

Abstract
Control of the genome-wide type I error rate (GWER) is an important issue in association mapping and linkage mapping experiments. For the latter, different approaches, such as permutation procedures or Bonferroni correction, were proposed. The permutation test, however, cannot account for population structure present in most association mapping populations. This can lead to false positive associations. The Bonferroni correction is applicable, but usually on the conservative side, because correlation of tests cannot be exploited. Therefore, a new approach is proposed, which controls the genome-wide error rate, while accounting for population structure. This approach is based on a simulation procedure that is equally applicable in a linkage and an association-mapping context. Using the parameter settings of three real data sets, it is shown that the procedure provides control of the GWER and the generalized genome-wide type I error rate (GWERk).
Keywords: association mapping, genome-wide type I error rate, linkage mapping, mixed model, Monte Carlo simulation, parametric bootstrap
Introduction
Of central importance for marker-assisted selection is the estimation of positions and effects of quantitative trait loci (QTL). Two of the most commonly used tools for estimating the position of QTL are classical linkage mapping (Lander and Botstein, 1989) and association mapping (Bodmer, 1986; Thornsberry et al., 2001; Yu et al., 2006; Sun et al., 2010). The difference between both methods is that in linkage mapping, there are only a few opportunities for recombination to occur within families and pedigrees with known ancestry. This results in a relatively low mapping resolution (Flint-Garcia et al., 2003). By contrast, for association mapping, historical recombination and natural genetic diversity of the different populations lead to a higher mapping resolution (Ersoz et al., 2008; Zhu et al., 2008).
The resolution of association mapping depends on the structure and degree of linkage disequilibrium across the genome. Linkage disequilibrium caused by population structure and familial relatedness lead to false positive results if not controlled correctly in the statistical analysis (Pritchard et al., 2000; Yu et al., 2006). Genetic and non-genetic factors, like recombination, drift and selection, affect the structure of linkage disequilibrium (Stich et al., 2005). To overcome these problems and to reduce the effect of the population structure, several procedures have been proposed, including the logistic regression ratio test (Q model) (Pritchard et al., 2000; Thornsberry et al., 2001), linear mixed models with effects for subpopulations (Breseghello and Sorrels, 2006) and a unified mixed model approach (QK model) (Yu et al., 2006). In the QK mixed model, Bayesian clustering (Pritchard et al., 2000) is used to estimate probabilities for subpopulation membership (matrix Q), which are used to fit fixed effects, whereas random effects are fitted with covariance proportional to the relative kinship matrix K (Hardy and Vekemans, 2002). Both Q and K account for population structure when scanning for marker trait association (Yu et al., 2006).
One major concern in the context of both linkage and association mapping studies is the statistical power and the control of false positive associations (type I error rate). A false positive association occurs when a significant QTL is declared where none really exists. A genome-wide type I error occurs if at least one false QTL is declared. In both linkage and association mapping, multiple testing needs to be accounted for to control the genome-wide type I error rate (GWER).
Different methods were proposed for linkage mapping to control the GWER. Traditionally, the type I error rate has been controlled by a Bonferroni correction. This correction is conservative and sacrifices statistical power because it cannot exploit the correlation structure among the multiple tests. Several alternative analytical methods have been proposed (Davies, 1977; Lander and Botstein, 1989; Feingold et al., 1993; Rebai et al., 1994; Dupuis and Siegmund, 1999; Piepho, 2001; Li and Ji, 2005) that exploit the correlation structure of multiple tests on the same chromosome.
A further approach to control the GWER commonly used in linkage mapping is the permutation test of Churchill and Doerge (1994) and Doerge and Churchill (1996). This approach depends on no distributional assumption and is characterized by simplicity and applicability to different experimental populations. In this approach, the trait values are permutated relative to the genotypic data. A disadvantage of the permutation test procedure is the computational workload. To compute a critical threshold for a GWER of 0.01, 10 000 permutations of the trait values are necessary, in which for a GWER of 0.05, 1000 permutations are recommended (Churchill and Doerge, 1994).
