakamura S, Yamamura Y. (2002) Effects of nicotine chewing gum on UPDRS score and P300 in early-onset parkinsonism. Hiroshima J Med Sci 51:33–39 [PubMed] [Google Scholar]
- Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. (2002) Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther 302:197–204 [DOI] [PubMed] [Google Scholar]
- Morens DM, Grandinetti A, Reed D, White LR, Ross GW. (1995) Cigarette smoking and protection from Parkinson's disease: false association or etiologic clue? Neurology 45:1041–1051 [DOI] [PubMed] [Google Scholar]
- Moretti M, Mugnaini M, Tessari M, Zoli M, Gaimarri A, Manfredi I, Pistillo F, Clementi F, Gotti C. (2010) A comparative study of the effects of the intravenous self-administration or subcutaneous minipump infusion of nicotine on the expression of brain neuronal nicotinic receptor subtypes. Mol Pharmacol 78:287–296 [DOI] [PubMed] [Google Scholar]
- Morin N, Grégoire L, Gomez-Mancilla B, Gasparini F, Di Paolo T. (2010) Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in parkinsonian monkeys. Neuropharmacology 58:981–986 [DOI] [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. (2006) alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol 70:755–768 [DOI] [PubMed] [Google Scholar]
- Morozova N, O'Reilly EJ, Ascherio A. (2008) Variations in gender ratios support the connection between smoking and Parkinson's disease. Mov Disord 23:1414–1419 [DOI] [PubMed] [Google Scholar]
- Mugnaini M, Garzotti M, Sartori I, Pilla M, Repeto P, Heidbreder CA, Tessari M. (2006) Selective down-regulation of [125I]Y(0)-alpha-conotoxin MII binding in rat mesostriatal dopamine pathway following continuous infusion of nicotine. Neuroscience 137:565–572 [DOI] [PubMed] [Google Scholar]
- Mukhin AG, Gündisch D, Horti AG, Koren AO, Tamagnan G, Kimes AS, Chambers J, Vaupel DB, King SL, Picciotto MR, et al. (2000) 5-Iodo-A-85380, an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol Pharmacol 57:642–649 [DOI] [PubMed] [Google Scholar]
- Mulle C, Choquet D, Korn H, Changeux JP. (1992) Calcium influx through nicotinic receptor in rat central neurons: its relevance to cellular regulation. Neuron 8:135–143 [DOI] [PubMed] [Google Scholar]
- Muñoz A, Li Q, Gardoni F, Marcello E, Qin C, Carlsson T, Kirik D, Di Luca M, Björklund A, Bezard E, et al. (2008) Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain 131:3380–3394 [DOI] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. (2003) Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 63:332–341 [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Rasmussen BA, Perry DC. (2003) Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther 307:1090–1097 [DOI] [PubMed] [Google Scholar]
- Nishino N, Kitamura N, Hashimoto T, Tanaka C. (1993) Transmembrane signalling systems in the brain of patients with Parkinson's disease. Rev Neurosci 4:213–222 [DOI] [PubMed] [Google Scholar]
- O'Neill MJ, Murray TK, Lakics V, Visanji NP, Duty S. (2002) The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Targets CNS Neurol Disord 1:399–411 [DOI] [PubMed] [Google Scholar]
- O'Reilly EJ, McCullough ML, Chao A, Henley SJ, Calle EE, Thun MJ, Ascherio A. (2005) Smokeless tobacco use and the risk of Parkinson's disease mortality. Mov Disord 20:1383–1384 [DOI] [PubMed] [Google Scholar]
- O'Sullivan SS, Williams DR, Gallagher DA, Massey LA, Silveira-Moriyama L, Lees AJ. (2008) Nonmotor symptoms as presenting complaints in Parkinson's disease: a clinicopathological study. Mov Disord 23:101–106 [DOI] [PubMed] [Google Scholar]
- Obeso JA, Olanow CW, Nutt JG. (2000) Levodopa motor complications in Parkinson's disease. Trends Neurosci 23:S2–S7 [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G. (2010) Missing pieces in the Parkinson's disease puzzle. Nat Med 16:653–661 [DOI] [PubMed] [Google Scholar]
- Olanow CW. (2004) The scientific basis for the current treatment of Parkinson's disease. Annu Rev Med 55:41–60 [DOI] [PubMed] [Google Scholar]
- Overton PG, Clark D. (1997) Burst firing in midbrain dopaminergic neurons. Brain Res Brain Res Rev 25:312–334 [DOI] [PubMed] [Google Scholar]