Although permutation testing is the standard method in linkage mapping, it is not applicable in an association mapping context because permutation would destroy any correlations between trait and population structure (Aulchencko et al., 2007). This would be inappropriate because a valid test must control for any such structure. Furthermore, analytical methods as proposed for linkage mapping are not available for association mapping.
Another error rate that has been used for linkage mapping and association mapping is the false discovery rate (FDR). Loosely speaking, FDR is the ratio of false positives among detections. This approach was proposed by Benjamini and Hochberg (1995) and for genome-wide studies by Storey and Tibshirani (2003). The popularity of the FDR stems from the fact that it leads to more liberal thresholds than the GWER. Chen and Storey (2006), however, have shown that it is difficult to interpret the FDR when applied to genome-wide linkage scans, because the FDR counts multiple true discoveries as being distinct even though they are from the same underlying gene (De Silva and Ball, 2007). As the marker density applied for association mapping studies will dramatically increase in the near future (Donnelly, 2008), the FDR does not seem to be an appropriate error rate concept for association studies. For this reason, we will not consider it further.
Use of the GWER can lead to conservative tests, if there are numerous QTL. Control of the GWER requires that not a single false positive result occurs among all tests, and it may be argued that this requirement is too stringent in the presence of many QTL. Therefore, Chen and Storey (2006) proposed to relax the definition of GWER by allowing a small number k>0 of false positives, the so-called generalized genome-wide k-error rate (GWERk). The usual GWER corresponds to k=0.
In this study a new approach for controlling both GWER and GWERk is proposed. This method, which is based on simulation, is equally applicable in linkage and association mapping. In the simulation procedure, S random samples from the same multivariate normal distribution are generated under the null hypothesis. For each sample, the test statistic is calculated for each QTL. The critical value, which is used as threshold for controlling GWERk, is given by the α-quantile of the simulated distribution of S values of the (k+1)th smallest P-value. The simulation reflects both the population structure and the correlation of tests. The performance of the method is assessed for three different real data sets regarding different GWERk (k=0, 1, 2 and 5).
Materials and methods
Plant materials, phenotypic data and molecular markers
To assess the performance of our method, we used three empirical data sets that were described in detail by Stich et al. (2008) (winter wheat) and by Stich and Melchinger (2009) (sugar beet and rapeseed).
Winter wheat
A total of 303 winter wheat genotypes (Triticum aestivum) developed by KWS Lochow GmbH (Bergen-Wohlde, Germany) was used for this study. The entries were evaluated for grain yield in a series of five breeding trials at four to six locations, with the number of entries per trial ranging from 36 to 110. All 303 inbreds were fingerprinted by KWS Lochow GmbH following standard protocols with 36 simple sequence repeat markers and one single nucleotide polymorphism marker. The 37 marker loci were randomly distributed across 19 of the 21 wheat chromosomes.
Sugar beet
A total of 178 sugar beet inbreds (Beta vulgaris) of the pollen parent heterotic pool of the KWS SAAT AG (Einbeck, Germany) were analyzed. The test-cross progenies of these entries with an inbred of the seed parent heterotic pool were evaluated in a series of plant breeding trials. Data were recorded among others for beet yield. All entries were fingerprinted with 59 simple sequence repeat markers and 41 single nucleotide polymorphism marker, both randomly distributed across the sugar beet genome. The fingerprinting was done by the KWS SAAT AG following standard protocols.
Rapeseed
A total of 136 rapeseed (Brassica napus) inbreds of the Norddeutsche Pflanzenzucht Hans-Georg Lembke KG (Holtsee, Germany) were studied. All entries were evaluated in a series of field trials, in which data were collected for thousand-kernel weight. Furthermore, all entries were fingerprinted with 59 genome-wide distributed simple sequence repeat markers by Saaten-Union Resistenzlabor GmbH (Hovedissen, Germany) following standard protocols.