- Papke RL, Porter Papke JK. (2002) Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br J Pharmacol 137:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A. (1990) Extrinsic connections of the basal ganglia. Trends Neurosci 13:254–258 [DOI] [PubMed] [Google Scholar]
- Parent A, Sato F, Wu Y, Gauthier J, Lévesque M, Parent M. (2000) Organization of the basal ganglia: the importance of axonal collateralization. Trends Neurosci 23:S20–S27 [DOI] [PubMed] [Google Scholar]
- Parker SL, Fu Y, McAllen K, Luo J, McIntosh JM, Lindstrom JM, Sharp BM. (2004) Up-regulation of brain nicotinic acetylcholine receptors in the rat during long-term self-administration of nicotine: disproportionate increase of the alpha6 subunit. Mol Pharmacol 65:611–622 [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group (2006) Randomized placebo-controlled study of the nicotinic agonist SIB-1508Y in Parkinson disease. Neurology 66:408–410 [DOI] [PubMed] [Google Scholar]
- Pauly JR, Marks MJ, Gross SD, Collins AC. (1991) An autoradiographic analysis of cholinergic receptors in mouse brain after chronic nicotine treatment. J Pharmacol Exp Ther 258:1127–1136 [PubMed] [Google Scholar]
- Perez XA, Bordia T, McIntosh JM, Grady SR, Quik M. (2008) Long-term nicotine treatment differentially regulates striatal alpha6alpha4beta2* and alpha6(nonalpha4)beta2* nAChR expression and function. Mol Pharmacol 74:844–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez XA, O'Leary KT, Parameswaran N, McIntosh JM, Quik M. (2009) Prominent role of alpha3/alpha6beta2* nAChRs in regulating evoked dopamine release in primate putamen; effect of long-term nicotine treatment. Mol Pharmacol 75:938–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Dávila-García MI, Stockmeier CA, Kellar KJ. (1999) Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther 289:1545–1552 [PubMed] [Google Scholar]
- Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, Kellar KJ. (2007) Chronic nicotine differentially regulates alpha6- and beta3-containing nicotinic cholinergic receptors in rat brain. J Pharmacol Exp Ther 322:306–315 [DOI] [PubMed] [Google Scholar]
- Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH. (1995) Alteration in nicotine binding sites in Parkinson's disease, Lewy body dementia and Alzheimer's disease: possible index of early neuropathology. Neuroscience 64:385–395 [DOI] [PubMed] [Google Scholar]
- Pezzoli G, Zini M. (2010) Levodopa in Parkinson's disease: from the past to the future. Expert Opin Pharmacother 11:627–635 [DOI] [PubMed] [Google Scholar]
- Philip NS, Carpenter LL, Tyrka AR, Price LH. (2010) Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology (Berl) 212:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. (2008) It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol 84:329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M. (2008) Neuroprotection via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer's and Parkinson's disease. Front Biosci 13:492–504 [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. (2003) Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci 6:501–506 [DOI] [PubMed] [Google Scholar]
- Placzek AN, Zhang TA, Dani JA. (2009) Age dependent nicotinic influences over dopamine neuron synaptic plasticity. Biochem Pharmacol 78:686–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poewe W. (2009) Treatments for Parkinson disease–past achievements and current clinical needs. Neurology 72:S65–S73 [DOI] [PubMed] [Google Scholar]
- Poisik OV, Shen JX, Jones S, Yakel JL. (2008) Functional alpha7-containing nicotinic acetylcholine receptors localize to cell bodies and proximal dendrites in the rat substantia nigra pars reticulata. J Physiol 586:1365–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis RB, Goriounova NA, Couey JJ, Mansvelder HD. (2009) Nicotinic actions on neuronal networks for cognition: general principles and long-term consequences. Biochem Pharmacol 78:668–676 [DOI] [PubMed] [Google Scholar]