Statistical analyses
Phenotypic data analyses
In the study of Stich et al. (2008) the empirical type I error rates of association mapping approaches, which were based on adjusted entry means calculated by a two-step analysis, were only slightly higher than that of approaches in which the phenotypic data analysis and the association analysis were performed in one step (one-step analysis) (also see Möhring and Piepho, 2009). We therefore calculated adjusted entry means (winter wheat and rapeseed) or entry means (sugar beet) in the first step (for more details, see Stich et al., 2008; Stich and Melchinger, 2009) for each entry under consideration. These estimates were then used in a second step for the association analyses.
Population structure analyses
For each of the three above mentioned data sets, the kinship matrix K was calculated based on the available marker data using the software package SPAGeDi (Hardy and Vekemans, 2002), in which negative kinship values between entries were set to 0. We used the first p principal components of an allele frequency matrix (PC-matrix) instead of the Q matrix of STRUCTURE (Pritchard et al., 2000), as previous studies suggested that both methods are comparable with respect to adherence to the nominal α level, but the former requires much less computational effort (Yu et al., 2006; Zhao et al., 2007). The explained variance of the first p principal components was about 25% (Stich and Melchinger, 2009).
Method for controlling GWER
To scan the genome for QTL in linkage mapping or association mapping, we use a mixed linear model to represent the phenotypic data. At each putative QTL position/marker, we test the null hypothesis of no QTL effect. Under this hypothesis, the null model for genotype means can be written as
where y′=(y1, y2,…,yG), yi is the mean of the i-th genotype (i=1,…,G), β0 is a vector of fixed effects, X0 is the corresponding design matrix and e is a random residual. In association mapping, X0 might represent the probabilities of subpopulation membership (Q matrix) or PC-matrix of allele frequencies and, possibly, cofactors accounting for major background QTL, whereas e models genetic correlation due to coancestry and identically distributed noise, that is, var(e)=V=2AσA2+Iσ2, where A is the numerator relationship matrix. Alternatively, A could be replaced by the kinship matrix K (Yu et al., 2006), which was done in this study. For the rapeseed data set, e models var(e)=V=Iσ2, because A was similar to I and no changes were visible in the log likelihoods when fitting the full model including A.
To test the null hypothesis at the qth putative position (q=1, 2, …, Z), we augment the null model by
where aq is the vector of fixed genetic effects at the qth putative position and Wq is the associated design matrix. Notably, the dimension of aq may vary among markers, depending on the genetic model and the number of alleles per marker. Furthermore, we need to cater for the possibility that marker information may be missing, especially in association mapping, in which imputation is not straightforward. The approach taken in this study is to simply discard records of individuals with missing information at the qth marker when testing the qth marker, meaning that different subsets of the data will be used for different markers. We therefore add a subscript q also to the data vector y, writing yq. Thus, yq contains all records with complete data at the qth marker. Consequently, the design matrix Wq will have rows only for observations in yq. The marker-specific data vector may be formally defined as follows. Let B be a G × Z indicator matrix of zeros and ones, with rows corresponding to genotypes and columns to markers, reflecting the missing data pattern and let Dq be computed by diag(bq), deleting all rows that have zeros only where bq is the qth column of B. We then have
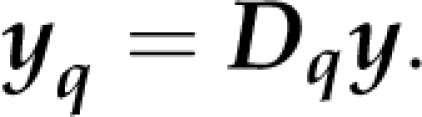 |
Dq selects from y all observations that have complete data for the qth marker. The reduced data vector yq has variance
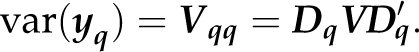 |
The full model can be written compactly as
where Xq=(DqX0,Wq) and β′q=(β′0,α′q). The null hypothesis at the qth position can be stated as
where Hq is a suitable matrix of known constants. The size and form of Hq depend on the putative position q, for example, on the number of marker alleles. Furthermore, the null hypothesis pertains to αq only, that is, Hq=(0q H̃q), where 0q is a null matrix with number of columns corresponding to those of DqX0 and H̃q states the null hypothesis pertaining to αq. For example, when H0 states equality of all additive allele effects at a locus, then H̃q=(In(q), −1n(q)), where n(q) equals the number of marker alleles minus one. Thus,
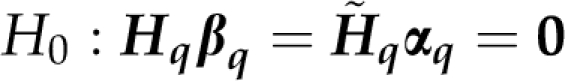 |
When V is known, the Wald statistic
where β̂q=(X′qV−1qqXq)−X′qVqq−1yq, has an exact central χ2-distribution with rank(Hq) degrees of freedom. In practice, V needs to be estimated from the data based on the null model (1). In this case, one may use the Kenward–Roger method to approximate the distribution of Tq. Provided the number of genotypes G is not small, Equation (5) will have an approximate χ2-distribution. We expect the approximation to be very accurate in most practical cases, so long as the number of genotypes is not very small (for example, <50).
Simulation of the joint distribution of T1, T2, …, TZ
It is convenient to re-write Tq as
where 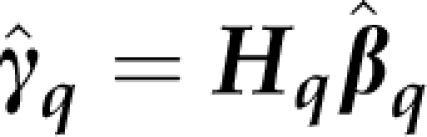 and
and
Under the global null hypothesis the joint distribution of
is multivariate normal with zero mean and variance–covariance matrix
 |
where Mqq′=cov(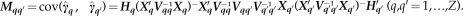 . This result is explained in more detail in the Appendix. Notably, when q=q′, then Equation (9) simplifies to Equation (7).
. This result is explained in more detail in the Appendix. Notably, when q=q′, then Equation (9) simplifies to Equation (7).
For simulating 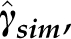 , it is convenient to compute Equation (9), obtain a decomposition
, it is convenient to compute Equation (9), obtain a decomposition 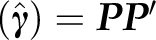 , where the number of columns in P equals the rank r of var (
, where the number of columns in P equals the rank r of var (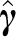 ), store P in memory during iterations, and at each iteration simulate
), store P in memory during iterations, and at each iteration simulate 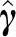 as
as 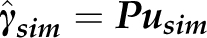 , where usim is a vector of r-independent standard normal deviates. We can use the singular value decomposition
, where usim is a vector of r-independent standard normal deviates. We can use the singular value decomposition
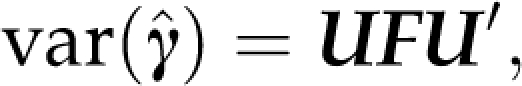 |
where F is a diagonal matrix, first diagonal elements of which are the r non-zero singular values of var (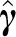 ), whereas the remaining ones are zero. We can then choose
), whereas the remaining ones are zero. We can then choose
P=(U√F)r, where (M)r is given by the first r columns of M.
To compute a critical threshold for the Wald tests controlling the GWER at level α, we may generate S random samples γ̂sim from this same multivariate normal distribution. For each sample, we compute the corresponding test statistics Tq (q=1,…,Z). As test statistics Tq may involve hypotheses with differing degrees of freedom for different q, we convert each Tq to the point-wise P-value pq based on a χ2 distribution with degrees of freedom equal to rank(Hq). Conversion to P-values allows us to use the same rejection region for all QTL (Storey, 2002). Subsequently we determine the minimum of pq across positions (pq(min)). The critical value is given by the α-quantile of the simulated distribution of S values of pmin.
The approach can be extended further using the GWERk approach of Chen and Storey (2006), which defines a genome-wide error to occur when more than k point-wise tests are falsely declared significant. In this more general case, the (k+1)th lowest pq across positions is determined in each simulation run. Notably the ordinary GWER corresponds to k=0.
Simulation study
The performance of the above method is verified by simulation. As the method for determining the threshold is also based on simulation, there are two levels of simulation: (1) an inner simulation that generates the thresholds for a given data set, and (2) an outer simulation that generates data to be analyzed by a mixed model.
The simulation scheme can be described as follows:
Do i=1 to n (n=number of outer loops)
(a) Generate a data set ysim from a multivariate normal distribution with zero mean, using restricted maximum likelihood estimates of V of a real data set.