- Pucak ML, Grace AA. (1994) Evidence that systemically administered dopamine antagonists activate dopamine neuron firing primarily by blockade of somatodendritic autoreceptors. J Pharmacol Exp Ther 271:1181–1192 [PubMed] [Google Scholar]
- Pradhan AA, Cumming P, Clarke PB. (2002) [125I]Epibatidine-labelled nicotinic receptors in the extended striatum and cerebral cortex: lack of association with serotonergic afferents. Brain Res 954:227–236 [DOI] [PubMed] [Google Scholar]
- Quarta D, Naylor CG, Barik J, Fernandes C, Wonnacott S, Stolerman IP. (2009) Drug discrimination and neurochemical studies in alpha7 null mutant mice: tests for the role of nicotinic alpha7 receptors in dopamine release. Psychopharmacology (Berl) 203:399–410 [DOI] [PubMed] [Google Scholar]
- Quik M, Bordia T, Forno L, McIntosh JM. (2004) Loss of alpha-conotoxinMII- and A85380-sensitive nicotinic receptors in Parkinson's disease striatum. J Neurochem 88:668–679 [DOI] [PubMed] [Google Scholar]
- Quik M, Campos C, Parameswaran N, Langston JW, McIntosh JM, Yeluashvili M. (2010) Chronic nicotine treatment increases nAChRs and microglial expression in monkey substantia nigra after nigrostriatal damage. J Mol Neurosci 40:105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Chen L, Parameswaran N, Xie X, Langston JW, McCallum SE. (2006a) Chronic oral nicotine normalizes dopaminergic function and synaptic plasticity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. J Neurosci 26:4681–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Cox H, Parameswaran N, O'Leary K, Langston JW, Di Monte D. (2007a) Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Ann Neurol 62:588–596 [DOI] [PubMed] [Google Scholar]
- Quik M, Huang LZ, Parameswaran N, Bordia T, Campos C, Perez XA. (2009) Multiple roles for nicotine in Parkinson's disease. Biochem Pharmacol 78:677–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, McIntosh JM. (2006) Striatal alpha6* nicotinic acetylcholine receptors: potential targets for Parkinson's disease therapy. J Pharmacol Exp Ther 316:481–489 [DOI] [PubMed] [Google Scholar]
- Quik M, O'Neill M, Perez XA. (2007b) Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci 28:229–235 [DOI] [PubMed] [Google Scholar]
- Quik M, Parameswaran N, McCallum SE, Bordia T, Bao S, McCormack A, Kim A, Tyndale RF, Langston JW, Di Monte DA. (2006b) Chronic oral nicotine treatment protects against striatal degeneration in MPTP-treated primates. J Neurochem 98:1866–1875 [DOI] [PubMed] [Google Scholar]
- Quik M, Polonskaya Y, Gillespie A, Jakowec M, Lloyd GK, Langston JW. (2000b) Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol 425:58–69 [DOI] [PubMed] [Google Scholar]
- Quik M, Polonskaya Y, Gillespie A, K Lloyd G, Langston JW. (2000a) Differential alterations in nicotinic receptor alpha6 and beta3 subunit messenger RNAs in monkey substantia nigra after nigrostriatal degeneration. Neuroscience 100: 63–72 [DOI] [PubMed] [Google Scholar]
- Quik M, Polonskaya Y, Kulak JM, McIntosh JM. (2001) Vulnerability of 125I-alpha-conotoxin MII binding sites to nigrostriatal damage in monkey. J Neurosci 21:5494–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Polonskaya Y, McIntosh JM, Kulak JM. (2002) Differential nicotinic receptor expression in monkey basal ganglia: effects of nigrostriatal damage. Neuroscience 112:619–630 [DOI] [PubMed] [Google Scholar]
- Quik M, Sum JD, Whiteaker P, McCallum SE, Marks MJ, Musachio J, McIntosh JM, Collins AC, Grady SR. (2003) Differential declines in striatal nicotinic receptor subtype function after nigrostriatal damage in mice. Mol Pharmacol 63:1169–1179 [DOI] [PubMed] [Google Scholar]
- Quik M, Vailati S, Bordia T, Kulak JM, Fan H, McIntosh JM, Clementi F, Gotti C. (2005) Subunit composition of nicotinic receptors in monkey striatum: effect of treatments with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or L-DOPA. Mol Pharmacol 67:32–41 [DOI] [PubMed] [Google Scholar]
- Rahman S, Zhang J, Corrigall WA. (2004a) Local perfusion of nicotine differentially modulates somatodendritic dopamine release in the rat ventral tegmental area after nicotine preexposure. Neurochem Res 29:1687–1693 [DOI] [PubMed] [Google Scholar]