(b) Determine threshold based on simulation with S runs of the inner loops, using ysim and X0 and Wq (q=1, …, Z) from real data set.
(c) Evaluate significance tests for scan of ith simulated data set ysim and determine the (k+1)th ordered P-value across the positions.
End
Determine the threshold P-value for GWERk=α as the α-quantile of the n(k+1)th ordered P-values.
To start a simulation, we analyze a real data set under the global H0 based on model (1), obtain an estimate of V and then compute its Cholesky decomposition according to V=LL′. In each run of the outer loop, we then simulate data under the global H0 as
where v is a vector of independent standard normal deviates. The same L is used in all iterations of the outer loop, so L needs to be stored throughout the whole simulation.
Results
The proposed method for controlling the GWERk (Chen and Storey, 2006) was tested on three empirical data sets of commercial plant-breeding programs.
The threshold computation and the analysis of the PC-K mixed model were repeated 1000 times, meaning there were 1000 inner simulations and 1000 outer simulations. Notably, for a test to be declared significant, the P-value had to remain below the threshold P-value. At a nominal error rate of 5%, a 95% prediction interval for the observed error rate has lower limit of 3.65% and upper limit of 6.35% when 1000 runs converged. Thus, an observed error rate should not exceed 64 cases or fall below 36 cases of the 1000 simulations if tests control α exactly. For the sugar beet data set only 978 outer simulations converged. The 95% prediction interval therefore has a lower limit of 3.63% and an upper limit of 6.37%. We also computed Bonferroni-adjusted prediction intervals based on the 12 cases studied (Table 1 ). For 1000 runs and for the 978 runs of the sugar beet data set, the Bonferroni adjusted limits are 30 and 70, respectively. The empirical error rates for the GWERk are given in Table 1.
Table 1. Empirical levels in counts and percent of converged cases for the GWERk (k=0, 1, 2, and 5) at nominal level of 5% for the three different data sets and the prediction interval of upper and lower limit.
| Data set | GWERk | Empirical level (absolute count) | Empirical level (in %) | Prediction interval (95%) for absolute rejection frequency (in %) | Bonferroni-adjusted prediction interval (95%) for absolute rejection frequency (in %) |
|---|---|---|---|---|---|
| Wheat | 0 | 60 | 6.0 | 3.649–6.351 | 3.027–6.973 |
| Wheat | 1 | 49 | 4.9 | 3.649–6.351 | 3.027–6.973 |
| Wheat | 2 | 36 | 3.6 | 3.649–6.351 | 3.027–6.973 |
| Wheat | 5 | 29 | 2.9 | 3.649–6.351 | 3.027–6.973 |
| Sugar | 0 | 60 | 6.1 | 3.634–6.366 | 3.006–6.993 |
| Sugar | 1 | 53 | 5.4 | 3.634–6.366 | 3.006–6.993 |
| Sugar | 2 | 46 | 4.7 | 3.634–6.366 | 3.006–6.993 |
| Sugar | 5 | 23 | 2.4 | 3.634–6.366 | 3.006–6.993 |
| Rapeseed | 0 | 63 | 6.3 | 3.649–6.351 | 3.027–6.973 |
| Rapeseed | 1 | 50 | 5.0 | 3.649–6.351 | 3.027–6.973 |
| Rapeseed | 2 | 33 | 3.3 | 3.649–6.351 | 3.027–6.973 |
| Rapeseed | 5 | 27 | 2.7 | 3.649–6.351 | 3.027–6.973 |
The nominal GWER could be maintained for the winter wheat data set of KWS Lochow. For GWERk=0 in 6.0% of simulations, the critical threshold was higher than the P-values of the PC-K mixed models. The threshold for GWERk=0 was 0.00139, which is higher than the Bonferroni-corrected threshold (0.00135). The extension of Chen and Storey (2006) led to further reduction of times the critical threshold was higher than the P-values of the PC-K mixed models. The critical threshold was passed for GWERk=1 in 4.9% of the simulations, for GWERk=2 in 3.6%, and for the GWERk=5 in 2.2% of the simulations (Table 1).
For the sugar beet data set of KWS, the nominal GWER could be kept. In 6.1% of the simulations for GWERk=0, the threshold was higher than the P-values of the PC-K mixed model. The threshold for GWERk=0 was 0.00052 and therefore higher than the threshold corrected by the Bonferroni method (0.00050). Furthermore, for the modified GWERk with k=1, 2 and 5 the nominal rate of 5% could be maintained. In 5.4% of the simulations, a type I error occurred for GWERk=1, in 4.7% of the simulations for the GWERk=2 and in 2.4% of the simulations for the GWERk=5 (Table 1).
Our method could also satisfactorily control the nominal GWER for the third data set. For the rapeseed data set of Norddeutsche Pflanzenzucht, the threshold for the GWERk=0 was higher than the P-values of the PC-K mixed model in 6.3% of the simulations. The threshold for GWERk=0 was 0.000937 and therefore also higher than the Bonferroni-corrected threshold that had the value 0.000847. For the GWERk=1, the empirical error rate was 5.0% for GWERk=2 it was 3.3%. The empirical GWERk=5 was 2.7% (Table 1).
Discussion
Error rates for controlling the multiple testing in linkage and association mapping experiments include the FDR, which was proposed by Benjamini and Hochberg (1995) and Storey and Tibshirani (2003), and the GWER and its extension GWERk, which was proposed by Chen and Storey (2006). For linkage mapping, different approaches were proposed, which control the GWER, like the Bonferroni correction, the permutation procedure (Churchill and Doerge, 1994; Doerge and Churchill, 1996) and several analytical methods for specific population structures (Davies, 1977; Lander and Botstein, 1989; Feingold et al., 1993; Rebai et al., 1994; Dupuis and Siegmund, 1999; Piepho, 2001). Thus, at present there do not seem to be tailor-made methods for controlling GWER for association mapping experiments. This study has proposed a simulation-based approach for controlling the type I error rate, which includes the information of the population structure. The approach is akin to that proposed by Edwards and Berry (1987) in the context of multiple mean comparisons in linear models, and it is also similar in spirit to the method of Zou et al. (2004) in the context of linkage mapping. The simulation approach can also be regarded as a parametric bootstrap procedure (Efron and Tibshirani, 1993). The simulations of the proposed method based on the three commercial plant breeding data sets have shown that the calculated thresholds provide reasonable, slightly conservative control of the genome-wide type I error rate.
An advantage of our proposed method over the permutation procedure of Churchill and Doerge (1994) is that the information of the population structure is accounted for in our threshold computation. The associations between trait and population structure are not destroyed like for the permutations procedure. Aulchenko et al., 2007 proposed an approach, in which residuals from a mixed model fit ignoring markers, but corrected for family effects are used for the permutations test. The method was developed in an animal breeding context for genetically homogeneous populations, but its principles could be applied to the more general setting considered here. Residuals from a mixed model fit will typically display correlation and heteroscedasticity arising from the estimation of model effects, which may affect the performance of the method. Our procedure does not have these limitations, because the null distribution is simulated rather than computed from permutations.
Li and Ji (2005), Seaman and Müller-Myhsok (2005) and Conneely and Boehnke (2007) suggested methods to adjust the P-value regarding the correlation structure of the markers. These approaches are therefore similar to our approach; but they do not account for population structure. Moreover, the approaches of Seaman and Müller-Myhsok (2005) and Conneely and Boehnke (2007) need imputation, if there are missing values in the marker data. The occurrence of missing values can be handled without imputation by our proposed method.
For the three data sets used in this study, the computation time for one approximate threshold was 1 min and 23 s for the rapeseed data set up to 9 min and 20 s for the sugar beet data set (Intel Pentium Dual central processing unit, 2.20 GHZ, 1.95 GB random access memory). The computational time depends on the number of markers and on the number of genotypes. The computational time increases mainly due to the generation of the matrix M, if there are more markers. Furthermore, the computational time is increased by the number of genotypes because mixed model analysis takes longer time. The computational time could be reduced, if necessary, by performing threshold computation separately for each chromosome and using a Bonferroni correction across chromosomes (Piepho, 2001). Moreover, when the number of markers by far exceeds the number of genotypes, it will be computationally more efficient to simulate data y instead of test statistics Tq (Supplementary Information).