- Rahman S, Zhang J, Engleman EA, Corrigall WA. (2004b) Neuroadaptive changes in the mesoaccumbens dopamine system after chronic nicotine self-administration: a microdialysis study. Neuroscience 129:415–424 [DOI] [PubMed] [Google Scholar]
- Raisman-Vozari R, Girault JA, Moussaoui S, Feuerstein C, Jenner P, Marsden CD, Agid Y. (1990) Lack of change in striatal DARPP-32 levels following nigrostriatal dopaminergic lesions in animals and in parkinsonian syndromes in man. Brain Res 507:45–50 [DOI] [PubMed] [Google Scholar]
- Reuben M, Clarke PB. (2000) Nicotine-evoked [3H]5-hydroxytryptamine release from rat striatal synaptosomes. Neuropharmacology 39:290–299 [DOI] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, De Biasi M. (2010) The ubiquitin-proteasome system regulates the stability of neuronal nicotinic acetylcholine receptors. J Mol Neurosci 40:177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci 7:583–584 [DOI] [PubMed] [Google Scholar]
- Ritz B, Rhodes SL. (2010) After half a century of research on smoking and PD, where do we go now? Neurology 74:870–871 [DOI] [PubMed] [Google Scholar]
- Ritz B, Ascherio A, Checkoway H, Marder KS, Nelson LM, Rocca WA, Ross GW, Strickland D, Van Den Eeden SK, Gorell J. (2007) Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol 64:990–997 [DOI] [PubMed] [Google Scholar]
- Robertson HA. (1992) Dopamine receptor interactions: some implications for the treatment of Parkinson's disease. Trends Neurosci 15:201–206 [DOI] [PubMed] [Google Scholar]
- Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, et al. (2007a) Pharmacological profile of the alpha4beta2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 52:985–994 [DOI] [PubMed] [Google Scholar]
- Rollema H, Coe JW, Chambers LK, Hurst RS, Stahl SM, Williams KE. (2007b) Rationale, pharmacology and clinical efficacy of partial agonists of alpha4beta2 nACh receptors for smoking cessation. Trends Pharmacol Sci 28:316–325 [DOI] [PubMed] [Google Scholar]
- Ryan RE, Ross SA, Drago J, Loiacono RE. (2001) Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. Br J Pharmacol 132:1650–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylander D, Iderberg H, Li Q, Dekundy A, Zhang J, Li H, Baishen R, Danysz W, Bezard E, Cenci MA. (2010) A mGluR5 antagonist under clinical development improves l-DOPA-induced dyskinesia in parkinsonian rats and monkeys. Neurobiol Dis 39:352–361 [DOI] [PubMed] [Google Scholar]
- Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ. (2005) Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron 46:595–607 [DOI] [PubMed] [Google Scholar]
- Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ, Grady SR. (2007) Pharmacology of alpha-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol 71:1563–1571 [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. (2004) Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 65:1526–1535 [DOI] [PubMed] [Google Scholar]
- Samii A, Nutt JG, Ransom BR. (2004) Parkinson's disease. Lancet 363:1783–1793 [DOI] [PubMed] [Google Scholar]
- Sandor NT, Zelles T, Kiss J, Sershen H, Torocsik A, Lajtha A, Vizi ES. (1991) Effect of nicotine on dopaminergic-cholinergic interaction in the striatum. Brain Res 567:313–316 [DOI] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Hervé D, Greengard P, Fisone G. (2007) Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci 27:6995–7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe WM. (2009) nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms. Biochem Pharmacol 78:658–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH. (2009) Neurobiology and treatment of Parkinson's disease. Trends Pharmacol Sci 30:41–47 [DOI] [PubMed] [Google Scholar]
- Schapira AH, Emre M, Jenner P, Poewe W. (2009) Levodopa in the treatment of Parkinson's disease. Eur J Neurol 16:982–989 [DOI] [PubMed] [Google Scholar]
- Schilström B, Rawal N, Mameli-Engvall M, Nomikos GG, Svensson TH. (2003) Dual effects of nicotine on dopamine neurons mediated by different nicotinic receptor subtypes. Int J Neuropsychopharmacol 6:1–11 [DOI] [PubMed] [Google Scholar]