Acknowledgments
We thank the breeding companies KWS, KWS Lochow and Norddeutsche Pflanzenzucht for providing the data sets within the GABI BRAIN project. This study was supported by the GABI GAIN project (Grant no FKZ0315072C).
Appendix
Let yq=Dqy and yq′=Dq′y. Then
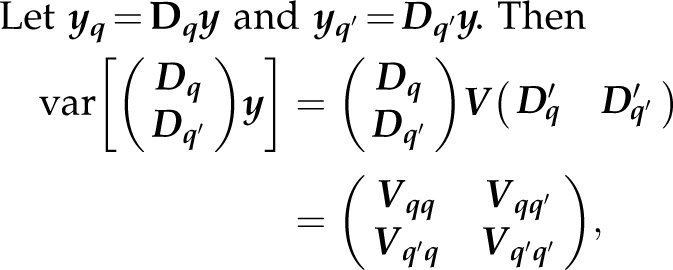 |
where
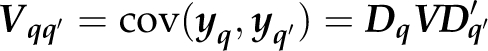 |
Similarly, noting that
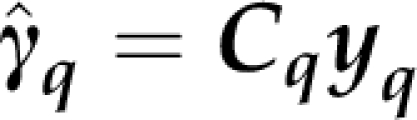 |
with Cq=Hq(X′qV−1qqXq)−X′qV−1qq, we have
and
 |
where Mqq′=CqVqq′Cq′′. Inserting the expression for Cq we find
 |
and
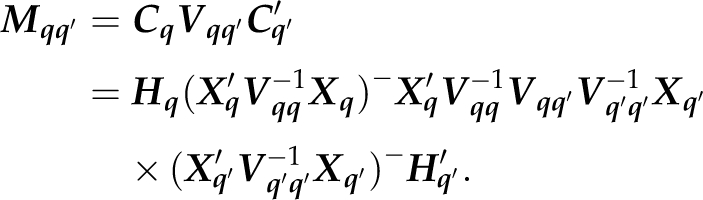 |
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177:577–585. doi: 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;85:289–300. [Google Scholar]
- Bodmer WF. Human genetics: the molecular challenge. Cold spring harbour symp. Quant Biol. 1986;51:1–13. doi: 10.1101/sqb.1986.051.01.003. [DOI] [PubMed] [Google Scholar]
- Breseghello F, Sorrels ME. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics. 2006;172:1165–1177. doi: 10.1534/genetics.105.044586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Storey JD. Relaxed significance criteria for linkage analysis. Genetics. 2006;173:2371–2381. doi: 10.1534/genetics.105.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conneely KN, Boehnke M. So many correlated tests, so little time! Rapid adjustment of P-values for multiple correlated tests. Am J Hum Genet. 2007;81:1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies RB. Hypothesis testing when a nuisance parameter is present only under the alternative. Biometrika. 1977;64:247–254. doi: 10.1111/j.0006-341X.2005.030531.x. [DOI] [PubMed] [Google Scholar]
- De Silva HN, Ball RD.2007Linkage disequilibrium mapping conceptsIn: Oraguzie NC, Rikkerink EHA, Gardiner SE, De Silva HN (eds).Association Mapping in Plants Springer: New York, NY, USA [Google Scholar]
- Doerge RW, Churchill GA. Permutation tests for multiple loci affecting a quantitative character. Genetics. 1996;142:285–294. doi: 10.1093/genetics/142.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–731. doi: 10.1038/nature07631. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Siegmund D. Statistical methods for mapping quantitative trait loci from a dense set of markers. Genetics. 1999;151:373–386. doi: 10.1093/genetics/151.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D, Berry J. The efficiency of simulation-based multiple comparisons. Biometrics. 1987;43:913–928. [PubMed] [Google Scholar]
- Efron B, Tibshirani RJ. An introduction to the bootstrap. Chapman & Hall, London; 1993. [Google Scholar]
- Ersoz ES, Yu J, Buckler ES.2008Applications of linkage disequilibrium and association mapping in maizeIn: Kriz A, Larkins B (eds).Molecular Genetic Approaches to Maize Improvement Springer: Dordrecht, The Netherlands [Google Scholar]
- Feingold EP, Brown PO, Siegmund D. Gaussian models for genetic linkage analysis using complete high-resolution maps of identity by descent. Am J Hum Genet. 1993;53:234–251. [PMC free article] [PubMed] [Google Scholar]
- Flint-Garcia SA, Thornsberry JM, Buckler ES. Structure of linkage disequilibrium in plants. Ann Rev Plant Biol. 2003;54:357–374. doi: 10.1146/annurev.arplant.54.031902.134907. [DOI] [PubMed] [Google Scholar]