- Schneider JS, Pope-Coleman A, Van Velson M, Menzaghi F, Lloyd GK. (1998) Effects of SIB-1508Y, a novel neuronal nicotinic acetylcholine receptor agonist, on motor behavior in parkinsonian monkeys. Mov Disord 13:637–642 [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Kellar KJ. (1983) Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science 220:214–216 [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Lehmann J, Kellar KJ. (1984) Presynaptic nicotinic cholinergic receptors labeled by [3H]acetylcholine on catecholamine and serotonin axons in brain. J Neurochem 42:1495–1498 [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. (1993) Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13:596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seipel AT, Yakel JL. (2010) The frequency-dependence of the nicotine-induced inhibition of dopamine is controlled by the alpha7 nicotinic receptor. J Neurochem 114:1659–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples CG, Kaiser S, Soliakov L, Marks MJ, Collins AC, Washburn M, Wright E, Spencer JA, Gallagher T, Whiteaker P, et al. (2000) UB-165: a novel nicotinic agonist with subtype selectivity implicates the alpha4beta2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J Neurosci 20:2783–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simosky JK, Stevens KE, Freedman R. (2002) Nicotinic agonists and psychosis. Curr Drug Targets CNS Neurol Disord 1:149–162 [DOI] [PubMed] [Google Scholar]
- Smith AD, Bolam JP. (1990) The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci 13:259–265 [DOI] [PubMed] [Google Scholar]
- Smith Y, Kieval JZ. (2000) Anatomy of the dopamine system in the basal ganglia. Trends Neurosci 23:S28–S33 [DOI] [PubMed] [Google Scholar]
- Söderman A, Mikkelsen JD, West MJ, Christensen DZ, Jensen MS. (2011) Activation of nicotinic alpha(7) acetylcholine receptor enhances long term potentiation in wild type mice but not in APP(swe)/PS1ΔE9 mice. Neurosci Lett 487:325–329 [DOI] [PubMed] [Google Scholar]
- Soliakov L, Wonnacott S. (1996) Voltage-sensitive Ca2+ channels involved in nicotinic receptor-mediated [3H]dopamine release from rat striatal synaptosomes. J Neurochem 67:163–170 [DOI] [PubMed] [Google Scholar]
- Soto-Otero R, Méndez-Alvarez E, Hermida-Ameijeiras A, López-Real AM, Labandeira-García JL. (2002) Effects of (−)-nicotine and (−)-cotinine on 6-hydroxydopamine-induced oxidative stress and neurotoxicity: relevance for Parkinson's disease. Biochem Pharmacol 64:125–135 [DOI] [PubMed] [Google Scholar]
- Staley JK, Krishnan-Sarin S, Cosgrove KP, Krantzler E, Frohlich E, Perry E, Dubin JA, Estok K, Brenner E, Baldwin RM, et al. (2006) Human tobacco smokers in early abstinence have higher levels of beta2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci 26:8707–8714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JP, Donnelly-Roberts D, Briggs CA, Anderson DJ, Gopalakrishnan M, Xue IC, Piattoni-Kaplan M, Molinari E, Campbell JE, McKenna DG, et al. (1997) ABT-089 [2-methyl-3-(2-(S)-pyrrolidinylmethoxy)pyridine]: I. A potent and selective cholinergic channel modulator with neuroprotective properties. J Pharmacol Exp Ther 283:235–246 [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Jr., Nairn AC, Greengard P. (1995) Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14:385–397 [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. (2004) DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol 44:269–296 [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Raymond LA, Huganir RL. (1999) Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res 33:49–78 [DOI] [PubMed] [Google Scholar]
- Tepper JM, Bolam JP. (2004) Functional diversity and specificity of neostriatal interneurons. Curr Opin Neurobiol 14:685–692 [DOI] [PubMed] [Google Scholar]
- Thacker EL, O'Reilly EJ, Weisskopf MG, Chen H, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. (2007) Temporal relationship between cigarette smoking and risk of Parkinson disease. Neurology 68:764–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth E, Sershen H, Hashim A, Vizi ES, Lajtha A. (1992) Effect of nicotine on extracellular levels of neurotransmitters assessed by microdialysis in various brain regions: role of glutamic acid. Neurochem Res 17:265–271 [DOI] [PubMed] [Google Scholar]
- Tsuang D, Larson EB, Li G, Shofer JB, Montine KS, Thompson ML, Sonnen JA, Crane PK, Leverenz JB, Montine TJ. (2010) Association between lifetime cigarette smoking and lewy body accumulation. Brain Pathol 20:412–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuneki H, Klink R, Léna C, Korn H, Changeux JP. (2000) Calcium mobilization elicited by two types of nicotinic acetylcholine receptors in mouse substantia nigra pars compacta. Eur J Neurosci 12:2475–2485 [DOI] [PubMed] [Google Scholar]
- Tumkosit P, Kuryatov A, Luo J, Lindstrom J. (2006) Beta3 subunits promote expression and nicotine-induced up-regulation of human nicotinic alpha6* nicotinic acetylcholine receptors expressed in transfected cell lines. Mol Pharmacol 70:1358–1368 [DOI] [PubMed] [Google Scholar]
- Ungless MA, Cragg SJ. (2006) A choreography of nicotinic receptors directs the dopamine neuron routine. Neuron 50:815–816 [DOI] [PubMed] [Google Scholar]
- Unwin N. (2003) Structure and action of the nicotinic acetylcholine receptor explored by electron microscopy. FEBS Lett 555:91–95 [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, et al. (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA 102:491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieregge A, Sieberer M, Jacobs H, Hagenah JM, Vieregge P. (2001) Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled study. Neurology 57:1032–1035 [DOI] [PubMed] [Google Scholar]
- Villafane G, Cesaro P, Rialland A, Baloul S, Azimi S, Bourdet C, Le Houezec J, Macquin-Mavier I, Maison P. (2007) Chronic high dose transdermal nicotine in Parkinson's disease: an open trial. Eur J Neurol 14:1313–1316 [DOI] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. (1989) Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol 284:314–335 [DOI] [PubMed] [Google Scholar]
- Walsh H, Govind AP, Mastro R, Hoda JC, Bertrand D, Vallejo Y, Green WN. (2008) Up-regulation of nicotinic receptors by nicotine varies with receptor subtype. J Biol Chem 283:6022–6032 [DOI] [PubMed] [Google Scholar]
- Wanamaker CP, Green WN. (2007) Endoplasmic reticulum chaperones stabilize nicotinic receptor subunits and regulate receptor assembly. J Biol Chem 282:31113–31123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Pickel VM. (2002) Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. J Comp Neurol 442:392–404 [DOI] [PubMed] [Google Scholar]
- Wang HL, Morales M. (2009) Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur J Neurosci 29:340–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sherwood JL, Miles CP, Whiffin G, Lodge D. (2006) TC-2559 excites dopaminergic neurones in the ventral tegmental area by stimulating alpha4beta2-like nicotinic acetylcholine receptors in anaesthetised rats. Br J Pharmacol 147:379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JM, Cockcroft VB, Lunt GG, Smillie FS, Wonnacott S. (1990) Methyllycaconitine: a selective probe for neuronal alpha-bungarotoxin binding sites. FEBS Lett 270:45–48 [DOI] [PubMed] [Google Scholar]
- Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ. (2000) 125I-alpha-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol 57:913–925 [PubMed] [Google Scholar]
- Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC, Marks MJ. (2002) Involvement of the alpha3 subunit in central nicotinic binding populations. J Neurosci 22:2522–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner A, Fuhrer C. (2006) Regulation of nicotinic acetylcholine receptors by tyrosine kinases in the peripheral and central nervous system: same players, different roles. Cell Mol Life Sci 63:2818–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Chang HT, Kitai ST. (1990) Firing patterns and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci 10:508–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Groves PM. (1980) Fine structure and synaptic connections of the common spiny neuron of the rat neostriatum: a study employing intracellular inject of horseradish peroxidase. J Comp Neurol 194:599–615 [DOI] [PubMed] [Google Scholar]