- Hardy OJ, Vekemans X. SPAGeDI: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2:618–620. [Google Scholar]
- Lander ES, Botstein D. Mapping Mendelian factors underlying quantitative traits using RFLP markers. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- Möhring J, Piepho HP. Comparison of weighting in two-stage analysis of plant breeding trials. Crop Sci. 2009;49:1977–1988. [Google Scholar]
- Piepho HP. A quick method for computing approximate thresholds for quantitative trait loci detection. Genetics. 2001;157:425–432. doi: 10.1093/genetics/157.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. Am J Hum Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebai A, Goffinet B, Mangin B. Approximate thresholds of interval mapping tests for QTL detection. Genetics. 1994;138:235–240. doi: 10.1093/genetics/138.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman SR, Müller-Myhsok B. Rapid simulation of P-values for product methods and multiple-testing adjustment in association studies. Am J Hum Genet. 2005;76:399–408. doi: 10.1086/428140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich B, Melchinger AE. Comparison of mixed-model approaches for association mapping in rapeseed, potato, sugar beet, maize, and Arabidopsis. BMC Genomics. 2009;10:94. doi: 10.1186/1471-2164-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich B, Melchinger AE, Frisch M, Maurer HP, Heckenberger M, Reif JC. Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theor Appl Genet. 2005;111:723–730. doi: 10.1007/s00122-005-2057-x. [DOI] [PubMed] [Google Scholar]
- Stich B, Möhring J, Piepho HP, Heckenberger M, Buckler ES, Melchinger AE. Comparison of mixed-model approaches for association mapping. Genetics. 2008;178:1745–1754. doi: 10.1534/genetics.107.079707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J R Stat Soc Ser B Stat Methodol. 2002;64:479–498. [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Zhu C, Kramer MH, Yang SS, Song W, Piepho HP, et al. Comparing different R2 statistics for mixed model association mapping. Heredity. 2010;105:333–340. doi: 10.1038/hdy.2010.11. [DOI] [PubMed] [Google Scholar]
- Thornsberry JM, Goodmann MM, Doebley J, Kresovich S, Nielsen D, Buckler ES., IV Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet. 2001;28:286–289. doi: 10.1038/90135. [DOI] [PubMed] [Google Scholar]
- Yu J, Pressoir G, Briggs WH, Vroh BiI, Yamasaki M, Doebley JF, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- Zhao J, Paulo MJ, Jamar D, Lou P, Van Eeuwijk F, Bonnema G, et al. Association mapping of leaf traits, flowering time, and phytate content in Brassica rapa. Genome. 2007;50:963–973. doi: 10.1139/g07-078. [DOI] [PubMed] [Google Scholar]
- Zhu C, Gore M, Buckler ES, Yu J. Status and prospects of association mapping in plants. Plant Genome. 2008;1:5–19. [Google Scholar]
- Zou F, Fine JP, Hu J, Lin DY. An efficient resampling method for assessing genome-wide statistical significance in mapping quantitative trait loci. Genetics. 2004;168:2307–2316. doi: 10.1534/genetics.104.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.