- Windels F, Kiyatkin EA. (2003) Modulatory action of acetylcholine on striatal neurons: microiontophoretic study in awake, unrestrained rats. Eur J Neurosci 17:613–622 [DOI] [PubMed] [Google Scholar]
- Witten IB, Knudsen PF, Knudsen EI. (2010) A dominance hierarchy of auditory spatial cues in barn owls. PLoS One 5:e10396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnacott S. (1997) Presynaptic nicotinic ACh receptors. Trends Neurosci 20:92–98 [DOI] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. (2003) Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 23:3176–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Nashmi R, McKinney S, Cai H, McIntosh JM, Lester HA. (2009) Chronic nicotine selectively enhances alpha4beta2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J Neurosci 29:12428–12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Fan H, Musachio JL, Wei ZL, Chellappan SK, Kozikowski AP, Kellar KJ. (2006) Sazetidine-A, a novel ligand that desensitizes alpha4beta2 nicotinic acetylcholine receptors without activating them. Mol Pharmacol 70:1454–1460 [DOI] [PubMed] [Google Scholar]
- Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. (1999) Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci 2:13–17 [DOI] [PubMed] [Google Scholar]
- Yin R, French ED. (2000) A comparison of the effects of nicotine on dopamine and non-dopamine neurons in the rat ventral tegmental area: an in vitro electrophysiological study. Brain Res Bull 51:507–514 [DOI] [PubMed] [Google Scholar]
- Yu ZJ, Wecker L. (1994) Chronic nicotine administration differentially affects neurotransmitter release from rat striatal slices. J Neurochem 63:186–194 [DOI] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. (2003) Glutamate spillover in the striatum depresses dopaminergic transmission by activating group I metabotropic glutamate receptors. J Neurosci 23:10585–10592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. (2004) Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci 7:581–582 [DOI] [PubMed] [Google Scholar]
- Zhang L, Doyon WM, Clark JJ, Phillips PE, Dani JA. (2009a) Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum. Mol Pharmacol 76:396–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang L, Liang Y, Siapas AG, Zhou FM, Dani JA. (2009b) Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J Neurosci 29:4035–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CJ, Noack C, Brackmann M, Gloveli T, Maelicke A, Heinemann U, Anand R, Braunewell KH. (2009) Neuronal Ca2+ sensor VILIP-1 leads to the upregulation of functional alpha4beta2 nicotinic acetylcholine receptors in hippocampal neurons. Mol Cell Neurosci 40:280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 4:1224–1229 [DOI] [PubMed] [Google Scholar]
- Zhou FM, Wilson CJ, Dani JA. (2002) Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol 53:590–605 [DOI] [PubMed] [Google Scholar]
- Zhou FW, Jin Y, Matta SG, Xu M, Zhou FM. (2009) An ultra-short dopamine pathway regulates basal ganglia output. J Neurosci 29:10424–10435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Lee M, Guan F, Agatsuma S, Scott D, Fabrizio K, Fienberg AA, Hiroi N. (2005) DARPP-32 phosphorylation opposes the behavioral effects of nicotine. Biol Psychiatry 58:981–989 [DOI] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C. (2002) Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 22:8785–8789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Carbone AL, Moroni M, Bermudez I, Mogg AJ, Folly EA, Broad LM, Williams AC, Zhang D, Ding C, et al. (2008) Sazetidine-A is a potent and selective agonist at native and recombinant alpha 4 beta 2 nicotinic acetylcholine receptors. Mol Pharmacol 73:1838–1843 [DOI] [PubMed] [Google Scholar]